




1 Laboratory of CNS Injury and Molecular Therapy, JFK Neuroscience Institute, Hackensack Meridian JFK University Medical Center, Edison, NJ 08820, USA
Abstract
In this review, we discuss the possibility and feasibility of nuclear factor erythroid 2-related factor 2 (Nrf2) as a therapeutic target to minimize the devastating effects of a brain injury. To complete this review, comprehensive literature searches were conducted in MEDLINE, PubMed, Embase, and PsycINFO databases for English scientific peer-reviewed articles through December 2022. This short review addressed the different sources of oxidative stress and its effects on blood-brain barrier (BBB) dysfunction, mitochondrial damage, and changes in a variety of inflammatory molecules associated with central nervous system (CNS) injury. At last, we explained the potential efficacy of the Nrf2 transcription factor in reducing oxidative stress-mediated secondary damages after a CNS injury. The role of CPUY192018, an inhibitor of Nrf2-Keap1 protein-protein interaction in protecting the injured brain cells is given as evidence of Nrf2’s role in activating antioxidant genes. Overall, the scope of Nrf2 in developing therapeutic interventions for a variety of pathophysiological conditions associated with CNS injury-induced free radical/inflammatory signaling is acknowledged. Nrf2 has a widespread application in basic and clinical neuroscience for understanding and treating free radical/inflammatory signaling disorders, including neurological diseases. The development of innovative therapeutic strategies using Nrf2-inducing agents can be applied to reduce the complications of TBI before advancing it to posttraumatic stress disorder (PTSD).
Graphical Abstract
Keywords
- Nrf2
- oxidative stress
- reactive oxygen species
- antioxidants
- brain injury
- blood-brain barrier
- antioxidant response elements
Traumatic brain injury (TBI) is a sudden injury that often leads to permanent
disabilities and impairment of cognitive and emotional functions. It has been
estimated that approximately 2% of the USA population is living with some degree
of disability as a result of TBI. While the primary injury of trauma can cause
direct damage to neuronal structures, the mechanical tissue deformation triggers
secondary injury leading to the blood-brain barrier (BBB) damage, edema,
increased intracranial pressure, inflammation, and cell death [1]. Secondary
injury is the progression of TBI as a long-term neurological problem by affecting
physically, cognitively, and emotionally which often leads to a permanent
disability of the victim [2]. The etiology of secondary injury is primarily due
to oxidative stress. Oxidative stress causes neuroinflammation, edema formation,
BBB damage, cell death, and finally leads to cognitive impairments in TBI.
Immediately after a brain injury, a huge quantity of inflammatory cytokines such
as interleukin-1beta (IL-1
Oxidative stress is the key player in the secondary injury of TBI [10]. It is
the imbalance of oxidants and antioxidants that causes biochemical and
physiological damage to the cells. A free radical is an atom or a molecule that
has at least one unpaired electron and is formed during a redox reaction. These
free radicals are highly reactive unstable molecules capable of independent
existence and attacking cell components [17]. Reactive oxygen species (ROS) and
reactive nitrogen species (RNS) are the two types of free radicals generated
after a brain injury. ROS is the byproduct of oxygen metabolism, and these
include peroxides, superoxide (O
Oxidative stress has a momentous function in the progression of the
pathophysiology after TBI [10]. It is the main source of secondary injury-induced
neuroinflammatory and cell death pathophysiology. In fluid percussion injury
(FPI) and mild blast injury animal models, we have established that oxidative
stress has a vital function in the pathophysiology of CNS injury [1, 5, 6, 12]. We
have observed that oxidative stress activates matrix metalloproteinases (MMPs),
reduces tight junction protein expression, and causes BBB damage and induction of
inflammatory signaling pathways [1]. We also demonstrated the role of oxidative
stress in causing neuroinflammation and cell death by activating Ca
Oxidative stress-induced mitochondrial damage has been studied in the
pathogenesis of TBI. Oxidative stress induces mutations in the mitochondrial DNA,
impairs the mitochondrial respiratory chain, and alters Ca
Mitochondria are the central participants in cell death in the pathophysiology
of TBI. In TBI-induced mechanisms of apoptosis, mitochondrial membrane,
Ca
 Fig. 1.
