






 , Ronaldo Cunha Coelho 2, Jeane de Oliveira Moura 1,2, Igor Ferreira do Nascimento 2, Miklos Maximiliano Bajay 3, Leonardo Castelo Branco Carvalho 4, Fábio Barros Britto 1, José Lindenberg Rocha Sarmento 1, Danielle Maria M. Ribeiro Azevêdo 1,5, Salvatore Mastrangelo 6, Layonel Alves de Sousa 7, Adriana Mello de Araújo 1,5
, Ronaldo Cunha Coelho 2, Jeane de Oliveira Moura 1,2, Igor Ferreira do Nascimento 2, Miklos Maximiliano Bajay 3, Leonardo Castelo Branco Carvalho 4, Fábio Barros Britto 1, José Lindenberg Rocha Sarmento 1, Danielle Maria M. Ribeiro Azevêdo 1,5, Salvatore Mastrangelo 6, Layonel Alves de Sousa 7, Adriana Mello de Araújo 1,51 Center for Agricultural Sciences, Federal University of Piaui, 64049-550 Teresina, Piauí, Brazil
2 Federal Institute of Piauí, Campus Teresina-Central, 64000-040 Teresina, Piauí, Brazil
3 Department of Fisheries Engineering and Biological Sciences, State University of Santa Catarina, Center for Higher Education in the Southern, 88790-000 Laguna, Santa Catarina, Brazil
4 Socioenvironmental and Water Resources Institute, Federal Rural University of the Amazon, 66075-110 Belém, Pará, Brazil
5 Brazilian Agricultural Research Corporation, Embrapa Meio-Norte, 64008-780 Teresina, Piauí, Brazil
6 Dipartimento Scienze Agrarie, Alimentari and Forestali, University of Palermo, Viale delle Scienze, 90133 Palermo, Italy
7 Department of Veterinary Medicine, Center for Agroveterinary Sciences, State University of Santa Catarina, 88520-000 Lages, Santa Catarina, Brazil
Abstract
The conservation of genetic resources is crucial for maintaining biological diversity and ensuring the sustainability of animal production. Characterizing regions of homozygosity (ROHs), heterozygosity-rich regions (HRRs), and levels of inbreeding is crucial for the management and conservation of livestock populations. In Brazil, little is known about these genomic parameters in Marota goats. Thus, this study aimed to evaluate these characteristics to support conservation strategies for these breeds.
Genomic analyses were conducted using the Illumina Goat SNP50 BeadChip to investigate ROH, HRR, and to estimate the inbreeding coefficient derived from ROHs (FROH) in Marota and Anglo-Nubian goats. This study evaluated 192 individuals from three distinct herds. ROHs were classified according to their length; genes located within both ROH and HRR regions were identified and functionally annotated to explore their potential biological significance.
Genomic analysis identified a total of 22,872 ROHs in the studied goats. The average number of ROHs per individual varied between breeds, with Anglo-Nubian goats exhibiting an average of 74.73 ROHs, while Marota goats had an average of 173.85 ROHs. Inbreeding coefficients based on ROHs in Marota goats varied widely (0.040–0.283), reflecting recent reductions in effective population size. Functional annotation revealed that several genes with overlapping ROH and HRR islands, such as ZBTB11, IL18, and LPO, were significantly enriched in immune-related pathways. Additionally, candidate genes associated with adaptive traits, such as TEX12, YPEL5, CAPN14, and GALNT14, were identified and were linked to embryonic development, body growth, lipid metabolism, and brain function.
The ROHs and HRRs analyses in Marota and Anglo-Nubian goats revealed distinct patterns of genomic inbreeding and potential adaptive genetic regions. These findings emphasize the genetic distinctiveness of the Marota breed and underscore the importance of incorporating genomic data into conservation planning. The identification of candidate genes in islands with ROHs and HRRs provides valuable insights for guiding conservation priorities and managing genetic diversity in Brazilian goat populations.
Keywords
- local goat population
- genomic regions
- inbreeding
- heterozygosity
Among livestock species, goats (Capra hircus) play a crucial role in economic development, contributing significantly to rural production, particularly in milk production, and to rural subsistence by providing additional income to families in developing countries [1, 2, 3]. In the Brazilian context, rural activity with small ruminants is divided between large producers and small breeders, with goats being a notable presence, adapted to the semi-arid tropics [4, 5].
Indigenous or local breeds generally preserve higher genetic diversity than commercial populations, since their development has relied on long-standing selection practices distinct from those applied to intensively bred lines. In Brazil, several goat breeds adapted to the semi-arid tropical climate serve as important reservoirs of genes associated with tolerance to harsh environments, parasites, and diseases, thereby enriching overall genetic diversity [6, 7].
Recognizing and characterizing these genetic resources is crucial for formulating policies that ensure their conservation and sustainable use. Managing livestock genetic diversity represents a demanding yet urgent challenge, given its fundamental role in safeguarding biodiversity. As case studies, we considered two Brazilian goat breeds: the Anglo-Nubian, a commercial line, and the Marota, a locally adapted population [4, 7].
The Marota goat is a local breed from the semi-arid region, a small, hardy type currently at risk of extinction, maintained by government institutions and partners in northeastern Brazil [4, 8]. This population constitutes a valuable reservoir of genetic diversity, as it carries genes linked to resilience against climate and environmental stressors, as well as resistance to parasites and diseases, attributable to its long evolutionary history of adaptation to adverse conditions [4, 6, 8]. Although the Marota has been present in Brazil for centuries, its genetic diversity and population structure remain insufficiently characterized [4, 7].
Genomic analyses of these resources are therefore essential to guide policies for their conservation and sustainable management. In this framework, genome-wide assessments, particularly those based on runs of homozygosity (ROH), offer valuable information about molecular evolution, patterns of adaptation, and inbreeding levels estimated through the inbreeding coefficient based on runs of homozygosity (FROH) in both commercial and locally adapted goat populations [9, 10, 11, 12].
Autozygosity occurs when alleles inherited from a common ancestor are expressed in a homozygous state, which can arise under different genetic scenarios. Elevated levels of inbreeding (F) are known to reduce genetic variation, compromise individual performance via inbreeding depression, and threaten population viability in the long term [13, 14]. Among the available approaches, genomic inbreeding coefficients derived from ROH are regarded as highly informative [9, 15]. Moreover, ROH analyses allow the identification of genomic segments likely shaped by selection and linked to traits specific to individual populations [14, 15, 16].
