



1 BioLab, Instituto Universitario de Bio-Orgánica "Antonio González" (IUBO-AG), Centro de Investigaciones Biomédicas de Canarias (CIBICAN), Universidad de La Laguna, C/ Astrofísico Francisco Sánchez 2, 38206 La Laguna, Spain.
Abstract
In the scenario of drug discovery, the challenge is to fully understand and elucidate the mechanism of action to identify, with high resolution, the molecular determinant(s) targeted by the drug and responsible for its pharmacological activity. Cancer offers scientists an almost infinite arena of signaling pathways, targets and small molecules for therapeutic intervention. Among the multiple chemotherapeutic strategies to combat cancer, synthetic lethality remains underexplored. Casein kinase 1 ε (CK1ε) is a serine/threonine protein kinase that has been described as a synthetic lethal partner of the Wnt/β-catenin signaling pathway. Despite its potential as a desirable therapeutic target, only two selective inhibitors are available: PF-4800567 and GSD0054. Until the discovery of GSD0054, CK1ε inhibitors have been considered candidate drugs exclusively in psychopharmacology. In this review, we focus on three key points which we consider essential to define the scope of CK1ε as a synthetic lethal partner and its inhibitors as chemotherapeutics: the therapeutic relevance of this kinase, the scarce availability of selective inhibitors (due to the high homology with its sibling isoform CK1δ ), and the constraint of existing computational tools. This paper represents the first review covering the potential of CK1ε as a druggable target for cancer treatment.
Keywords
- Anticancer drugs
- casein kinase 1 epsilon
- selective inhibitors
- synthetic lethality
An ideal pharmacological approach to combat cancer is the development of targeted compounds directed exclusively and specifically to tumor cells, keeping intact healthy cells. That is, to eradicate cancer cells while avoiding collateral damage, which brings in the current concept of synthetic lethality. Tumor cells develop from cancer stem cells, while acquiring genetic alterations. Two genes are regarded synthetic lethal when the presence of loss of function mutations in either gene promotes cell viability, whereas the presence of inactivating mutations in both gene results in cell death [1]. Targeting synthetic lethal partner proteins encoded by mutated cancer genes represents an opportunity to specifically kill cancer cells bearing these loss of function mutations, while sparing healthy cells (Fig. 1). A major hurdle in clinical cancer therapeutics is that most oncogenes are not easily accessible for inhibition by small drug molecules. Applying the concept to anticancer therapy, these undruggable genes could be tackled indirectly by targeting their corresponding synthetic lethal counterpart [2]. The clinical relevance of synthetic lethality is evident, however the approach has not been widely addressed in a comprehensive manner due to the lack of knowledge of the molecular basis, and especially synthetic lethal genes involved in tumor development, progression and metastasis.
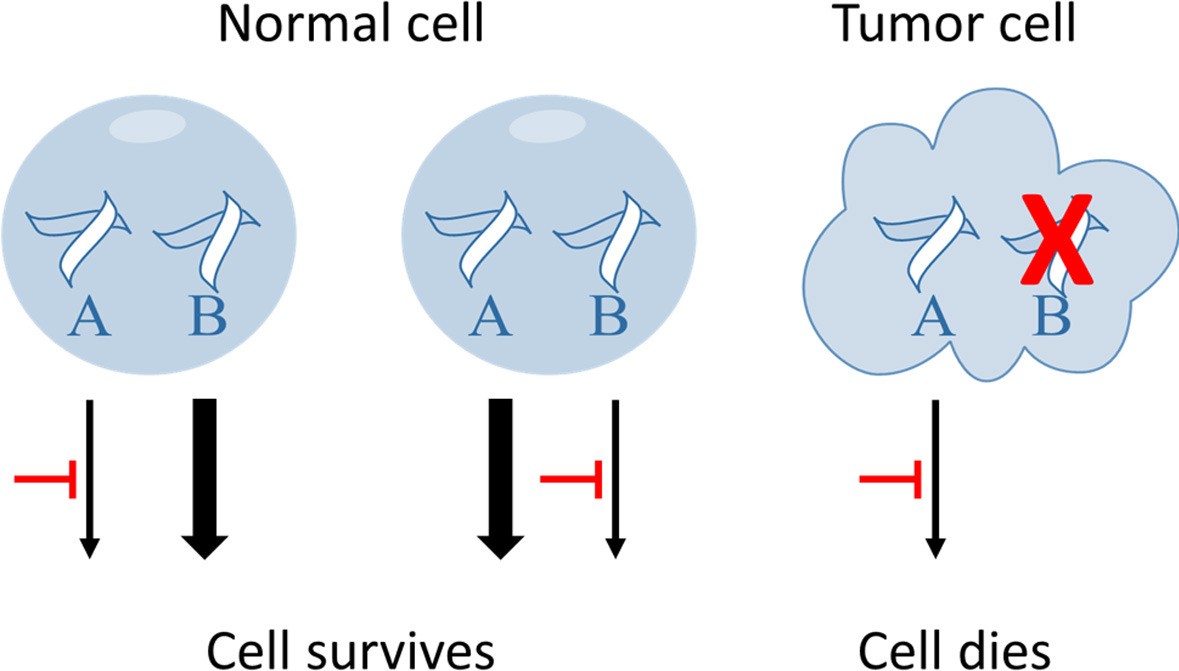 Fig. 1.