Fig. 1.Schematic presentation of oxidative stress-induced in
mitochondrial damage, neuroinflammation, BBB dysfunction, and neurodegeneration
in TBI. Reactive oxygen species (ROS) and reactive nitrogen species (RNS) are
the main sources of oxidative stress in brain injury. ROS/RNS activates MMPs,
TGF-
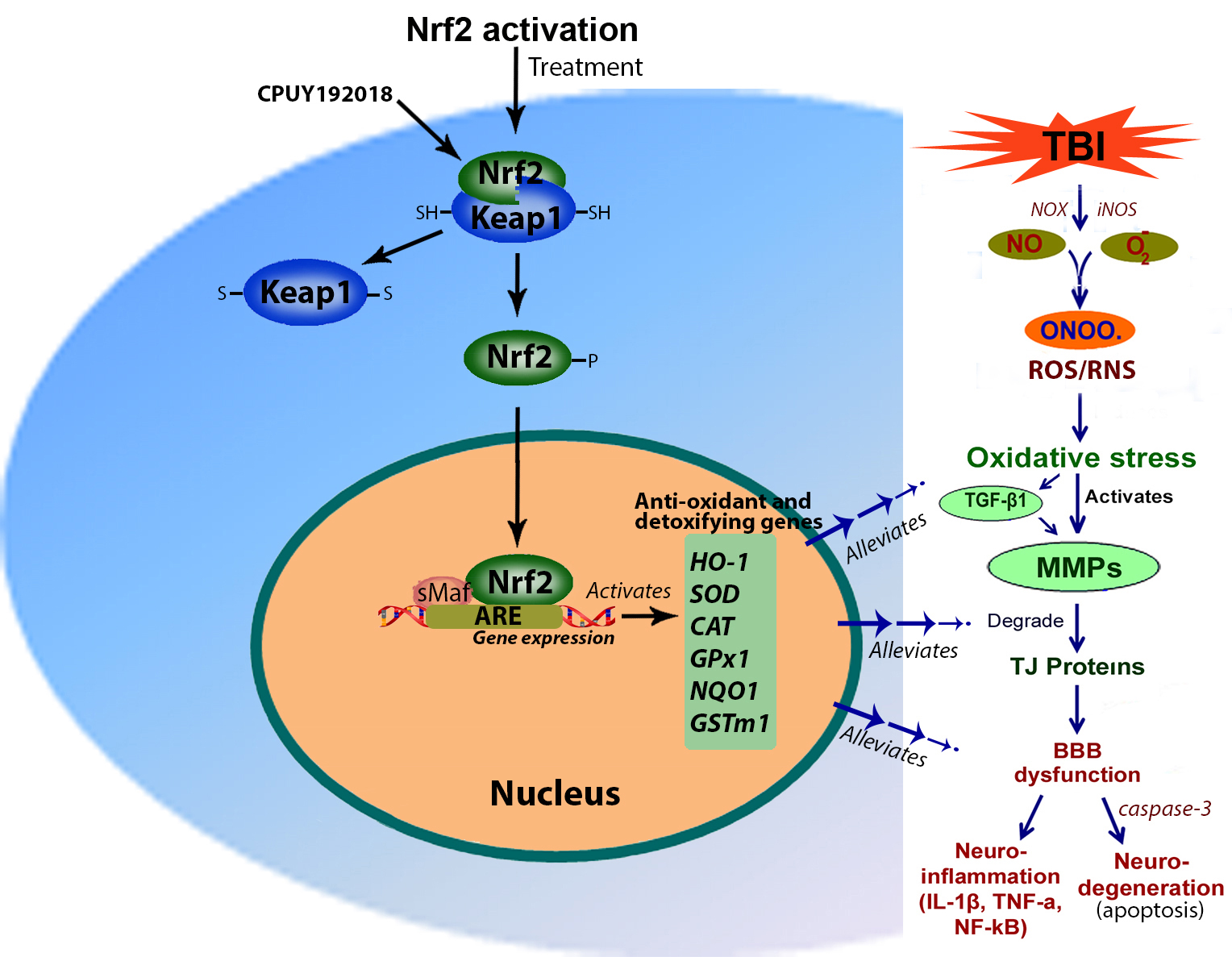
Recently, substantial research has been focused on the development of antioxidant therapies against oxidative stress for neuroprotection in TBI [32, 33, 34, 35]. Considering the importance of developing anti-oxidant therapy, we studied the role and mechanisms of Nrf2 in regulating anti-oxidant genes and thereby mitigating oxidative stress after TBI [12]. In 2017, Ding et al. [36] demonstrated that the Nrf2 pathway provides neuroprotection following TBI via regulation of the ubiquitin-proteasome system (UPS). In another study, Cui et al. [37] used calcitriol, an active form of vitamin D, for promoting the autophagic process and activated Nrf2 signaling by reducing the expression of Keap1 and enhancing Nrf2 translocation, thereby mitigating TBI-induced oxidative damage. Similarly, a few studies have been conducted by targeting Nrf2 pathways for neuroprotection in TBI [38, 39, 40, 41, 42, 43, 44, 45]. Therefore, the activation of antioxidant-promoting transcription factor Nrf2 alleviates oxidative stress-induced neurological damage associated with TBI, including activation of matrix metalloproteinases (MMPs), neuroinflammation, and neurodegeneration [12].