Considering recombination dynamics, genomic regions with extended homozygosity often result from selection, favoring advantageous alleles in surrounding loci. Therefore, ROH analysis has been employed to explore signatures of selection in various species, including both ruminants and non-ruminants [12, 17, 18].
Heterozygosity-rich regions (HRRs), which represent genomic stretches with high heterozygosity, can provide valuable information about population structure and demographic history. These regions may also harbor genes linked to adaptive responses, including heat stress tolerance, immune defense, reproductive success, and other fitness-related traits [15, 19].
Here, we investigated ROH and HRR patterns in the Marota breed, a locally adapted goat population, and in the commercial Anglo-Nubian breed, using single nucleotide polymorphism (SNP) genotyping data. We estimated genomic inbreeding using the ROH-based coefficient (FROH) and identified candidate genes that overlap with the ROH and HRR regions. This approach provides new insights into genetic diversity and population structure, supporting strategies for breed conservation and improvements in production systems.
The study was carried out in the state of Piauí, located in the Northeast region of Brazil and bordering the states of Maranhão, Ceará, Tocantins, Bahia, and Pernambuco (Fig. 1). The territory of Piauí has a total area of 251,529 km2 and hosts a significant goat population, including the third largest goat population in Brazil [20].
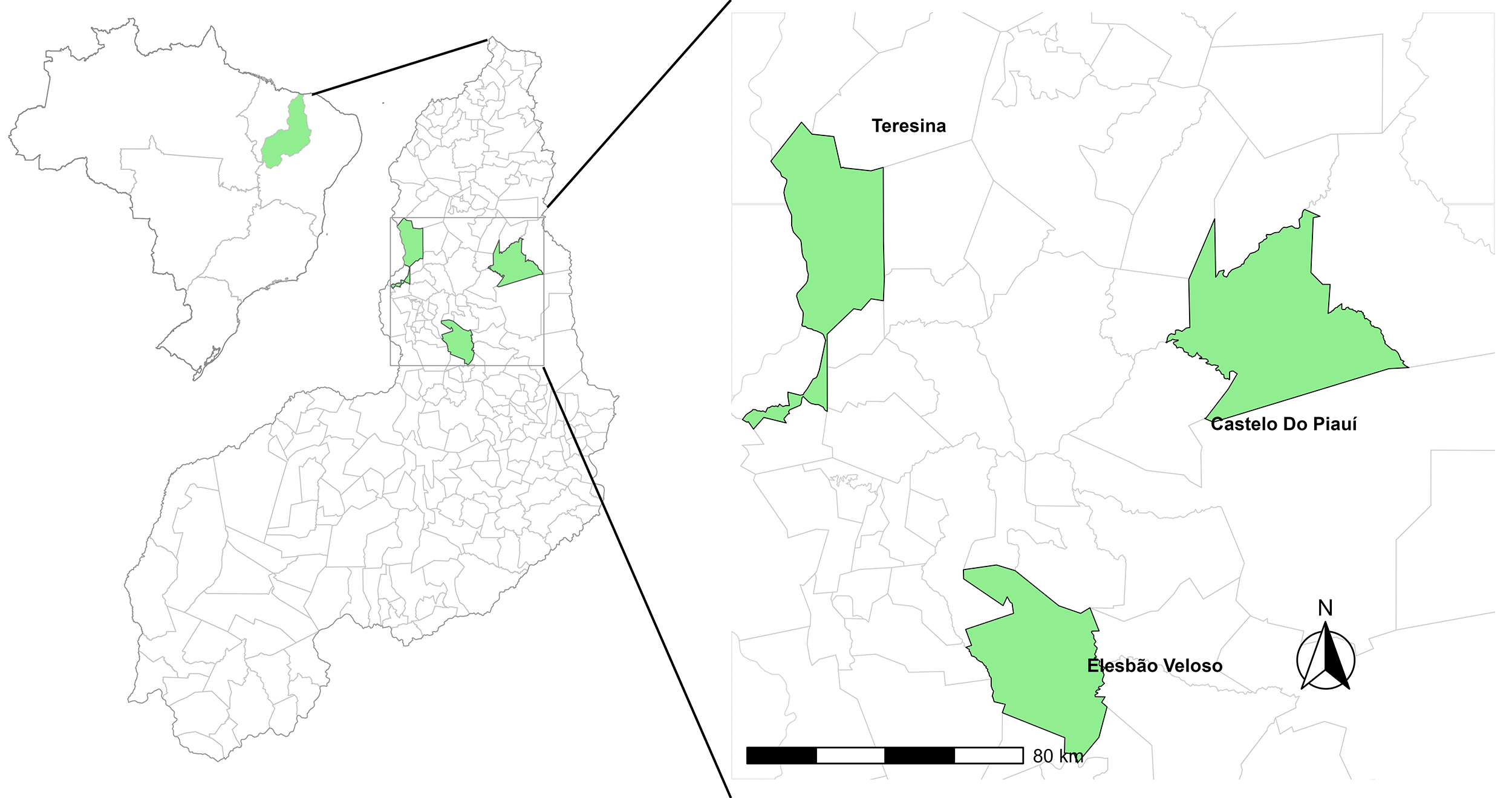 Fig. 1.
Fig. 1.
Geographical and phenotypic distribution of two goat populations in Piauí, Brazil.
The data used in the study consisted of 192 individuals classified into two breeds of different origins, both present in the semi-arid region with multiple herds. These herds included rare local breeds, namely Marota goats (n = 86) and Anglo-Nubian goats (n = 106). The Anglo-Nubian individuals were collected from the herd maintained by the Federal University of Piauí (UFPI) in Teresina, Piauí, Brazil (05°02’39.95”S, 42°47’03.70”W). The Marota individuals were sampled from two sources: the conservation herd maintained by Embrapa Meio-Norte, located in Castelo do Piauí, Piauí, Brazil (05°19’20”S, 41°33’09”W), as well as from a private herd in Elesbão Veloso, Piauí, Brazil (06°12’07”S, 42°08’25”W). The geographical locations of these herds are shown in Fig. 1.