Fig. 1.Targeting synthetic lethal partners of mutated cancer genes specifically kill cancer cells bearing the inactivating mutations but spare normal cells. In the example shown here, pathways A and B are both intact in normal cells, whereas pathway A is defective in cancer cells. Inhibitors targeting pathway A will cause normal cells to survive through pathway B, whilst tumor cells cannot recover and succumb to this drug treatment scheme. In contrast, inhibitor of pathway B had no differential effect on both types of cells.
The breast cancer gene 1-poly(adenosine diphosphate-ribose) polymerase (BRCA1-PARP) pair has become the paradigm for a novel class of rational targeted cancer therapeutics based on the synthetic lethality concept. Mutations in the BRCA1 gene increase the risk of developing breast or ovarian cancers. PARP inhibitors are able to specifically eradicate cells containing BRCA1 mutations, while leaving normal cells intact [3]. The myelocytomatosis (MYC) oncogene oncogene family of transcription factors embodies another example of undruggable gene products. They are widely involved in the regulation of cell proliferation, differentiation, apoptosis and cancer but do not constitute valid therapeutic targets. Recently, an RNA interference study has shown the possibility of using 48 genes as MYC synthetic lethal [4]. In said study, the gene CSNK1E (encoding the protein CK1ε) was validated in vivo as a synthetic lethal partner of MYC. CK1ε belongs to a family of Ser/Thr protein kinases involved in various signal transduction pathways such as the Wnt signaling pathway, the circadian rhythm, the export of transcription factors from the nucleus to cytoplasm, DNA repair and transcription. In relation to cancer, CK1ε behaves as a synthetic lethal partner of Wnt/β-catenin [5, 6], is involved in cell proliferation [7] and its overexpression has been linked with poor prognosis in patients with oral [8] and stomach cancers [9].
The intrinsic resistance to cytotoxic agents via the activation of the Wnt/β-catenin signaling pathway characterizes some types of cancer including pancreatic cancer [10] or breast cancer [11]. In addition, chemotherapeutics provoke alterations in Wnt/β-catenin signaling pathway that have been responsible for drug resistance in ovarian cancer patients [12, 13] and multiple myeloma [14]. These studies highlight the Wnt/β-catenin pathway as a potential therapeutic target for cancer. However, the original expectation of CK1ε as a synthetic lethal partner of Wnt/β-catenin did not lead to the development of suitable chemotherapeutics. CK1ε bears a high homology with CK1δ and most of the anticancer studies reported in the literature had been conducted with the so-called dual inhibitors.
In the current minireview, we focus on three key points we consider essential to define the scope of CK1ε as a synthetic lethal partner and its inhibitors as cancer chemotherapeutics. Firstly, the therapeutic relevance, which is open to debate with respect to its exact role in cancer, denoted by diverse studies that provided, in first glance, inconsistent results [15-22]. A second consideration is in terms of the scarce availability of selective inhibitors of CK1ε, which might explain, at least in part, the contradictions observed. Finally, the constraint of computational tools to design selective inhibitors.