The BBB is a highly selective semipermeable barrier that selectively prevents solutes, toxins, and microorganisms from blood circulation to the brain. It constitutes various cells including endothelial cells, astrocytes, neurons, and pericytes, and forms a neurovascular unit. The BBB damage causes the transmigration of blood cells to the brain and enhances neuroinflammation leading to several neurological disorders and diseases and other brain dysfunctions. BBB damage is the main hallmark of several neurological disorders or diseases including Alzheimer’s disease, stroke, multiple sclerosis, HIV-1 encephalitis, and TBI. In addition, in drug or alcohol abuse, oxidative stress has a vital role in compromising BBB integrity. In TBI, oxidative stress causes BBB dysfunction by downregulating tight junction proteins and causes BBB permeability and leakage of blood cells to the brain part. Oxidative stress also causes structural damage or loss of the cells in the neurovascular unit. Endothelial dysfunction, neuronal damage, and pericyte loss are hallmarks among them. Pericytes are mainly responsible for the structural integrity of BBB and blood flow. Loss of pericyte leads to neurovascular damage, impaired capillary perfusion and blood flow, neuroinflammation, and neurodegeneration. In a diabetic retinopathy study, the protein DJ-1 scavenges oxidants and prevents pericyte damage in the retinal capillary by activating antioxidants via the Nrf2 signaling pathway [46]. Recently, in TBI, we reported that pericyte loss impairs angiogenesis and results in BBB damage and neurovascular dysfunction [47]. Therefore, we suggest that activating the Nrf2 signaling pathway would be a therapeutic strategy for protecting the brain from TBI-induced neurological impairments. There are few reports on the activation of Nrf2 and BBB protection after TBI. A systemic administration of sulforaphane, an isothiocyanate abundant in cruciferous vegetables (e.g., broccoli) increases the expression of Nrf2-driven genes in brain tissue and microvessels. Postinjury sulforaphane-induced activation of Nrf2-driven genes attenuates endothelial cell death and tight junction protein loss and reduces BBB permeability [48].
Nrf2 is a basic region leucine-zipper transcription factor major regulator of cellular antioxidant and anti-inflammatory defense mechanisms. It belongs to the Cap’N’collar family of proteins [14] and plays a vital task in modulating the cells toward different cellular stress responses as it activates the transcription of a myriad of genes coding for cellular antioxidants, detoxification enzymes, and other cell-protective proteins [14]. Nrf2 has an important role in ameliorating cytotoxicity and inflammation that are characteristically associated with oxidative stress-induced degenerative and chronic disease. Within the cytoplasm of most cells including neurons, astrocytes, and endothelial cells, the Nrf2 protein is under stringent control by the adapter protein Kelch-like ECH- Kelch-like ECH-associated protein 1 (Keap1) [49]. During normal physiological circumstances, Nrf2 binds to Keap1 and degrades by ubiquitination and proteasome-mediated degradation. Free radicals disturb Nrf2-Keap1 interaction and Nrf2 separates and is released to the nucleus. In the nucleus, Nrf2 joins with Maf proteins and regulates the expression of genes by binding to the ARE region of anti-oxidant genes [49]. This leads to the upregulation of endogenous scavenging enzymes. These include the cellular antioxidants heme-oxygenase 1 (HO-1), catalase, superoxide dismutase (SOD), glutathione modulators, and oxidoreductase like nicotinamide adenine dinucleotide phosphate (NADPH): quinone oxidoreductase 1 (Fig. 2). Compared to chemical antioxidants, activation of endogenous enzymes arises with relatively low or no side effects. Moreover, being a broad-spectrum antioxidant transcription factor, Nrf2 can modulate several genes [14, 49]. It has been substantiated that Nrf2 knockout mice are much more prone to inflammation than wild types and this has been substantiated through in vitro studies [50]. In our very recent study, we examined the expression of Nrf2 and its phosphorylated form and the role of Nrf2 in protecting the brain from oxidative stress, neuroinflammation, and apoptotic cell death after FPI [12]. In several other CNS pathophysiological complications such as ischemia, hemorrhage, and other neurodegenerative diseases, oxidative stress has been implicated with Nrf2 dysfunction, and activation of Nrf2 would be the desired method to reduce the pathogenesis of these disorders. For this reason, Nrf2 has the potential for developing a new therapeutic strategy for mitigating CNS injury-induced brain damage.