From each animal, approximately 3 mL of blood were obtained by jugular vein puncture using a vacuum collection system. Samples were stored in tubes containing sodium fluoride as a glycolysis inhibitor and ethylenediaminetetraacetic acid (EDTA) as an anticoagulant to preserve cell integrity and sample quality. Genomic deoxyribonucleic acid (DNA) was extracted following the manufacturer’s protocol of the AxyPrep Blood Miniprep Kit (AP-MN-BL-GDNA-50) [4, 5].
Genotyping was performed with the Illumina Goat SNP50 BeadChip, which includes 53,347 SNP markers distributed across the genome. The array was developed using Illumina Infinium technology and processed on the iScan system. All procedures followed the manufacturer’s instructions (Illumina, San Diego, CA, USA) [4, 5].
To evaluate genotypic data quality, we applied PLINK v1.9 (Shaun Purcell in Boston, MA, USA) [21]. SNP positions were aligned to the ARS1 reference genome (https://www.ncbi.nlm.nih.gov/datasets/genome/GCA_000317765.1/). Quality control steps included removing variants located on sex chromosomes or unmapped contigs. Unlike many filtering pipelines, no minimum minor allele frequency (MAF) threshold was imposed to preserve rare alleles and ensure accurate detection of homozygous segments. SNPs with a call rate below 0.95 were excluded [15, 22]. After filtering, the dataset comprised 43,133 SNPs across 192 goats.
We employed the R package detectRUNS (v.0.9.6, R Core Team, Vienna, Austria) to identify ROHs and HRRs, applying both the sliding-window approach and the consecutive-runs method (CR) [21, 23, 24]. The complete script can be found in the Supplementary Material.
The following sliding-window parameters were used for ROH detection: a minimum of 20 SNPs per window (minSNP); tolerance of one heterozygous SNP within a window (maxOppWindow = 1); and up to one missing genotype per window (maxMissWindow = 1). Additional run-related thresholds were applied as follows: the maximum distance between consecutive SNPs was set to 250 kb (maxGap = 250,000 bp); the minimum ROH length considered was 1 Mb; SNP density was required to be at least one per 70 kb; and a minimum of 5% overlapping windows was imposed [15, 22, 25, 26, 27, 28, 29].
Chromosomal coverage by ROHs was estimated according to the formula proposed by Al-Mamun et al. (2015) [30], in which the average ROH length per chromosome (across carriers) is divided by the chromosome size and expressed as a percentage [27, 30, 31].
ROHs were further classified into five length categories (0–2, 2–4, 4–8,
8–16, and
The genomic inbreeding coefficient (FROH) was calculated as described by McQuillan et al. (2008) [13]. Specifically, FROH corresponds to the total length of ROHs (LROH) divided by the length of the autosomal genome (LAUTO = 2464.80 Mb for goats) [13, 33, 34].
To identify the genomic regions most frequently associated with ROH, the
percentage occurrence of each SNP within ROHs was estimated by counting the
