




1 Pathology Informatics, Bioinformatics, and Data Science, Department of Pathology and Laboratory Medicine, Children's Hospital Los Angeles, Los Angeles, CA 90027, USA
Abstract
Leigh syndrome (LS), first reported in 1951, is the most common primary mitochondrial disease. The overarching term, Leigh Syndrome Spectrum (LSS) was proposed by a ClinGen Expert Panel to encompass the wide continuum of neurodegenerative and non-neurologic manifestations which were associated with classic LS and Leigh-Like Syndrome (LLS). Notably, LSS typically presents developmental regression or delay by two years of age, with about 20% of cases presenting as late-/adult-onset forms after 2 years. Historically defined by clinical, biochemical, and neuropathological findings, the genetic basis of LSS has been elucidated through the use of Sanger and next-generation sequencing (NGS), resulting in the discovery of over 120 causative genes. Moreover, LSS can be caused by mutations in both nuclear-encoded genes and mitochondrial DNA (mtDNA), with overlapping clinical characteristics that occur at similar frequencies. This review aims to summarize the clinical and onset characteristics of LSS, genetic testing-aided diagnosis criteria, and the development of treatments. Furthermore, this review organizes the years since the first reports of gene and mutation discoveries into four consecutive eras: Clinical-Biochemical Era (1990–1999), Early Genomics Era (2000–2009), NGS Revolution Era (2010–2019), and Modern Era (2020–Present). Thus, using this framework, this review chronicles the evolution of LSS molecular genetics and treatment development, highlighting the shift from supportive care to targeted therapies driven by modern technologies. Cornerstone experimental models, such as the Ndufs4 -/-knockout mouse and patient-derived induced pluripotent stem cells (iPSCs), have facilitated mechanistic studies and drug repurposing screens, including the identification of sildenafil as a potential therapeutic agent, which has led to medical improvements in patients. Current advances in gene editing, including mitochondrial single-base editors such as eTd-mtABE and mitoBEs, are enabling gene therapy with precise introduction and correction of LS-causing variants in rat and mouse models. On the preventative front, Mitochondrial Replacement Therapy (MRT), guided by precise maternal mtDNA genotyping, has been successfully applied in clinical practice, allowing mothers carrying LSS-causing mtDNA variants to have healthy babies free of the LS manifestation. Collectively, these advances in gene discovery, genetic diagnosis, sophisticated disease modeling, rapid screening of small molecule drugs, precise gene editing for gene therapy, and innovative treatment strategies, such as MRT, are ushering in an era of precision medicine for LSS.
Keywords
- Leigh Syndrome (LS)
- Leigh Syndrome Spectrum (LSS)
- targeted therapy
- precision medicine
- mitochondrial replacement therapy (MRT)
- gene therapy
- mitochondrial DNA (mtDNA)
- Ndufs4-/- knockout mouse
Leigh syndrome (LS) (OMIM: 25600, Orphanet ID 506) was originally described by Archibald Denis Leigh in 1951 as Subacute Necrotizing Encephalomyelopathy [1], which was later renamed Leigh syndrome. LS is the most frequent manifestation of primary mitochondrial diseases (PMDs), with a prevalence of approximately 1 in 40,000 live births [2]. LS is a devastating disease with a highly heterogeneous symptom spectrum and disease course. This first reported case carried hallmark symptoms for LS, including early-onset development of necrotizing lesions in the grey nuclei of the brain stem, subthalamic region, and basal ganglia; bilateral optic atrophy; deafness; and bilateral spasticity [3].
In earlier years, clinical and research work into LS was largely observational, limited to physical and biochemical characterization with little dissection of the molecular genetic basis due to the lack of DNA sequencing techniques. In the 1990s, the advent of Sanger sequencing technique enabled the gene discovery through family segregation study and positional cloning. The causative genes and mutations were discovered in both mitochondrial genome DNA (mtDNA) [4] and nuclear genome [5], which gradually led to the discovery of dozens classic LS genes by the early 2000s. High-throughput next generation sequencing (NGS) technology, introduced in 2007 by Illumina, revolutionized the speed and width of LS gene discovery, enabling the broad availability of genetic testing products covering LS diagnosis for patients across the world. NGS-based patient tests led not only to more classic LS gene discovery, but also frequent Leigh-Like Syndrome (LLS) gene discovery where atypical symptoms may disqualify the patients being strictly diagnosed with classic LS and thus the genes may be overlooked and molecular diagnosis missed. The quick accumulation of LLS cases and genes with variable overlapping symptoms with classic LS cases led to the introduction of the overarching nomenclature, Leigh Syndrome Spectrum (LSS), by Mito-GCEP Expert Panel [6].
Like other PMDs, LS is caused by mitochondrial dysfunction due to mutations in mitochondrial DNA (mtDNA)-encoded genes or nuclear-encoded genes. There are 14 subtypes (child nodes) according to disease ontology tree at MSeqDR (Fig. 1, and URL: https://mseqdr.org/leigh.php). French-Canadian Type Leigh syndrome (LSFC, OMIM: 220111) is a subtype prevalent in the Saguenay-Lac-Saint-Jean region of Quebec, Canada, caused by population’s founder mutations in the LRPPRC gene. Another Leigh syndrome subtype is Maternally Inherited Leigh Syndrome (MILS) caused by mtDNA mutations inherited from the mothers, but this designation is rarely used considering that many causative mtDNA variants are de novo rather than inherited.