As a member of the CK1 family, CK1ε regulates a wide variety of cellular processes including RNA metabolism, response to DNA damage, DNA replication, circadian rhythms and cellular signaling [23-25]. One of these signaling cascades, the Wnt pathway, has been related to oncogenic processes when it is deregulated and β-catenin accumulates [23]. CK1ε promotes oncogenic transformation depending on β-catenin, as seen when direct inhibition of β-catenin with no modifications over CK1ε results in the absence of oncogenic transformation. Moreover, the ability of CK1ε to stimulate the synthesis of proteins through inactivation of 4EBP1, behaving as an activator of mRNA translation, has been previously described [7]. According to these results, high levels of CK1ε expression have been found in different types of cancers including ovarian [26], gastrointestinal tract [27] and breast [5]. Moreover, a search in Oncomine-a specific database that contains microarray expression levels of different genes, comparing normal versus affected tissues-showed that CSNK1E is up-regulated in malignant tissues when compared to the normal counterpart tissues [28]. More recently, it was reported that CK1ε was expressed at the highest level among six CK1 isoforms in glioblastoma and enriched in high-grade glioma [29]. Similarly, the use of inhibitors against CK1ε, blocked tumor cell growth, while sparing normal astrocytes. Hence, targeting CK1ε is currently emerging as an interesting therapeutic approach for cancer treatment.
In contrast, other studies reported that a significant number of patients with a high level of CK1ε expression are the ones that have better prognosis and survival [8, 22, 30]. Nevertheless, this source of contradiction must be properly interpreted. In most cases, the latter studies focus on specific types of cancers or even performed their analysis in specific subsets of patients. Although said studies reflect differences in survival depending on the expression levels of CK1ε, they all confirm the participation of this enzyme as a promoter of tumor cell growth. Discrepancies found in the literature may be explained by several factors. Among them, the lack of kinase activity [31] could explain a better prognosis in patients with overexpression of the mutated version of the protein. Additionally, the balance between wild-type and myristoylated CK1ε - reported as responsible of transforming normal cells into cancer cells [26] was not considered.
CK1ε is involved in other non-oncogenic pathways like the noncanonical Wnt route [31, 32]. Some of these studies were not performed in β-catenin dependent cell lines, in which alterations of the Wnt pathway would not alter the cell status. Furthermore, cells transfected with CK1ε are more sensitive to ultraviolet (UV) radiation in a significant manner [26]. This could be explained by the high rate of proliferation that triggers the overexpression of CK1ε, sensitizing cells to radiation. The use of specific RNAi against CK1ε has shown a decrease in cell proliferation in β-catenin-dependent tumor cell lines, suggesting that CK1ε promotes oncogenic transformation via deregulation of β-catenin degradation [5]. Moreover, knockdown of CK1ε has decreased cancer cell growth in several studies [7, 33], not only by interrupting the Wnt signaling pathway but by acting on other targets including the clock protein PERIOD2 [28].
The evidence that shRNAs against CK1ε, but not CK1δ , are able to attenuate cancer cell growth [5] prompted the search for new therapeutic agents. Drugs against CK1ε represent a topic of interest in the past years and remain a relevant field of study in the research of new chemotherapeutics for cancer treatment.
Despite the potential of CK1ε as a druggable target for cancer treatment, the actual development of inhibitors has been rather poor. The difficulty lies in the similarity between CK1ε and its sibling isoform CK1δ . Both kinases exhibit 98% and 53% amino acid homology in their kinase and C-terminal domains, respectively [34]. All reported inhibitors (Fig. 2) interact with the kinase domain. This makes it difficult to find selectivity towards a given isoform. In fact, the literature describes only two selective inhibitors of CK1ε (ICK1ε), GSD0054 [35, 36] and PF-4800567 [37]; as well as two selective inhibitors of CK1δ (ICK1δ ), SR-3029 [38] and LH846 [39] (Table 2). All the other reported compounds behave mainly as dual CK1δ /ε inhibitors (ICK1δ /ε). Representative examples of dual inhibitors are PF-670462 [40], PF-5006739 [41], IC261 (SU5607) [42], and a small library of thiazoles was reported by Bischof and coworkers [43].