number of times each SNP appeared in a ROH and dividing this value by the number
of animals of each breed. SNP frequencies (%) within the detected ROHs were
calculated for each breed and plotted against SNP positions on the autosomes.
Following previous studies (Hervás-Rivero et al., 2024 [35]; Wirth et al.,
2024 [36]; Bertolini et al., 2018 [37]), ROH islands were defined as
genomic regions
HRRs were identified using the consecutive runs (CR) approach, applying the following thresholds: (i) a minimum of 10 SNPs per segment; (ii) up to five homozygous SNPs allowed; (iii) up to five missing SNPs tolerated; (iv) minimum segment length of 1 Mb; and (v) maximum distance between adjacent SNPs of 1 Mb [25].
In general, HRRs are expected to be shorter and less frequent (
Information on the annotated genes within the highly homozygous (ROH islands) and heterozygous (HRR islands) genomic regions detected in Marota and Anglo-Nubian goats was obtained from the Genome Data Viewer tool provided by national center for biotechnology information (NCBI) (https://www.ncbi.nlm.nih.gov/datasets/genome/GCF_001704415.2/) and GeneCards (http://www.genecards.org); last accessed on May 31, 2024. The Capra hircus genome assembly GCA_000317765.1 was used as a reference. Additionally, an extensive literature search was conducted.
A total of 22,872 ROHs were identified in the 192 samples analyzed. The number of ROHs on each goat chromosome is displayed in Fig. 2, which illustrates the plausible correlation between the number of ROH fragments and the length of each chromosome.
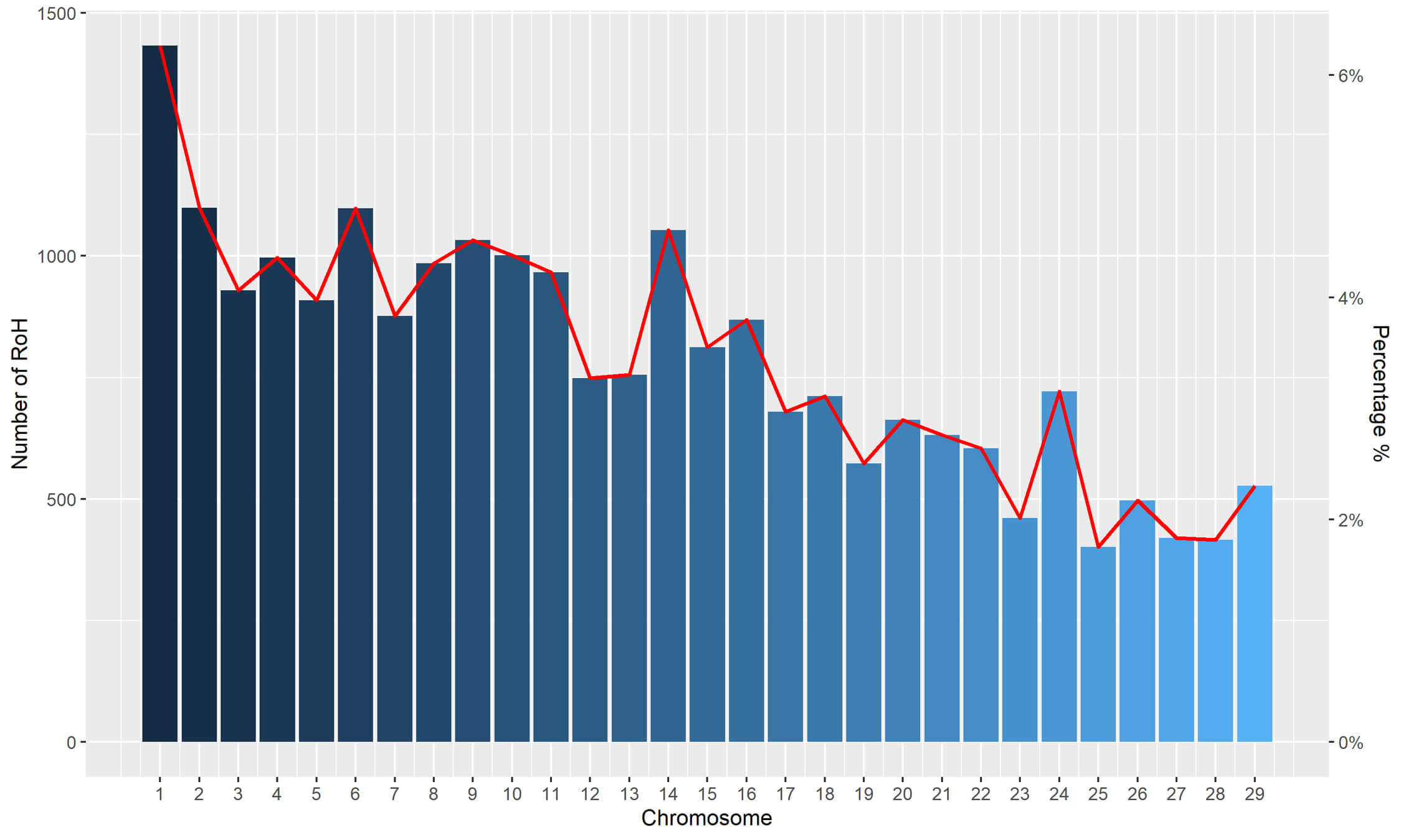 Fig. 2.
Fig. 2.
Number of runs of homozygosity (ROHs) per chromosome (bars) and average percentage of each chromosome covered by ROHs (lines) for all goats.
ROH segments were identified in both breeds, with mean lengths ranging from 2.01 Mb in the Marota breed to 2.07 Mb in the Anglo-Nubian breed. The average number of ROHs varied from 74.73 in the Anglo-Nubian breed to 173.85 in the Marota breed. The maximum individual ROH length was observed in the Marota breed (14.55 Mb). Marota goats also showed the highest average number of ROHs per individual (173.85), with a maximum of 269 ROHs, while the Anglo-Nubian breed exhibited the lowest values (1.0 Mb and 4.0 Mb, respectively; Table 1).
| Breed | *n | ROH length | ROH number | ||||
| Mean | *Min | *Max | Mean | Min | Max | ||
| Marota | 86 | 2.01 |
1.00 | 14.55 | 173.85 |
56 | 269 |
| Anglo-Nubian | 106 | 2.07 |
1.00 | 12.87 | 74.73 |
4 | 270 |
*‘n’ represents the number of individuals. *‘Min’ denotes the minimum values observed in estimations of individual animals within each breed, while *‘Max’ indicates the maximum values observed within each breed.
The average genomic inbreeding coefficient (FROH) for the Anglo-Nubian breed was 0.0627, with a range from 0.003 to 0.323. In contrast, the Marota breed exhibited higher FROH values, with an average of 0.1419 and a range from 0.040 to 0.283 (Fig. 3).
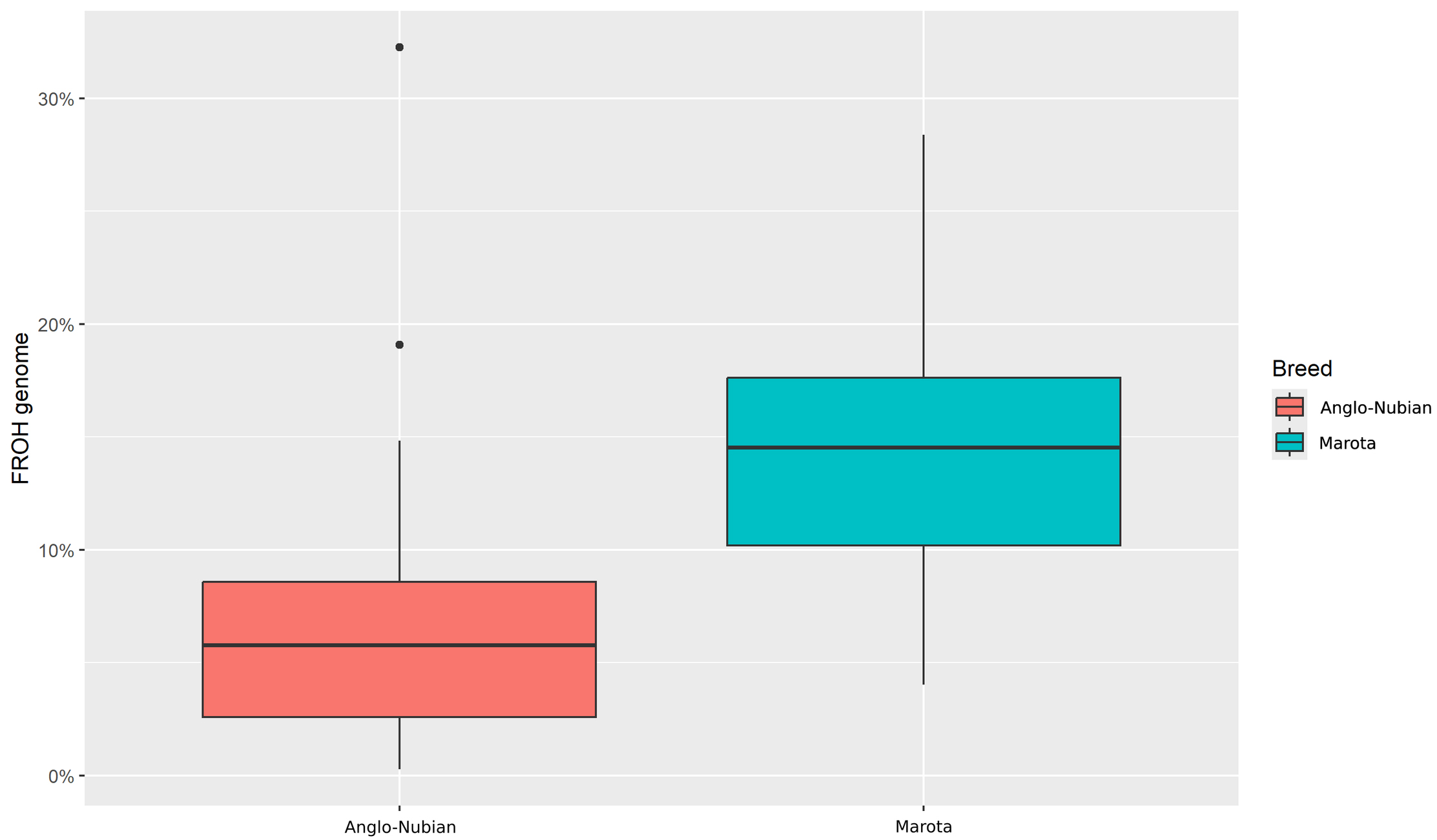 Fig. 3.