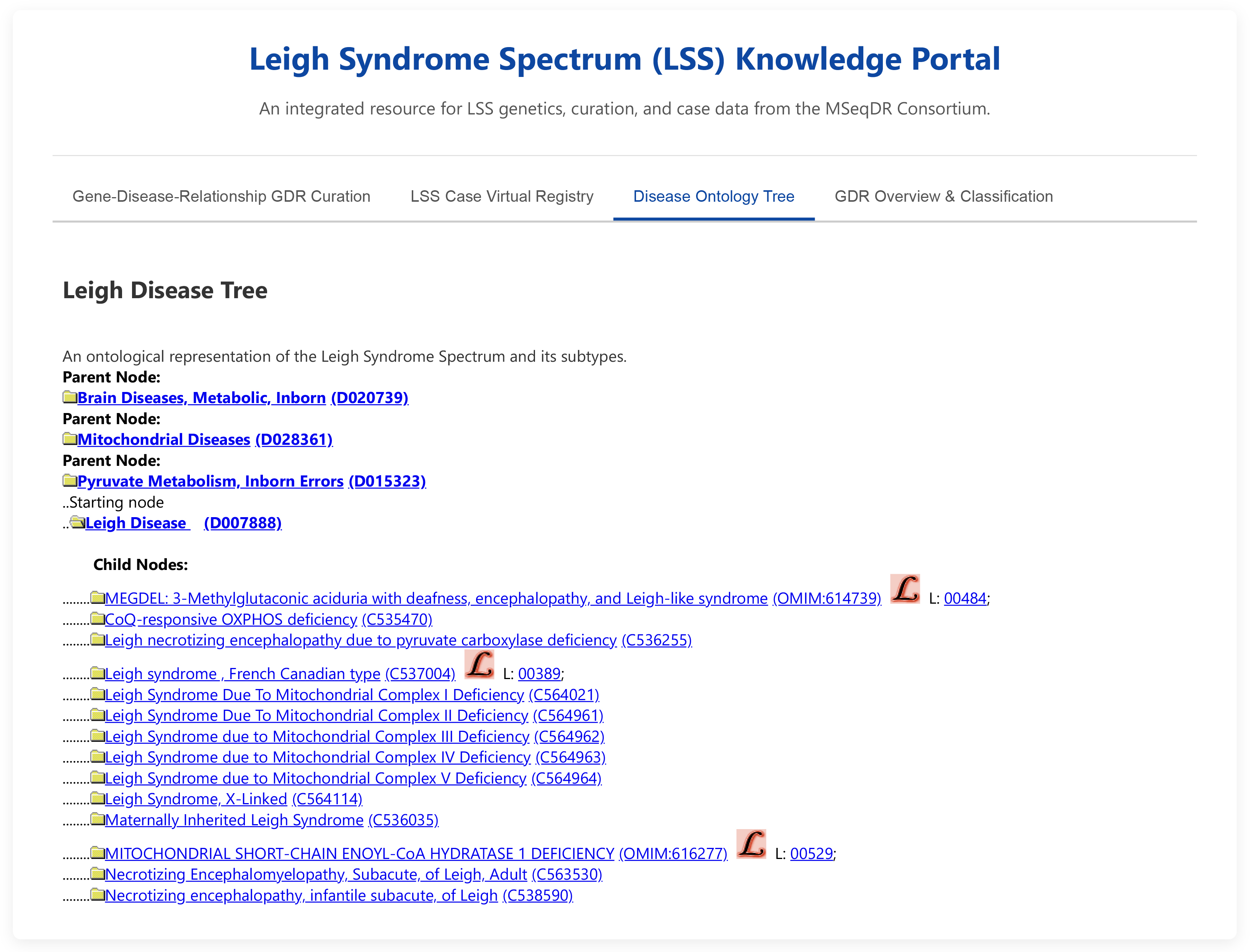 Fig. 1.
Fig. 1.
Screenshot of leigh syndrome spectrum (LSS) knowledge portal.
Leigh-Like Syndrome (LLS): Some patients may not meet the strict diagnosis criteria for classic LS including the requirement to display all the hallmark symptoms. The disease name Leigh-Like Syndrome (LLS) is used for such cases whose phenotypes resemble classic Leigh syndrome but may fail to meet some of the classic diagnostic criteria such as the characteristic brain lesions are present with atypical clinical features [6, 7]. Biotinidase deficiency is also regarded as LLS in cases with some LS hallmark manifestations [8].
Leigh Syndrome Spectrum (LSS): Given the overlap between LS and LLS, the Clinical Mito-GCEP Expert Panel proposed the overarching term, LSS, to be the disease entity accounting for the highly variable manifestations from LS to LLS [6]. This effort represents the first time these clinical entities have been redefined by the mitochondrial disease community in 25 years. The Mito-GCEP classified 113 genes on the basis of their LSS disease causality, classifying 31 genes as “definitive”, 38 genes as “moderate”, and 45 genes as “Limited” or “Disputed” to disease relationships (GDR) for LSS [6].
The clinical characteristics of Leigh Syndrome Spectrum (LSS), whether caused by mutations in nuclear-encoded genes (nuclear-LSS) or mitochondrial DNA mutations (mtDNA-LSS), are largely overlapping and present as a continuum of progressive neurodegenerative disorders. Most of the common symptoms present at similar frequencies between the two causes, with the notable exceptions of muscle weakness (30% vs 75%) and dysphagia (25% vs 80% fact) which are both more common in mtDNA-LSS patients [9, 10].
The primary symptoms are neurologic and occur at largely similar frequencies between both nuclear-LSS and mtDNA-LSS forms. Developmental delay and/or regression are core symptoms of LSS. Motor system involvements include hypotonia and movement disorders such as dystonia and chorea, spasticity, and cerebellar ataxia. Additionally, seizures/epilepsy is a common manifestation, and peripheral neuropathy can occur in patients.
Importantly, brain stem dysfunction and lesions may lead to a variety of critical issues. These include respiratory abnormalities such as apnea, central hypoventilation or hyperventilation, and irregular respiration, as well as bulbar problems which often lead to abnormal swallowing (dysphagia)—in up to 80% mtDNA-LSS and 25% in nuclear-LSS, and speech difficulties (dysarthria). Other brain stem-related signs include ophthalmoparesis and abnormalities of thermoregulation.
Neuroimaging is crucial for LSS diagnosis and typically reveals characteristic patterns. The hallmark finding is the presence of bilateral and typically symmetric lesions in the central nervous system. On T2-weighted magnetic resonance imaging (MRI), these lesions appear as hyperintensities (bright signals). On computed tomography (CT), they may be seen as hypodensities. These lesions are most found in the basal ganglia, thalamus, and brain stem. The periaqueductal gray matter, medulla, and spinal cord can also be involved.