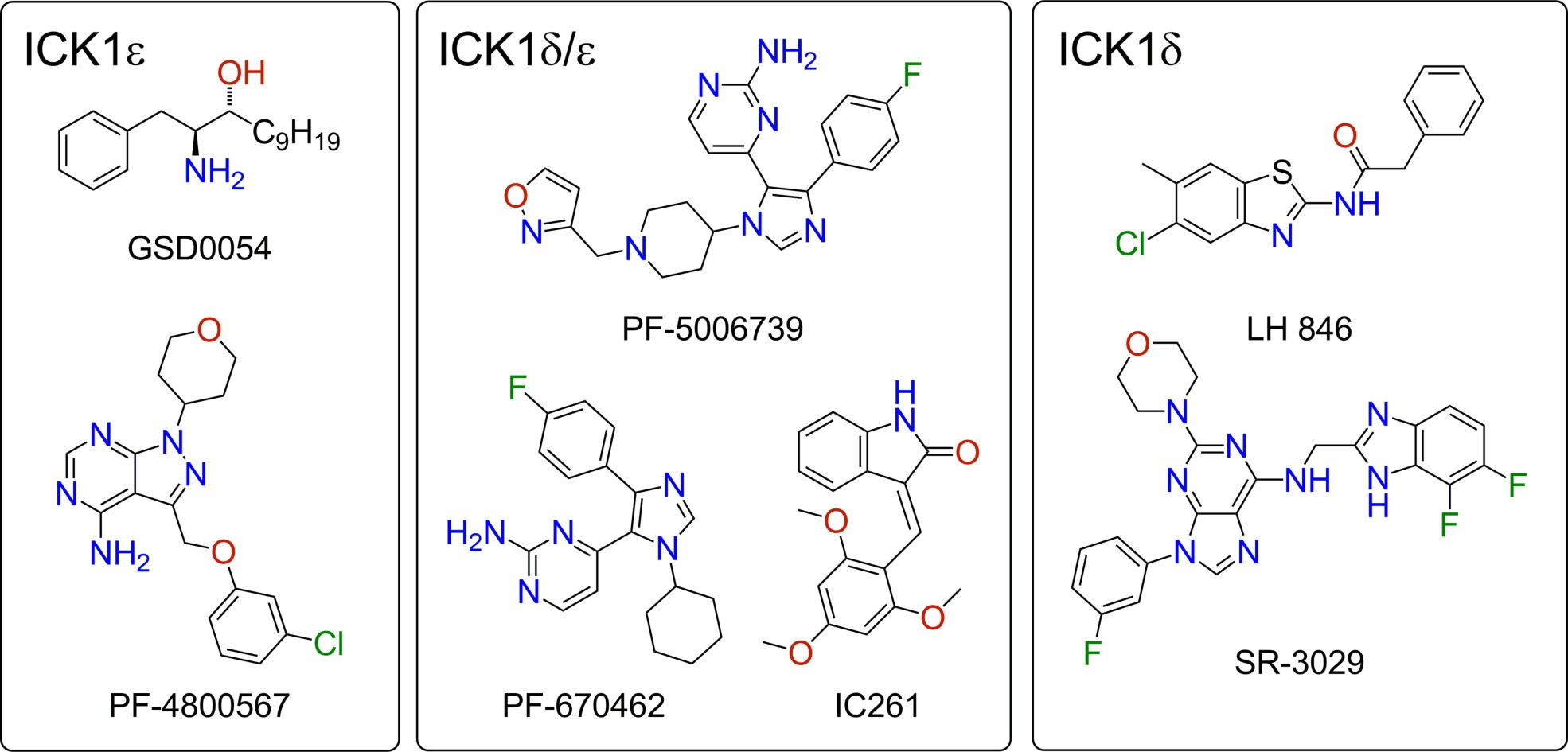 Fig. 2.
Fig. 2.Chemical structure of selective CK1ε inhibitors (ICK1ε), selective CK1δ inhibitors (ICK1δ ), and dual inhibitors (ICK1δ /ε).
PF-4800567 was the first compound reported to be a selective ICK1ε. This compound led to the conclusion that inhibition of CK1ε has little effect on the circadian clock [37]. Hence, inhibition of CK1ε with PF-4800567 is superfluous for establishing the disruption of circadian behavior, except at high concentrations that could also affect CK1δ [44, 45]. Considering its application in cancer therapy, PF-4800567 did not inhibit cell proliferation at 1 µM concentration in HEK293 (human embryonic kidney) and HT1080 (human fibrosarcoma) cells, even after 7 days of drug treatment [33]. To achieve growth inhibition, higher doses are required, although cell death does not occur. The half maximal inhibitory concentration (IC50) values in U87MG (human glioblastoma) cells after seven days of treatment was 28 µM [29]. Similarly, the growth inhibition 50% (GI50) values after 48 h of treatment against the human breast cancer cell lines T-47D and MDA-MB-453, as well as the human non-small cell lung cancer cell line SW1573 were: 79, 58, and 76 µM, respectively [36]. Overall, the potency of PF-4800567 a ICK1ε, does not correlate with its anti-proliferative activity. The hypothesis that CK1ε mediates activities of circadian rhythm gene products and β-catenin through independent mechanisms, might possibly explain the observed effects [29].