Fig. 3.
Box plots of genomic inbreeding (inbreeding coefficient based on runs of homozygosity, F𝐑𝐎𝐇) coefficients within the Anglo-Nubian and Marota goat breeds (Marota: n = 86; Anglo-Nubian: n = 106).
To investigate recent and past inbreeding in each breed, FROH was calculated for different length classes (Table 2).
| Class (Mb) | Marota* (n, %) | Anglo-Nubian (n, %) | ||
| 0–2 | 9813 | 65.60% | 4260 | 62.60% |
| 2–4 | 4962 | 28.50% | 2473 | 31.20% |
| 4–8 | 818 | 5.50% | 456 | 5.80% |
| 8–16 | 60 | 0.40% | 30 | 0.40% |
| 0 | 0.00% | 0 | 0.00% | |
| Total | 15,653 | 100% | 7219 | 100% |
* Marota is a local breed.
The distribution of ROH length classes is shown in Table 2. Both populations exhibited more ROHs between 1 and 2 Mb than in the other classes, with Marota showing the highest proportion (65.60%) and a very low proportion of ROHs above 8 Mb (0.4%). The Anglo-Nubian breed had the exact same proportion of ROHs above 8 Mb (0.4%).
To identify genomic regions potentially under selection and/or relevant for conservation, the frequency of SNPs contained in the runs was plotted on the autosomes of each breed (Fig. 4). Generally, adjacent SNPs with a proportion of ROH occurrences above the adopted threshold form genomic regions defined as ROH islands.
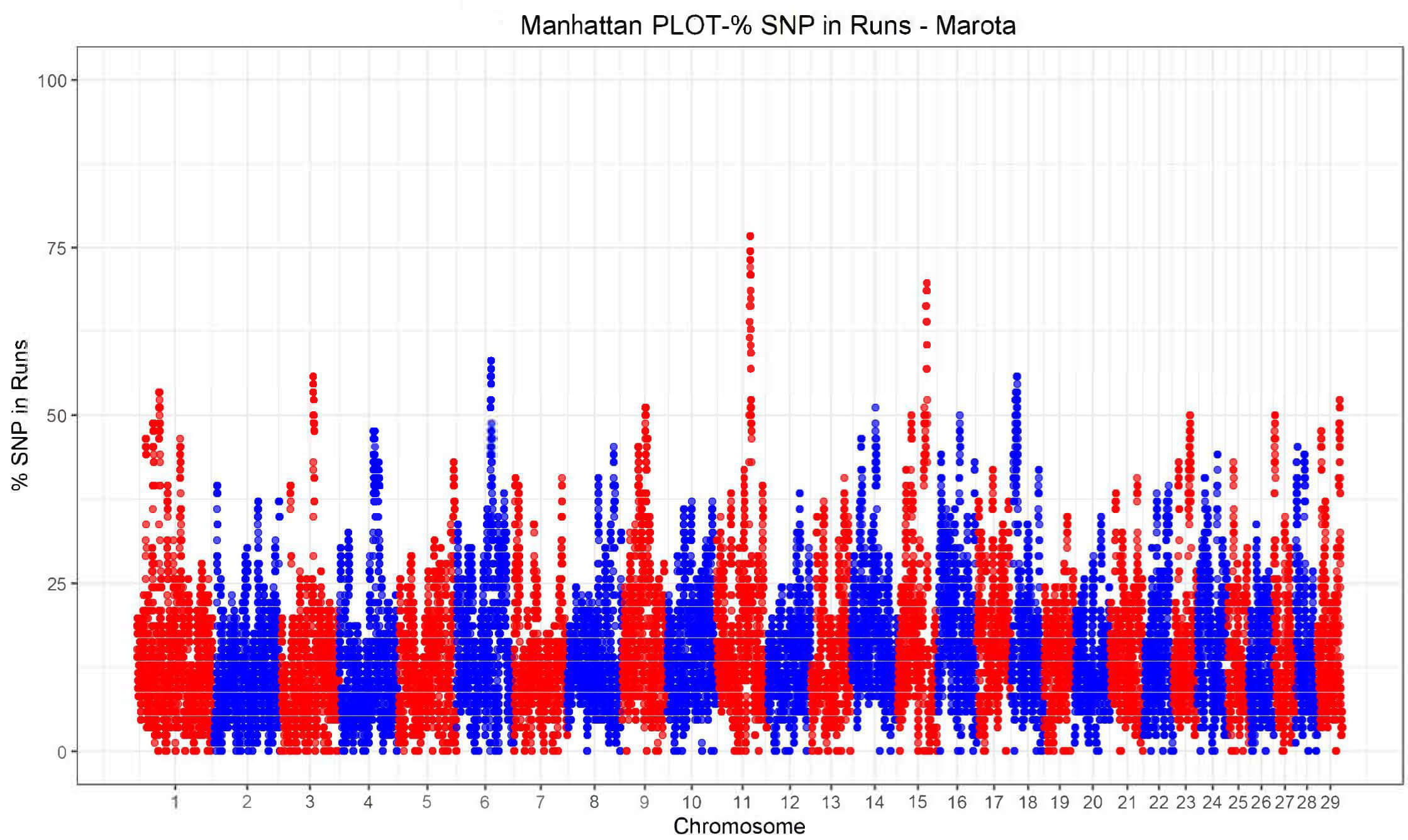 Fig. 4.