While LSS is primarily a neurologic disorder, various other systems can be affected, particularly in mtDNA-LSS. (1) Gastrointestinal manifestations that include poor weight gain and other gastrointestinal issues, (2) Cardiac involvement that may include cardiomyopathy and conduction defects, and (3) Other systems that include hepatic (liver), renal (kidney), and endocrine (hormonal) manifestations.
Leigh Syndrome Spectrum (LSS) is predominantly a group of diseases of infancy and early childhood. The onset for both nuclear-encoded (nuclear-LSS) and mitochondrial DNA-encoded (mtDNA-LSS) is typically in infancy or early childhood, often between the ages of 3 and 24 months. However, a significant minority of cases present later in childhood (late-onset) or even in adulthood (adult-onset), about 20% and 30% in two Chinese cohorts respectively [11, 12]. These late-onset forms often exhibit different clinical spectrums, better prognosis, and genetic associations compared to the classic early-onset cases. LSS onset frequently follows a metabolic challenge, such as an intercurrent viral illness, surgery, prolonged fasting, or vaccinations.
The disease often presents with sudden neurodevelopmental regression. This decompensation is commonly associated with elevated lactate levels in the blood and/or cerebrospinal fluid (CSF). While motor and developmental regression are hallmarks of early-onset Leigh syndrome, late-onset cases can present with a wider range of neurological symptoms, including ataxia, dystonia, seizures, and peripheral neuropathy [7, 11, 12, 13].
Mitochondrial gene-encoded Leigh Syndrome Spectrum (mtDNA-LSS) is maternally
inherited, for a minority of the patients (about 30%). About 40% of mtDNA-LSS
patients have mtDNA pathogenic variants affecting complex V subunit
MT-ATP6, with the most common variants being m.8993T
Nuclear gene-encoded Leigh Syndrome Spectrum (nuclear-LSS) accounts for about 70% of the cases, caused by over 120 LSS-related genes reported to date. Most of the genes are associated with autosomal recessive inheritance. There are only a few genes associated with LSS in autosomal dominant modes: DNM1L, OPA1, SLC25A4, and SSBP1, which are all de novo pathogenic variants. Interestingly, they are all involved in mitochondrial fission/fusion (DNM1L, OPA1, SLC25A4) and maintenance (SSBP1) [6, 10].
X-linked inheritance is reported for LSS caused by a heterozygous or hemizygous pathogenic variants in genes PDHA1, AIFM1, NDUFA1, and HSD17B10, where PDHA1 is the first discovered nuclear LSS gene [5].
Currently, the diagnosis of both mtDNA-LSS and nuclear-LSS can be established in a proband both fulfilling clinical criteria for LSS as detailed below and carrying one or two pathogenic or likely pathogenic variants. These variants in specific mtDNA or nuclear genes are listed in reference databases such as ClinVar and regional or disease society guidelines [6]. The variant genotypes and zygosity must be consistent with the expected mode of inheritance. The diagnosis of mtDNA-LSS includes identification of a heteroplasmic or homoplasmic pathogenic variant in one of the known causative mtDNA genes known to be involved in mtDNA-LSS. For nuclear-LSS, the pathogenic variants can be in more than 120 nuclear genes known to be associated with LSS under compatible autosomal recessive, autosomal dominant, or X-linked inheritance modes [6, 7].
Clinical criteria below for mtDNA-LSS and nuclear-LSS are adapted from the Mito-GCEP expert panel consensus [6].
Typical neuroradiologic findings: Must include the bilateral and typically symmetric hyperintensities on T2-weighted magnetic resonance imaging (MRI) or hypodensities on CT. Hyperintensities or hypodensities must be found in the brain stem and/or basal ganglia with or without bilateral. Alternatively, Hyperintensities or hypodensities must be found in the thalamus, cerebellum, subcortical white matter, and/or spinal cord.
AND at least one of the following characteristic neurologic clinical features: Developmental regression, developmental delay, or neurobehavioral/psychiatric features.
AND at least one of the following biochemical and/or mitochondrial abnormalities: Elevated concentration of lactate and/or glycine in relevant tissues, respiratory chain enzyme activity deficiency, pyruvate dehydrogenase complex in respective tissues, or mitochondrial fission/fusion defect.
For nuclear-LSS, molecular genetic testing approaches include comprehensive whole genome (WGS) or whole exome (WES) tests, PMD-targeted multi-gene panel tests, or single-gene tests. For mtDNA-LSS, molecular genetic testing may be an integral part—either as by-product or specially augmented with mitochondrial genome content—of the comprehensive WGS or WES tests or a pure whole mtDNA genome sequencing.
Molecular testing with LSS coverage is widely available across the world, and the choice of approach may be dependent on clinical suspicion, age of presentation, and availability of genomic testing. A search of NCBI Genetic Test Registry (GTR) website returned 271 tests from 32 labs across the world, using the following keyword filters: Test purpose = “Diagnosis” and Test method = “Sequence analysis of the entire coding region” (https://www.ncbi.nlm.nih.gov/gtr/all/tests/?term=Leigh%20syndrome&filter=testpurpose:diagnosis, accessed on July 22, 2025), many of them are multi-gene panels and some of them are single LSS gene tests. Understandably, WGS and WES can also cover the test for nuclear-LSS and most mtDNA-LSS.
The genetic study of LS began with the first description of the disease in 1951, yet molecular genetics only truly started in the 1980s after Sanger sequencing technology became available. We searched for all the PubMed entries with “Leigh syndrome” keywork in either the title or abstract retuned about 1100 matching publications for reviewed. With a comprehensive review of historical scientific progress in understanding and treating Leigh syndrome, we can divide the period between 1951 to 2025 into 4 chronologically sequential periods, with most periods spanning roughly a decade and characterized by distinct technology status regarding LS molecular genetics, clinical investigation, and treatment innovation. We then generated a chronicle narrative summary that describes the key advancements in each era including gene discoveries, the emergence of new animal and cell line models, and the evolution of therapy development and treatment strategies.