The second molecule reported as a selective ICK1ε is GSD0054, which behaves as a selective CK1ε inhibitor in enzymatic assays [35]. This small molecule does not interact with other kinases, as demonstrated with the DiscoverRX® KINOME® scan analysis of 456 kinases. The effects of GSD0054 on the disruption of cell cycle progression showed selective cell killing in T-47D breast cancer cells (β-catenin positive) but not in MDA-MB-453 cells (β-catenin negative) [36]. This effect was not observed in comparative studies using the ICK1ε PF-4800567, and the ICK1δ /ε PF-670462 and IC261.
The ICK1δ SR-3029 was identified by a high-throughput screen-ing (HTS) campaign targeting inhibitors of Wee1 degradation [46]. This compound exhibits a very potent activity against human melanoma cell line A375, displaying an IC50 value of 86 nM [38]. The five off-target kinases of SR-3029 do not seem to be responsible for the potent anti-proliferative effects that were demonstrated. Moreover, SR-3029 is highly selective, potent, and efficacious in multiple preclinical models of human breast cancer that overexpress CK1δ (MDA-MB-231, SKBR3, BT474), but had no effect on MCF-7 breast cancer cells [47]. However, the interaction of SR-3029 with both CK1ε and CK1δ, potently inhibited phorbol-induced tumorigenesis by blocking Wnt/β-catenin signaling in a mouse skin cancer model [48].
LH846 is the less studied inhibitor of the series with respect to cancer treatment. In fact, it emerged following the screening of a library of 500,000 drug-like compounds tested to identify small molecules that modulate clock function [22]. However, LH846 served to establish the role of CK1δ in yeast respiratory oscillations (which is related to circadian rhythms) [49], in the maturation of cytoplasmic 40S [50], and in genomic instability characterized by the accumulation of DNA damage and down-regulation of checkpoint kinase 1 [51].
A pharmaceutical company developed PF-670462 in the search for small molecules that could affect the circadian rhythm [40]. This small molecule displays a similar activity against CK1ε and CK1δ (Table 1). PF-670462 has been the tool compound to study the modulation of circadian rhythms [52] or the stimulant effects of methamphetamine [53], amongst others. This is possible since PF-670462, like its sibling compound PF-4800567, is unable to produce cell death [33, 36]. A third member of this family of small molecules is PF-5006739, which is a promising candidate to test the hypothesis of CK1δ /ε inhibition in treating multiple indications in the clinic such as attenuation of opioid drug-seeking behavior [41] or improving glucose homeostasis in obesity [54].
| Compound | Selectivity | Measure | CK1ε | CK1δ | Ref. |
|---|---|---|---|---|---|
| GSD0054 | ε | %Ctrl | 27% | 93% | [35, 36] |
| PF-4800567 | ε | IC50 | 32 nM | 711 nM | [37] |
| SR-3029 | δ | IC50 | 260 nM | 44 nM | [38] |
| LH846 | δ | IC50 | 1.3 µM | 290 nM | [39] |
| PF-670462 | δ /ε | IC50 | 7.7 nM | 14 nM | [40] |
| PF-5006739 | δ /ε | IC50 | 17 nM | 3.9 nM | [41] |
| IC261 | δ /ε | IC50 | 0.6-1.4 µM | 0.7-1.3 µM | [42] |
| Compound | CK1ε | CK1δ |
|---|---|---|
| GSD0054 | -20.6 | -18.3 |
| PF-4800567 | -22.4 | -6.7 |
| SR-3029 | -23.2 | -16.8 |
| LH846 | -18.7 | -18.1 |
| PF-670462 | -16.6 | -14.7 |
| PF-5006739 | -18.7 | -12.4 |
| IC261 | -17.3 | -10.6 |
IC261 represents a small molecule dual inhibitor that was described originally as intervening at mitosis events and triggering the p53-dependent mitotic checkpoint [42]. Thus, treatment of cells with IC261 leads to apoptosis and cell death. However, further studies confirmed that IC261 neither inhibited CK1δ /ε kinase activity nor blocked Wnt/β-catenin signaling in cancer cells. In fact, IC261 binds to tubulin with an affinity similar to colchicine and behaves as a potent inhibitor of microtubule polymerization. This activity seems responsible for many of the diverse biological effects of IC261, including cancer cell killing [33].