Fig. 4.
Manhattan plot of the proportion of times each SNP falls within a ROH in the Marota goat breed.
As shown in Fig. 4, a total of seven genomic regions characterized by a high occurrence of ROH, potentially relevant for selection, were identified. Applying the aforementioned 50% threshold, seven ROH islands were detected (Table 3), with the largest number found in the Marota breed.
| *CHR | Start (*BP) | End (*BP) | *n SNP | Genes |
| 1 | 45,076,527 | 45,783,017 | 12 | ABI3BP, SENP7, IMPG2, TRMT10C, pCNP, ZBTB11 |
| 6 | 69,566,353 | 70,525,889 | 17 | LNX1, GSX2, CHIQUE2, PDGFRA |
| 11 | 70,903,218 | 72,142,096 | 21 | PLB1, FOSL2, BRE, TRNAC-GCA, RBK5, MRPL33, SLC4A1AP, SUPT7L, GPN1, CCDC121, ZFN512, GCKR, FNDC4, IFT172, KRTCAP3, NRBP1, PCPM1G, ZFN513, SNX17, FEI2B4, GTF3C2, MPV17, UCN, TRIM54, DNAJ5G, SLC3AOA3, ATRAID |
| 11 | 67,889,566 | 70,479,268 | 48 | ANX4, GMCL1, SRNP27, MXD1, ASPRV1, PCBP1, C11H2ofr42, TIA1, PCYOX1, SNRPG, EHD3, CAPN14, GALNT14, CAPN13, LCLAT1, LBH, ALK, CLIP4, SRNP27, MXD1, ASPRV1, PCBP1, C11H2ofr42, TIA1, SNRPG, EHD3, TRNAG-CCC, CAPN14, GALNT14, TRNAG-UCC, CAPN13, YPEL5 |
| 15 | 57,947,094 | 60,035,602 | 34 | NCAM1, C15H11orf57, USP28, CLDN25, ZW10, DRD2, HTR3B, TMPRSS5, ANKK1, TTC12, PT, BCO2, TEX12, IL18, PIH1D2, DLAT, DIXDC1 |
| 18 | 11,115,584 | 12,091,788 | 18 | CDH13, HSBP1, MLYCD, OSGIN1, NECAB2, SLC38A8, MBTPS1, HSDL1, DNAAF1, TAF1C, ADAD2, KCNG4, WFDC1, TLDC1 |
| 18 | 8,548,476 | 9,214,650 | 15 | CDIL2, DYNLRB2, CMC2, CENPN, ATMIN, C18H16ofr46, GCSH, PKD1L2 |
*CHR, chromosome; *BP, base pairs; *n SNP, number of SNPs in the ROH island.
The autosomes most consistently associated with regions of high ROH occurrence were chromosomes 01, 03, 06, 11, 15, and 18. No ROH islands were identified in the Anglo-Nubian breed, which stood out as an exception (Table 3).
The analysis of HRR was conducted using the investigative method, yielding a total of 17,278 regions. Table 4 provides descriptive statistics for HRR according to breed.
| Breed | *n | HRR length | HRR number | ||||
| Mean | *Min | *Max | Mean | Min | Max | ||
| Marota | 86 | 1.18 |
1.00 | 2.98 | 67.24 |
48 | 108 |
| Anglo-Nubian | 106 | 1.18 |
1.00 | 2.63 | 108.44 |
41 | 360 |
*‘n’ represents the number of individuals. *‘Min’ denotes the minimum values observed in estimations of individual animals within each breed, while *‘Max’ indicates the maximum values observed within each breed. HRRs, heterozygosity-rich regions.
Regions occurring in at least 45% of animals were deemed HRR islands and were further examined for candidate genes and pathways. The specific HRR islands identified in Marota and Anglo-Nubian goats are reported in Table 5.
| Breed | *CHR | Start (*BP) | End (*BP) | *n SNP | Genes |
| Marota | 19 | 7,937,845 | 9,069,843 | 10 | MSI2, MRPS23, CUEDC1, VEZF1, SRSF1, DYNLL2, MKS1, LPO, MPO, TSPOAPI, SUPT4H1, RNF43, HSF5 |
| Marota | 13 | 35,082,374 | 35,788,791 | 8 | BAMBI, WAC, MPP7 |
| Anglo-Nubian | 03 | 164,779 | 1,669,699 | 18 | DUSP28, OTOS, ANKMY1, GPC1, COPS9, NDUFA10 |
| Anglo-Nubian | 27 | 18,840,355 | 19,909,422 | 11 | GTF2E2, SMIM18, RBPMS, DCTN6, MBOAT4, LEPROLTL1, SARAF, TRNAQ-CUG, TRNAE-UUC |
| Anglo-Nubian | 21 | 22,153,516 | 23,030,023 | 10 | HOMER2, CPEB1, AP3B2, FSD2, UAU, FAM103A1 |
| Anglo-Nubian | 15 | 80,435,546 | 81,371,836 | 9 | MRE11A, GRIA4, MSANTD4, KBTBD3, AASDHPPT, GPR83, TRNAG-UCC, IZUMO1R, ANKRD49 |
*CHR, chromosome; *BP, base pairs; *n SNP, number of SNPs covered by the HRR.
To provide a concise overview of the functional roles of the key genes discussed in this study, we included a supplementary table summarizing their biological functions and associated GO terms (Supplementary Table 1).
ROH analysis is a widely used tool to elucidate the demographic history of populations, as it provides precise information on genetic diversity, while the evaluation of the length of ROH makes it possible to detect past and recent genomic inbreeding in animals [32, 43, 44, 45, 46, 47].
In this study, the average number of ROHs in local goat breeds from Northeast Brazil ranged from 74.73 in Anglo-Nubian goats to 173.85 in Marota goats. These values are higher than those reported for Turkish (60 ROHs), Russian (approximately 73 ROHs), and Central Asian (90 ROHs) breeds [32, 37, 48]. In the context of American goats (average of 133 ROHs), only Anglo-Nubian goats showed lower values [37].