The initial decades of Leigh syndrome (LS) research was comprised mostly of clinical observations and foundational investigations in biochemistry and imaging. LS was first described as a distinct neuropathological entity as “subacute necrotizing encephalomyelopathy”, characterized by symmetrical lesions in the brainstem and basal ganglia. Early research focused on establishing LS as an inborn error of metabolism, with strong evidence pointing towards defects in cellular energy production.
Studies from the 1970s through the 1990s frequently linked LS to elevated lactate and pyruvate levels, suggesting a primary defect in pyruvate metabolism. The X-linked form of Leigh syndrome was inferred from an unexplained male/female ratio in Leigh’s syndrome of 1.83/1 [14]. These led to the identification of Pyruvate Dehydrogenase Complex (PDHC) deficiency as a significant cause, with mutations in the X-linked PDHA1 gene being the first nuclear gene mutation discovered as a cause of LSS [5, 15]. Deficiencies in mitochondrial respiratory chain complexes, particularly Cytochrome c Oxidase (Complex IV), were also reported [16], though the underlying genetic causes remained elusive. Functional studies were limited to biochemical assays on patient tissues, primarily muscle and fibroblasts.
The first mitochondrial DNA (mtDNA) mutation linked to LS, the m.8993T
| Decade period | Genes first discovered during the period (Year – Gene) |
| 1990–1999, the pre-genomic era of clinical and biochemical discovery | 8 genes: 1992 - MT-ATP6, 1993 - PDHA1, 1995 - SDHA, 1998 - NDUFS4, 1998 - NDUFS8, 1998 - SURF1, 1999 - NDUFV1, 1999 - NDUFS7 |
| 2000–2009, the early genomics era and expansion of genetic etiologies | 19 genes: 2000 - MT-TK, 2000 - MT-CO3, 2002 - BCS1L, 2003 - MT-ND6, 2003 - MT-ND5, 2003 - LRPPRC, 2004 - MT - ND3, 2004 - COX15, 2004 - NDUFS3, 2005 - NDUFS1, 2005 - NDUFAF2, 2006 - PDHX, 2006 - PDSS2, 2007 - SUCLA2, 2007 - MT-ND2, 2008 - NDUFA2, 2009 - MT-TW, 2009 - TACO1, 2009 – PDHB |
| 2010–2019, the next-generation sequencing revolution | 51 genes: 2010 - MTRFR, 2010 - NDUFAF5, 2010 - FOXRED1, 2011 - NDUFA10, 2011 - NDUFA12, 2011 - TPK1, 2011 - MTFMT, 2012 - MT-TI, 2012 - NDUFA9, 2012 - SERAC1, 2013 - TUFM, 2013 - LIPT1, 2013 - SLC19A3, 2013 - FBXL4, 2014 - GYG2, 2014 - PET100, 2014 - IARS2, 2014 - TSFM, 2014 - ECHS1, 2015 - NDUFV2, 2015 - NARS2, 2015 - GFM2, 2015 - COX8A, 2015 - HIBCH, 2016 - GTPBP3, 2016 - CDKL5, 2016 - NDUFB8, 2016 - SLC25A46, 2016 - SCO2, 2016 - MFF, 2016 - EARS2, 2016 - PARS2, 2016 - NDUFAF6, 2016 - ADAR, 2017 - OPA1, 2017 - ACY1, 2017 - PNPT1, 2017 - NDUFAF4, 2017 - NDUFAF3, 2017 - MRPS34, 2017 - NDUFS6, 2018 - ADAMTSL2, 2018 - ATP5MK, 2019 - CRAT, 2019 - NDUFAF8, 2019 - TIMMDC1, 2019 - NDUFS2, 2019 - TIMM50, 2019 - NDUFA1, 2019 - PUS1, 2019 - PTCD3 |
| 2020–present, the modern era of targeted therapies and sophisticated models | 38 genes: 2020 - SUOX, 2020 - SQOR, 2020 - COQ4, 2020 - HSD17B10, 2020 - RMND1, 2020 - TRMU, 2020 - NDUFA13, 2020 - NDUFC2, 2020 - NAXE, 2020 - VPS13D, 2020 - FASTKD2, 2020 - UQCRC2, 2021 - FDXR, 2021 - HPDL, 2022 - DNAJC30, 2022 - NFU1, 2022 - MRPL38, 2022 - SLC25A19, 2022 - L2HGDH, 2022 - DLD, 2022 - DLAT, 2022 - ETHE1, 2022 - PMPCB, 2022 - PMPCA, 2022 - MTO1, 2022 - ALDH5A1, 2022 - ATP5PO, 2022 - NDUFA4, 2022 - SUCLG1, 2022 - TARS2, 2023 - MRPL39, 2023 - TMEM126B, 2024 - NDUFA3, 2024 - MRPS36, 2024 - VARS2, 2024 - RNF213, 2024 - POLG, 2025 - FASTKD5 |
Towards the end of this era, the first nuclear genes encoding respiratory chain
components and assembly factors (complex II) were identified for gene
SURF1. The study used complementation assays in patient cell lines of
rodent/human rho0 hybrids (Table 2) to confirm that this nuclear gene caused LS. This led
to the landmark discovery of mutations in SURF1 as a major cause of
COX-deficient LS [18]. Of note, Rho-0 (
| Decade period | Models first reported | Model details | Model species/type | Citation |
| 1989*–1999 | Patient-derived Fibroblasts, Complementation Hybrids | Patient skin biopsies cultured to study biochemical defects. Cell fusion techniques (patient cells + mtDNA-less rho0 cells) used to distinguish nuclear vs. mitochondrial genetic origin. | Human cell lines | (King et al., PMID: 2814477, 1989; Miranda et al., PMID: 2540452, 1989) |
| 2000–2009 | Surf1 Knockout Mouse, E. coli model, Yarrowia lipolytica (yeast) model | Surf1 KO mice developed LS-like phenotype with COX deficiency, but not reduced survival of LS; Yeast models were created to study the functional consequences of specific human mutations (e.g., MT-ATP6, NDUFS7/8). | Mouse, Yeast | (Agostino et al., PMID: 12566387, 2003; Dell’agnello et al., PMID: 17210671, 2007; Kucharczyk et al., PMID: 19454486, 2009; Ahlers et al., PMID: 11004438, 2000) |
| 2010–2019 | Ndufs4-/- Knockout Mouse, Patient-derived iPSCs, Drosophila melanogaster model | Ndufs4-/- KO mouse became the cornerstone model for preclinical therapy testing; Patient-derived iPSCs were generated, allowing differentiation into disease-relevant cell types like neurons; Drosophila models were used to study genetic interactions and pathophysiology. | Mouse, Human iPSCs, Fruit Fly | (Quintana et al., PMID: 20534480, 2010; Galera-Monge et al., PMID: 27346203, 2016; Loewen et al., PMID: 29496745, 2018) |
| 2020–Present | iPSC-derived Cerebral Organoids, Conditional Knockout Mice, Zebrafish Models | iPSC-derived neural progenitor cells (NPCs) from LS patients for drug repurposing prioritized sildenafil and shown as effective treatment in 6 patients; 3D brain organoids created from patient iPSCs to model neurodevelopmental aspects; Conditional KOs (Ndufs4-/- in specific neurons) used to dissect cell-type-specific disease contributions; CRISPR/Cas9-generated surf1 knockout zebrafish models developed for high-throughput screening. | Human organoids, Mouse, Zebrafish | (Zink et al., preprint, 2025; Romero-Morales et al., PMID: 35792828, 2022; Gella et al., PMID: 32850799, 2020; Haroon et al., PMID: 36795052, 2023; Hyslop et al., 2025, PMID: 40673696 and McFarland et al., PMID: 40689593, 2025) |
*: The decade is extended by one year to 1989 to include the first rho-zero cell model [19].