Molecular docking is commonly used to predict interaction between proteins and small molecules. It predicts binding modes and interaction energies at the binding site and it is used to test novel molecules before chemical synthesis and experimental biological screening. Typically, the target of interest is treated as rigid whereas small molecules have full flexibility. Docking scores depend on the software and there are plenty of different scoring functions available. However, all of them provide an estimation of the protein-ligand complex stability.
The six human CK1 isoforms have a high degree of homology in their binding sites, with CK1δ and CK1ε sharing a remarkable 98% of amino acid identity. There are several crystal structures of both enzymes deposited in the Protein Data Bank (PDB), some of which co-crystalized with small molecule inhibitors. We ran some calculations to examine if docking methods could identify specific inhibitors of CK1δ and CK1ε and the results are shown in Table 2.
All compounds are predicted to form more stable complexes with CK1ε. The difference in stability with CK1δ complexes is significant for compounds PF-4800567, SR-3029, PF-5006739, and IC261.However, these results do not correlate well with experimental results of specificity. Therefore, one could suggest that docking methods are not suited to design or distinguish between specific inhibitors of either CK1δ or CK1ε. The scoring functions used in docking are very “forgiving”, in the sense that they predict better binding stability than what is obtained experimentally, and when binding sites are very similar, as is the case between CK1δ and CK1ε, they do not perform adequately. However, docking could be used to guide design of dual inhibitors of CK1δ and CK1ε. A method that could help designing specific inhibitors of either CK1δ or CK1ε is QSAR [55]. However, to implement such a method, one would need high quality data with experimentally validated binding affinities for each enzyme for a significant number of inhibitors. To the best of our knowledge, this data set is not available, and unsurprisingly it is very difficult to create specific inhibitors for these enzymes.
Diverse studies show that the inhibition (with RNAi or shRNAs) or the knockdown of CK1ε, result in a decrease in cell proliferation by interrupting the Wnt/β-catenin signaling pathway. These results confirm CK1ε as a druggable target for cancer treatment. However, the development of ICK1ε has not progressed as would be expected. When analyzing in detail the currently available inhibitors, we found that there are only two selective ICK1ε: a) PF-4800567, which does not induce cell kill and has been studied in relation to its ability to alter the circadian rhythm; and b) GSD0054, which is reported as capable of inducing selective cell killing of β-catenin-positive cells but not β-catenin-negative cells. From the available ICK1δ, the selectivity index against both isoforms is not as large to allow to consider that their biological effects are not related to intervention at the CK1ε level. Additionally, these ICK1δ were tested exclusively for their ability to modify the circadian clock but not as potential chemotherapeutics. In fact, the only inhibitor besides GSD0054 that has shown the ability to kill cancer cells is IC261. However, this compound exerts cytotoxicity by microtubule depolymerization, rather than through the inhibition of CK1ε. In summary, the contradictory results from one study to another could be attributed to several reasons: the studies have been conducted under diverse experimental conditions, the large similarity between CK1 isoforms could mask real effects, and the inhibitors employed in these studies are not selective enough or have off-target effects that could explain, at least in part, the observations. It is anticipated that focused studies including all the reported inhibitors could shed light on the scope of ICK1ε for the treatment of malignant and non-malignant disorders.
Considering the limitations described thus far, the future of CK1ε as a druggable target with clinical relevance in cancer treatment lies on the availability of selective ICK1ε. The challenge remains to develop new small molecules that could act as selective inhibitors and preferably without relevant off-target effects. ICK1ε GSD0054 represents the first small molecule that addresses those requirements, at least in part. Another important limitation that has to be overcome relates to computational approaches. Again, GSD0054 demonstrates that the existing computational methods are not valid to discern between highly similar isoforms and they do not allow to explain the experimental results. Consequently, new methods should be developed and implemented to avoid the limitations of the existing ones.
All authors wrote the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript.
The authors acknowledge the institutional support from Univer-sidad de La Laguna.
The authors declare no competing interests.