However, comparisons with Turkish, Russian, and Central Asian goats should be
interpreted cautiously given differences in SNP chip density and ROH definitions
across studies. Using standardized classes (0–2, 2–4,
Overall, differences in the number and characteristics of ROHs may reflect historical disparities in genomic structure. Highly selected breeds often exhibit a greater abundance of ROHs and broader coverage compared to local breeds [11, 31, 33, 50, 51, 52]. In our study, the local Marota breed exhibited a markedly higher number of ROHs compared to Anglo-Nubian goats. Bertolini et al. (2018) [37] identified a trend of increased homozygosity in local breeds, attributing this phenomenon to their small population size and geographical isolation.
This study provides new insights into genomic inbreeding levels in goat breeds from the Brazilian semi-arid region. Understanding inbreeding is critical for herd management, as it improves estimates of kinship, corrects errors in pedigrees, and reveals undocumented ancestral inbreeding. This approach is particularly beneficial for small, at-risk populations [6, 48, 52, 53].
The average FROH in Brazilian local Marota goats (0.1419) exceeded that of other goat breeds worldwide (FROH = 0.12). In contrast, Anglo-Nubian goats exhibited lower FROH values (0.0627) and demonstrated a lower degree of inbreeding compared to several goat breeds [48, 54, 55].
In the local breed, this situation could encourage reproduction between closely related individuals, leading to a significant prevalence of recessive genes in homozygous conditions due to inbreeding and genetic alterations. Consequently, they would become more susceptible to selective pressures [26, 37, 44, 56]. Therefore, closely monitoring the population of this goat breed is essential to prevent the loss of genetic resources and minimize the negative effects of harmful mutations, inbreeding depression, and reduced genetic diversity [4, 50].
Analyzing goat populations in institutional herds, a significant prevalence of
short ROH segments was observed—specifically, 21,508 segments (
Generally, comparing ROH results is not a simple task due to the variety of
criteria used in different studies. Our research revealed that in the Marota and
Anglo-Nubian breeds, the majority of ROHs are short or medium in size. Therefore,
our findings suggest that animals experienced ancestral inbreeding events due to
the absence of longer ROHs [12, 26, 56, 57]. Deniskova et al. (2021) [32]
reported similar findings (ROH
Previous studies have already provided formal evidence of population structure and genetic introgression in these breeds. In a study entitled “Population genomics and gene introgression in goat herds naturally adapted to Brazil” (Moura et al., 2019 [4]), genomic analyses including Principal Component Analysis (PCA), Fixation Index (FST), and Bayesian clustering approaches implemented in STRUCTURE and ADMIXTURE clearly demonstrated genetic differentiation between Marota and Anglo-Nubian goats (FST = 0.16), as well as signals of Anglo-Nubian introgression, particularly in private Marota herds.
These findings corroborate our present results based on ROHs and FROH (Supplementary Fig. 1), reinforcing the endangered status of the Marota breed. While management strategies have contributed to the preservation of Marota goats, the high level of inbreeding highlights the need for continuous monitoring and adaptive conservation practices [4, 44, 58].
Our genomic findings have direct implications for conservation. The elevated FROH observed in Marota goats (0.1419) highlights the need to prioritize individuals with lower inbreeding levels in breeding programs to mitigate inbreeding depression. Moreover, the identification of ROHs and HRRs harboring candidate genes involved in immunity, reproduction, and adaptation (e.g., IL18, TEX12, GMCL1) underscores the importance of maintaining heterozygosity at key adaptive loci. These insights can be integrated into breeding schemes in both official conservation nuclei and private herds, where different levels of Anglo-Nubian introgression have been documented (Moura et al., 2019 [4]), thereby providing a genomic basis for long-term strategies to preserve the adaptive potential and genetic integrity of the Marota breed.
Livestock farming is a human practice, influenced by various environmental factors. When environmental conditions are unfavorable, animals can adapt in various ways to cope with stressors. Therefore, biological adaptation is essential to ensure that animals are healthy and productive. In their DNA, it is possible to find marks of natural selection that arise in response to environmental pressures, and which can be identified through genomic and bioinformatics techniques [12, 19, 42, 58, 59, 60].
In this context, some goat breeds introduced to the Brazilian semi-arid region demonstrate adaptive evolution aimed at survival, reproduction, and production across different ecological zones. It is likely that their genomes have developed unique genetic characteristics, evolving to adapt to remarkably heterogeneous environments [4, 7, 61]. It is therefore expected that artificial and natural selection will act on animal genomic diversity, leaving positive or deleterious adaptive signatures [45, 46, 57, 62].
Analysis of ROHs and HRRs reveals, in the Marota and Anglo-Nubian breeds, some markers potentially under selection for adaptation to the semi-arid environment of Brazil. Our discoveries led to the identification of target genes related to adaptation and, more specifically, to the immune/inflammatory response, energy homeostasis, reproductive and production traits and heat stress [12, 19, 23, 58, 59, 61, 63].
However, it is important to highlight that no ROH islands were detected in the
Anglo-Nubian breed. This absence is likely related to its demographic history and
genetic composition. As an exotic and admixed population introduced into Brazil,
Anglo-Nubian goats have undergone continuous crossbreeding and artificial
selection, which increases genomic diversity and reduces the probability of long
homozygous segments being shared by most individuals (Peripolli et al.,
2017 [64]). In addition, the stringent thresholds applied in this study
(
Research into the specific functions of genes has unveiled an intricate panorama of biological processes, wherein genes play distinct and essential roles in cellular functions, contributing to a wide range of biological processes. For example, the GMCL1 and TEX12 genes are known to be involved in the regulation and/or participation of reproductive processes, specifically in sperm production [69, 70, 71, 72, 73].
The ZBTB11 gene, with its zinc finger integrase-like His-His-Cys-Cys (HHCC), serves as a crucial regulator in neutrophil differentiation, contributing to the immune defense system. Similarly, the IL18 gene expresses the IL-18 protein, a pro-inflammatory cytokine pivotal for immune response regulation, activating immune system cells like T lymphocytes and natural killer (NK) cells, thereby enhancing the production of inflammatory cytokines [74, 75].
The LBH and YPEL5 genes play specific roles in embryonic development, RNA processing, and cell cycle regulation, thereby influencing cell growth [76]. The data indicate that certain genes, subject to natural selection, are involved in a wide range of cellular metabolic processes, including protein cleavage and lipid homeostasis. These genes include PCYOX1, CAPN14, GALNT14, TRNAG-UCC, CAPN13, and LCLAT1 [77, 78, 79, 80]. Genes like CLDN25 and ZW10 actively participate in the regulation of cellular permeability, chromosomal segregation during cell division, and may play a role in cell growth [81, 82].