Therapeutic approaches during this period were entirely supportive, focusing on managing symptoms and metabolic crises with “mitochondrial cocktails” consisting of vitamins and cofactors such as thiamine, riboflavin, and coenzyme Q10, often with limited and anecdotal success (Table 3).
| Decade period | First reported treatments/Therapies | Therapy details | Citation |
| 1990–1999 | Ketogenic Diet, Thiamine (Vitamin B1), Dichloroacetate (DCA) | Supportive/Targeted: Ketogenic diet improved lesions and stabilized a PDHC-deficient patient; High-dose thiamine used for thiamine-responsive PDH deficiency; DCA investigated to lower lactate levels, with transient effects. | (Wijburg et al., PMID: 1641082, 1992; Naito et al., PMID: 9266390, 1997; Fujii, Kymura et al., PMID: 9440797, 1997) |
| 2000–2009 | Coenzyme Q10, Idebenone -a derivative of coenzyme Q-10 | Supportive/Targeted: CoQ10 showed a dramatic response in a CoQ10 deficiency case; Idebenone, a derivative of coenzyme Q-10 improved respiratory function. | (Van Maldergem et al., PMID: 12447928, 2002; Haginoya et al., PMID: 19101701, 2009) |
| 2010–2019 | EPI-743 (Vatiquinone), Rapamycin (mTOR inhibition), Hypoxia, Gene Therapy (preclinical) | Targeted: EPI-743 (antioxidant) entered Phase 2A clinical trial (NCT01370447), showing neurologic improvement; Rapamycin and hypoxia showed dramatic rescue in Ndufs4-/- mouse models; First AAV-based gene therapy concepts tested preclinically. | (Martinelli et al., PMID: 23010433, 2012; Johnson et al., PMID: 24231806, 2013; Ferrari et al., PMID: 28483998, 2017, Di Meo et al., PMID: 28753212, 2017) |
| 2020–Present | Small molecule drugs sildenafil and HypoxyStat, Gene Therapy (clinical trials), Leukocyte Depletion, Acarbose, Mitochondrial Transfer, Mitochondrial Replacement | Targeted: Human off-label compassionate treatment with sildenafil in six LS patients showed improvements in motor function and resistance to metabolic crises; HypoxyStat is well-tolerated and effective and improved neurologic outcomes in the LS mouse; Preclinical work showed mice gene therapy on SURF1 KO mice with human AAV9/hSURF1 is safe and effective; CSF1R inhibitors (leukocyte depletion), acarbose (modulating gut microbiome), and mitochondrial transfer therapy are effective in mouse models. | (Zink et al., preprint, 2025; Blume et al., PMID: 39965572, 2025; Ling et al., PMID: 34703839, 2021; Bitto et al., PMID: 37365290, 2023; Nakai et al., PMID: 39223312, 2024) |
The potential of sodium dichloroacetate (DCA) to lower lactate levels was explored. The lactic and pyruvic acid concentrations in serum and cerebrospinal fluid (CSF) were decreased by the oral DCA treatment in some patients but not in other patients, thus its clinical efficacy was controversial [22, 23].
The advent of Sanger sequencing technologies dramatically accelerated the pace of gene discovery. This decade saw the identification of numerous novel mutations in both mtDNA and nuclear DNA genes. The list of causative nuclear genes for Complex I deficiency expanded significantly to include NDUFS4, NDUFS3, NDUFS2, NDUFA2, and the assembly factor NDUFAF2 (Table 1). Mutations in other respiratory chain complexes and related pathways were also identified, including SDHA (Complex II), BCS1L (Complex III), SCO2 and COX10 (Complex IV assembly), SUCLA2 and SUCLG1 (Krebs cycle), and PDHB (PDHC).
A key development was the creation of yeast [24] model and the first animal model of mouse with knockout nuclear gene Surf1 [25]—though newer study questioned that this mode did not develop LS, rather displayed prolonged lifespan, which reflects the complexity in creating LS animal model [26] (Table 2, Ref. [19]). Cell-based models, primarily patient-derived fibroblasts, remained crucial for biochemical characterization and for confirming the pathogenicity of newly discovered genetic variants through complementation assays.
Regarding treatment options, this era remained focused on supportive care and the use of mitochondrial cocktails. However, the advancements in molecular testing and genetic mechanism study were leading to the emergence of the first targeted strategies based on specific genetic diagnoses (Table 3). For instance, thiamine and biotin supplementation were shown to be highly effective for biotin-thiamine responsive basal ganglia disease caused by mutations in the thiamine transporter gene SLC19A3 [27].