 Fig. 2.
Fig. 2.Mechanisms of Nrf2 activation. Nrf2 expresses within
the cytoplasm under normal physiological conditions and Keap1 regulates the
expression of Nrf2. The interaction or binding of Nrf2 with Keap1 degrades the
Nrf2 either by ubiquitination or proteasomal degradation. However, oxidative
stress activates Nrf2 and disrupts the Nrf2-Keap1 interaction and the Nrf2 is
converted to its phosphorylated form and released to the nucleus. In the nucleus,
Nrf2 binds with Maf and forms a dimer. This Nrf2-Maf dimer binds to the ARE
region of the antioxidant gene promoter and initiates their transcription.
Activation of antioxidant genes reduces TBI-induced oxidative stress,
TGF-
In our preliminary study, we determined the effect of injury in cortical mouse
neuronal cultures pretreated with CPUY192018 and subjected to 2 psi stretch
injury (mild in vitro injury). CPUY192018 is a potent inhibitor of Nrf2
and Keap1 binding with K
 Fig. 3.
Fig. 3.CPUY192018, an inhibitor of Nrf2-Keap1 protein-protein
interaction protects the brain by activating antioxidant genes. (A) The chemical
structure of CPUY192018. (B,C) Western blot analysis of GPx1 and HO-1 (B) and
4-HNE and 3-NT (C) and normalized with
Background information on the Nrf2 transcription system suggests that the most compelling strategy for improving Nrf2 activity is to stabilize this important but labile protein and facilitate its nuclear translocation [53]. Activation of the Nrf2 transcriptional system, a transcription enhancer of a variety of endogenous antioxidants, and detoxifying genes has been demonstrated as a viable method for the remediation of oxidative radicals by various investigators [33, 54, 55, 56]. Peptidergic drugs have the potential as a therapeutic alternative to improve recovery in TBI patients [57]. Nrf2 cell-penetrating peptides can activate the antioxidant genes by sustained stabilization and increased nuclear translocation of Nrf2. Compared to chemical antioxidants, peptide treatments have few if any side effects [58, 59] and show high therapeutic efficacy in clinical trials [60]. Moreover, the efficacy of the peptide in activating antioxidant/cell-defense pathways has been studied in different CNS injuries including spinal cord injury (SCI), and optic nerve injury. The development of innovative therapeutic strategies using Nrf2-inducing agents can be applied to reduce the complications of TBI before advancing it to posttraumatic stress disorder (PTSD).
This review has highlighted the role of oxidative stress in the pathogenesis of
TBI including the activation of inflammatory cytokines, TGF-
PMAM conceived and wrote the manuscript and created the figures. The author contributed to editorial changes in the manuscript. The author read and approved the final manuscript. The author has participated sufficiently in the work and agreed to be accountable for all aspects of the work.
The animal experiments were carried out in accordance with National Institutes of Health (NIH) ethical guidelines for the care of laboratory animals, and the Institutional Animal Care Use Committee (No. MM2201), Seton Hall University, South Orange, NJ.
Not applicable.
The New Jersey Commission on Brain Injury Research grant #CBIR19PIL010 (to P.M. Abdul-Muneer) and the Neuroscience Institute at JFK University Medical Center supported this work financially.
P. M. Abdul-Muneer is serving as one of the Guest editors of this journal. We declare that P. M. Abdul-Muneer had no involvement in the peer review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to Gernot Riedel and Hongmin Wang. The author declares no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.