Heterozygosity-rich regions exhibit high rates of heterozygosity, corresponding to a new concept introduced by Williams et al. (2016) [83]. HRRs are much less characterized than ROHs in livestock. These regions are identified as heterozygous genomic regions potentially associated with disease resistance, immunity, and adaptive processes, serving as a valuable genetic reservoir and providing additional understanding of goat genomes [19].
Heterozygosity loci are clustered in islands throughout the genome and are significantly rarer and shorter compared to ROH [19]. Several studies have reported divergent means of HRR per chromosome [12, 19, 84]. Variations in methods, animal populations, and SNP arrays may account for differences with our results, a trend similarly observed by Chessari et al. (2024) [19].
After determining the threshold of 45% for SNPs most frequently found in HRRs, 66 SNPs were identified beyond this threshold. These SNPs formed six regions across six chromosomes. Specifically, chromosomes 3, 13, 15, 19, 21, and 27 each displayed a single region containing genes. In these regions, we identified a total of 46 genes. Based on genetic functions obtained from http://www.genecards.org (last accessed on 04/07/2024), we conclude that the genes found in HRRs are important for adaptive biological processes.
Notable examples include 3 of the 46 genes with known and well-described functions. Particularly interesting is the MPO gene, located on chromosome 19, which encodes the enzyme myeloperoxidase. In humans, myeloperoxidase is involved in generating free radicals and hypochlorite, which are released by neutrophils during the inflammatory response to bacterial infections [85].
Similarly, the LPO gene encodes the enzyme lactoperoxidase, which plays a fundamental role in immune defense against bacterial infections by catalyzing the production of hypothiocyanous acid (HOSCN) from H2O2 and SCN–, a component of the antibacterial defense system present in the respiratory tract [86]. The WAC gene is also a vital component of the molecular mechanism that coordinates the cell’s response to genotoxic stress, ensuring genome integrity and maintaining cellular homeostasis through transcriptional regulation of p53 target genes [87].
Our findings align with previous research involving Russian cattle, particularly in identifying a single candidate region on chromosome 15 of the Anglo-Nubian goat, which matches the scan results for selective sweeps. Within this region, several genes are located, notably MSANTD4 and GRIA4. Both are potential contributors to the climate stress resistance phenotype, given their indirect functions in the cold shock response (MSANTD4) and body thermoregulation (GRIA4) [88].
Although this study identified candidate SNPs in genes associated with ROHs and HRRs (e.g., IL18, TEX12, GMCL1), further experimental validation is required to confirm these findings. Approaches such as Sanger sequencing or Kompetitive Allele-Specific PCR (KASP)/TaqMan assays in independent cohorts would strengthen the evidence for these variants and their role in adaptation and inbreeding. Future work will prioritize validation of SNPs with higher functional impact and occurrence within ROH/HRR islands.
The integration of genomic tools into Brazilian conservation and breeding programs may provide a practical framework to safeguard genetic diversity. Estimates of genomic inbreeding (FROH) allow the selection of animals with lower relatedness to minimize inbreeding, while ROH/HRR analyses highlight adaptive genes essential for resilience. These approaches, already demonstrated in goat populations worldwide [37, 44], could complement ongoing initiatives in Brazil, reinforcing strategies to conserve endangered local breeds such as the Marota [4].
ROH models revealed low levels of introgression and minor gene flow between the Anglo-Nubian and Marota breeds. Higher FROH values in the Marota breed severely affect its overall genetic diversity, demonstrating high levels of molecular inbreeding and classifying this population as endangered.
Some of the gene sequences within ROH and HRR islands are linked to adaptive pathways. Thus, the presence of ROH and HRR islands in the Marota genome highlights the evolutionary forces driving adaptation. The ROH and HRR analyses reinforce the strength of using genomic marker-based data to mitigate future loss of diversity and indicate a need for more powerful conservation programs.
The paper is listed as a preprint titled “Genomic analysis revealed hotspots of genetic adaptation and risk of disappearance in the Brazilian goat populations” on bioRxiv at: https://www.biorxiv.org/content/10.1101/2024.06.03.597192v1.
ROH, runs of homozygosity; HRR, heterozygosity-rich regions; SNP, single nucleotide polymorphism; FROH, inbreeding coefficient based on runs of homozygosity; EDTA, ethylenediaminetetraacetic acid; DNA, deoxyribonucleic acid; MAF, minor allele frequency; LAUTO, length of the autosomal genome; CR, consecutive runs; NCBI, National Center for Biotechnology Information; CHR, chromosome; BP, base pairs; NK cells, natural killer cells.
All data points generated or analyzed during this study are included in this article and there are no further underlying data necessary to reproduce the results. Further data can be obtained from the first author or the corresponding author.
FDS, RC, IN, JM, MB, LC, FB, JS, SM, DA and AA conceptualization. FDS, IN, JM, MB, LC, FB, JS, SM and AA methodology. DA, FDS, IN, JM, MB, LC, FB, JS, SM, AA and LS validation. DA, FDS, RC, IN, JM, MB, LC, FB, JS, AA and LS formal analysis. FDS and AA resources. FDS wrote the original draft. DA, FDS, RC, IN, JM, MB, LC, FB, JS, SM, AA and LS wrote, revised and edited. FDS, RC and AA visualization. FDS, MB, LC, FB, JS, SM and AA supervision. FDS and LS made the editorial changes. All authors viewed, read, re-viewed and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be ac-countable for all aspects of the work.
All animal procedures were conducted in accordance with the ethical standards of the institutional animal care committee and followed the Brazilian guidelines for the care and use of animals in research established by the National Council for the Control of Animal Experimentation (CONCEA). The study was approved by the Ethics Committee on Animal Use of the Federal University of Piauí (protocol no. 058/14).
We would like to express our gratitude to The Foundation of Support to Research of Piauí (FAPEPI).
This research received no external funding.
The authors declare no conflict of interest. Danielle Maria M. Ribeiro Azevêdo and Adriana Mello de Araújo are affiliates of Brazilian Agricultural Research Corporation; the judgments in data interpretation and writing was not influenced by this relationship.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/FBS43536.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.