The widespread adoption of Next-Generation Sequencing (NGS), particularly Whole Exome Sequencing (WES) and large multi-gene panels designed with LSS/PMD associated genes, revolutionized the diagnostic process for LSS. This high-throughput technology enabled the rapid and cost-effective identification of causative mutations in many patients for whom previous single-gene testing had been slow and lack sensitivity. This led to an explosion in the number of novel LSS-associated genes with dozens of new genes identified (Table 1, Supplementary Table 1), including those involved in mitochondrial translation (MRPS34, IARS2, NARS2, TSFM), coenzyme Q10 biosynthesis (COQ2, pDSS2), and novel respiratory chain assembly factors (NDUFAF3, NDUFAF4, NDUFAF5, NDUFAF6, ACAD9). Biotin-responsive Basal Ganglia disease is highlighted as a treatable differential diagnosis of Leigh syndrome [28]. The genetic landscape of LSS was revealed to be far more complex than previously imagined.
This period also saw the maturation of experimental models (Table 2), most notably the Ndufs4-/- knockout mouse [29], which recapitulated some core features of LS, including progressive neurodegeneration and brain stem lesions. This model became a cornerstone for investigating disease pathophysiology and for preclinical testing of potential therapies. Patient-derived induced pluripotent stem cells (iPSCs) emerged as a powerful tool, allowing researchers to generate disease-relevant cell types, such as neurons and cardiomyocytes, to study disease mechanisms in a human, patient-specific context [30]. Study on Ndufs4-/- knockout mouse led to the discovery that mTOR inhibition with rapamycin could significantly slow disease progression and extend lifespan [31]. This demonstrated that a mTOR signaling pathway could potentially be targeted for treating LS. Another discovery in this mouse model was that chronic hypoxia could both prevent and reverse established neurodegenerative disease [32].
These breakthroughs spurred early targeted clinical trials for LS (Table 3). The antioxidant EPI-743 (vatiquinone) was tested in a Phase 2a open-label trial, showing signs of neurological improvement in a small cohort of children [33]. These early trials, while small, marked a significant shift from purely supportive care towards incorporating mechanism-based therapeutic development. Of note, no positive results with EPI-743 have been published for Leigh Syndrome since 2012.
Piloting a new path of preventing LS occurrence, spindle-chromosome complex
transfer (ST) method was used in a woman carrying the m.8993T
The current era is characterized by a continued expansion of the genetic landscape of LSS, driven by WES, WGS, and RNA Sequencing. Adoption of the long read sequencing with PacBio and Nanopore technologies can reveal complicated variations that are previously not accessible with the short read NGS sequencing. Novel genes such as FASTKD5, NDUFC2, SQOR, and TMEM126B were shown to be linked to LS (Tables 1,4). The focus of research is now shifting from gene discovery towards understanding the complex cellular consequences of these mutations.
| Biochemical function and complex | Genes | Gene names*** | Gene Classification by MITO_GCEP as LSS (* Phase I) |
| Complex I | 35 | DNAJC30, FOXRED1, MT-ND1, MT-ND2, MT-ND3, MT-ND4, MT-ND5, MT-ND6, NDUFA1, NDUFA10, NDUFA12, NDUFA13, NDUFA2, NDUFA3, NDUFA9, NDUFAF2, NDUFAF3, NDUFAF4, NDUFAF5, NDUFAF6, NDUFAF8, NDUFB8, NDUFC2, NDUFS1, NDUFS2, NDUFS3, NDUFS4, NDUFS6, NDUFS7, NDUFS8, NDUFV1, NDUFV2, NUBPL, TIMMDC1, TMEM126B | Definitive/Moderate (26): |
| FOXRED1, MT-ND1, MT-ND2, MT-ND3, MT-ND4, MT-ND5, MT-ND6, NDUFA1, NDUFA13, NDUFA2, NDUFA9, NDUFAF2, NDUFAF5, NDUFAF6, NDUFAF8, NDUFB8, NDUFC2, NDUFS1, NDUFS2, NDUFS3, NDUFS4, NDUFS6, NDUFS7, NDUFS8, NDUFV1, NUBPL | |||
| Limited evidence (6): ** | |||
| MT-ND2, NDUFA10, NDUFA12, NDUFAF4, NDUFV2, TIMMDC1 | |||
| Complex II | 3 | SDHA, SDHAF1, SDHB | Definitive/Moderate (1): |
| SDHA | |||
| Limited evidence (1): | |||
| SDHAF1 | |||
| Complex III | 4 | BCS1L, TTC19, UQCRC2, UQCRQ | Definitive/Moderate (1): |
| TTC19 | |||
| Limited evidence (2): | |||
| BCS1L, UQCRQ | |||
| Complex IV | 15 | COA6, COX10, COX15, COX4I1, COX8A, LRPPRC, MT-CO1, MT-CO2, MT-CO3, NDUFA4, PET100, PET117, SCO2, SURF1, TACO1 | Definitive/Moderate (8): |
| COX10, LRPPRC, MT-CO1, MT-CO3, PET100, SCO2, SURF1, TACO1 | |||
| Limited evidence (8): | |||
| COX15, COX4I1, COX8A, MT-CO1, MT-CO2, MT-CO3, NDUFA4, PET117 | |||
| Complex V | 3 | ATP5MK, ATP5PO, MT-ATP6 | Definitive/Moderate (1): |
| MT-ATP6 | |||
| Biotin/Thiamine | 4 | BTD, SLC19A3, SLC25A19, TPK1 | Definitive/Moderate (3): |
| BTD, SLC19A3, TPK1 | |||
| Limited evidence (1): | |||
| SLC25A19 | |||
| Coenzyme Q10 | 5 | COQ4, COQ7, COQ9, HPDL, PDSS2 | Definitive/Moderate (2): |
| HPDL, PDSS2 | |||
| Limited evidence (1): | |||
| COQ9 | |||
| Defect of mitochondrial gene expression | 2 | MRPL3, MRPL39 | Definitive/Moderate (0): |
| Detoxification | 1 | NAXE | Limited evidence (1): |
| NAXE | |||
| Disorders of mitochondrial toxicity | 1 | L2HGDH | |
| Disorders of mitochondrial toxicity/Valine degradation | 1 | HIBCH | Definitive/Moderate (1): |
| HIBCH | |||
| Fission | 4 | DNM1L, MFF, MFN2, SLC25A46 | Definitive/Moderate (1): |
| MFF | |||
| Limited evidence (2): | |||
| DNM1L, SLC25A46 | |||
| Fusion/Fission | 1 | OPA1 | Definitive/Moderate (1): |
| OPA1 | |||
| Impaired mitochondrial protein synthesis | 2 | FARS2, PTCD3 | Definitive/Moderate (1): |
| FARS2 | |||
| Limited evidence (1): | |||
| PTCD3 | |||
| Lipid remodeling | 2 | SERAC1, TOMM7 | Definitive/Moderate (1): |
| SERAC1 | |||
| Lipoic acid | 3 | LIAS, LIPT1, MECR | Definitive/Moderate (2): |
| LIPT1, MECR | |||
| Limited evidence (1): | |||
| LIAS | |||
| Mitochondrial DNA maintenance | 1 | FBXL4 | Definitive/Moderate (1): |
| FBXL4 | |||
| Mitochondrial gene expression | 1 | MTRFR | |
| Mitochondrial stability, fission, clearance by mitophagy | 1 | VPS13D | Definitive/Moderate (-) |
| Limited evidence (1): | |||
| VPS13D | |||
| Mitochondrial translation | 19 | EARS2, GFM1, GFM2, GTPBP3, IARS2, MRPS34, MT-TI, MT-TK, MT-TL1, MT-TL2, MT-TV, MT-TW, MTFMT, MTRFR, NARS2, pNPT1, TARS2, TRMU, TSFM | Definitive/Moderate (15): |
| ARS2, GFM1, GFM2, GTPBP3, MRPS34, MT-TI, MT-TK, MT-TL1, MT-TL2, MT-TV, MT-TW, MTFMT, PNPT1, TRMU, TSFM | |||
| Limited evidence (7): | |||
| IARS2, MT-TI, MT-TL1, MT-TL2, MT-TW, NARS2, TARS2 | |||
| mtDNA depletion and/or multiple mtDNA deletions | 9 | MPV17, POLG, RNASEH1, RRM2B, SLC25A4, SSBP1, SUCLA2, SUCLG1, TWNK | Definitive/Moderate (2): |
| SUCLA2, SUCLG1 | |||
| Limited evidence (4): | |||
| POLG, RNASEH1, SLC25A4, SSBP1 | |||
| OXPHOS |
5 | ADAR, AIFM1, MORC2, RANBP2, SLC39A8 | Definitive/Moderate (1): |
| AIFM1 | |||
| Limited evidence (4): | |||
| ADAR, MORC2, RANBP2, SLC39A8 | |||
| OXPHOS |
1 | ECHS1 | Definitive/Moderate (1): |
| ECHS1 | |||
| Proteostasis | 2 | CLPB, LONP1 | Definitive/Moderate (0) |
| Limited evidence (2): | |||
| CLPB, LONP1 | |||
| Proteostasis/Defect of mitochondrial protein quality control | 4 | HTRA2, PMPCA, pMPCB, SPG7 | Definitive/Moderate (1): |
| PMPCB | |||
| Pyruvate dehydrogenase complex | 6 | DLAT, DLD, MPC1, PDHA1, pDHB, PDHX | Definitive/Moderate (5): |
| DLAT, DLD, PDHA1, PDHB, PDHX | |||
| Sulfide metabolism/Disorders of mitochondrial toxicity | 2 | ETHE1, SQOR | Definitive/Moderate (1): |
| ETHE1 | |||
| Limited evidence (1): | |||
| SQOR | |||
| To be assigned by function and complex | 8 | CRAT, FASTKD2, FASTKD5, GYG2, HSD17B10, KGD4, MTO1, NUP62 | Definitive/Moderate (1): |
| MTO1 | |||
| Limited evidence (1): | |||
| NUP62 |
Note: *: MITO_GCEP Phase I (McCormick et al. [6] 2023, PMID: 37255483); ** Italic font: Genes classified in Phase I as “Limited_Evidence” for LSS, MITO_GCEP Phase II is on-going to add more published cases and evidences to update the classification (Shen L, personal communication, July 2025). ***: Gene names include the 113 genes reviewed by Mito-GCEP, other LSS- causative genes reported in publications, and reported at ClinVar and HGMD reference databases.
Experimental models have become increasingly sophisticated. Human iPSC technology is being used to create 3D cerebral organoids that model features of LS brain development and pathology, revealing defects in corticogenesis [35]. Live animal models, particularly the Ndufs4-/- KO mouse, are continuously used to dissect the cell-type-specific contributions to the disease [36] and to explore novel therapeutic avenues, such as acarbose which can extend mice lifespan and suggested involvement of gut microbiome in disease pathogenesis [37].
The most exciting developments are in the therapeutic arena. The advancement in molecular genetics and mechanism study along with the preclinical successes of the previous decade have paved the way for more advanced and targeted strategies (Table 3). Gene therapy is a major new focus, with AAV-based vectors being developed to deliver functional copies of defective nuclear genes. Preclinical studies using AAV9 vectors to deliver SURF1 or NDUFS4 to mouse models have shown significant biochemical and clinical improvements [38, 39]. A double administration of self-complementary AAV9NDUFS4 prevents Leigh syndrome in Ndufs4-/- mice, leading to 9 months’ heathy lifespan [40]. An optimized mitoBEs single-base-editing could precisely edit and introduce LS-causing mtDNA variant mt-Atp6 T8591C into mouse and observed LS symptoms [41]. Another study used engineered mtDNA adenine base editor (eTd-mtABE), first introducing the pathogenic mtDNA variant into rat zygotes and later correcting the introduced variant at high efficiency—to 53% wild type allele—in a rat Leigh syndrome model [42].
A review of publications and clinical trial registries confirms multiple exciting avenues of development: several targeted therapies are in active clinical development, a few effective small molecule drugs are promising in compassionate prescription to patients, and a preventive treatment program using mitochondria replacement (MRT) achieved initial success in clinical practice. Further, mitochondrial transplantation preclinical studies successfully delivered healthy mitochondria to affected live animal model of Leigh syndrome and improved morbidity and mortality.
Mitochondrial replacement/donation therapy (MRT) with meiotic spindle transfer or pronuclear transfer has been developed. In such so-called three-person IVF techniques, the nuclear DNA from an oocyte or a zygote (the first two persons) carrying a pathogenic mtDNA variant is transferred into an enucleated donor cell (the third person) that provides wild-type mtDNA background.
In one early human trial, spindle-chromosome complex transfer (ST) method was
used on a female carrier of Leigh syndrome (mtDNA mutation 8993T
In two recent back-to-back publications, the studies used mitochondrial donation through pronuclear transfer on 22 patients carrying pathogenic mtDNA variants with homoplasmy or elevated heteroplasmy. There have since been 8 live births from 22 patients (36%), with all infants reported as being healthy at birth, with no or low levels of mtDNA heteroplasmy in blood, being 77% to 100% lower than levels of the maternal pathogenic mtDNA variants [43, 44] (ClinicalTrials.gov number: NCT04113447).
Put together, mitochondrial donation through meiotic spindle transfer or pronuclear transfer was effective in reducing the transmission of homoplasmic and heteroplasmic pathogenic mtDNA variants. MRT is currently only approved in the United Kingdom. Despite regulatory concerns about the probability of mitochondrion—nuclear incompatibility after introduction of mitochondria from a different origin into an embryo and concerns about a low probability of pathogenic variant heteroplasmy reversion, these precision therapies represent a new frontier in the treatment of Leigh syndrome, offering real and proven hope for disease-preventing interventions against mtDNA-LSS.
The mitochondrial transplantation approach delivers healthy mitochondria to affected cells. While still in early stages, preclinical studies have shown its promise in LS mouse models. The recent study by Nakai et al. [45] demonstrated that both bone marrow transplantation (which releases mitochondria into circulation) and direct administration of isolated healthy mitochondria could significantly improve morbidity and mortality in the Ndufs4-/- LS mouse model. Further, even cross-species administration of human mitochondria to Ndufs4-/- mice improves LS symptoms [45]. This pre-clinical success provides a strong rationale for exploring this strategy in humans to treat more mtDNA-caused mitochondrial diseases including mtDNA-LSS. Clinical trials for mitochondrial transplantation are underway for more diseases, and the positive preclinical data in LS mouse models make this a compelling research direction.
To date, no FDA-approved small molecule therapies exist for Leigh Syndrome Spectrum (LSS) or other primary mitochondrial diseases [46]. Nevertheless, several promising candidates are emerging, with early evidence drawn from preclinical in vivo studies in animal or organoid models, compassionate use in affected patients, and ongoing clinical trials.
iPSC-derived neural progenitor cells (NPCs) from LS patients was used for drug repurposing which prioritized sildenafil as an effective off-label compassionate treatment in 6 patients with outcome improvements [47].
A small-molecule form of hypoxia therapy that increases oxygen-hemoglobin affinity, HypoxyStat is well-tolerated and effective for pre-symptomatic and advanced disease treatment, improving neurologic outcomes in the LS mouse model [48].
An orally available small molecule that acts as an NAD+ modulator. One pre-clinical trial result indicates KL1333 is safe and well tolerated, supporting the efficacy of KL1333 in improving fatigue, functional strength, endurance [49]. A clinical trial is currently recruiting adult primary mitochondrial disease patients (NCT05650229) to evaluate drug efficacy (FALCON).
The striking preclinical success in the Ndufs4-/- mouse model has generated significant interest. Clinical trials exploring mTOR inhibitors for various mitochondrial diseases, including LS, are being considered and are listed in registries (e.g., NCT06843811, Sirolimus for Leigh syndrome for MELAS).
Despite promising early phase results, PTC Therapeutics announced in 2023 that the Phase 3 MIT-E trial for mitochondrial epilepsy did not meet their primary endpoints. Its future in Leigh syndrome treatment is uncertain (Press release: https://ir.ptcbio.com/news-releases/news-release-details/ptc-therapeutics-announces-results-mit-e-clinical-trial).
Daily CBD In Ndufs4-/- Leigh Syndrome mouse models significantly
delayed neurological and motor decline and extended the median lifespan from 57
to 70 days. CBD also improved phenotypes in patient-derived fibroblasts with
reduced PPAR
Over the past 4 decades of molecular genetic studies, over 120 LSS genes have been discovered and critically assessed for their GDRs [6], and several dozens of additional genes reported causative for LSS but remain pending systematic assessment with standard ClinGen framework (Table 4, Ref. [6]). Table 4 compiled 144 candidate LSS genes from publications and reference databases including ClinVar and the Human Gene Mutation Database (HGMD®) and organized them into 29 biochemical and functional categories. Over 1000 pathogenic variants from the nuclear and mitochondrial genomes have been reported in patients according to a recent search of the ClinVar and HGMD databases (data not shown). This wealth of rich clinical and genomics data underscores the need for comprehensive and regularly updated database-driven resources to disseminate the data and support ongoing research, improve genetic diagnosis, and aid in patient and provider education.
The diagnostic criteria for LSS, the approaches to gene discovery, and individual cases diagnosis practices by different clinicians have evolved over decades, which occasionally leads to inconsistencies across time and between healthcare centers. To standardize the evidence assessment for the LSS gene–disease relationship (GDR), the international ClinGen Mito-GCEP Expert Panel was established in 2017, with over 30 geneticists, neurologists, metabolic physicians, neuropathologists, bioinformaticians, researchers, and laboratory directors from 9 countries. Using a modified ClinGen framework for Gene–Disease Clinical Validity Curation, this multidisciplinary panel systematically evaluated 113 LSS genes for GDR. In the first phase, the panel classified 31 GDRs as “Definitive”, 38 as “Moderate” and 45 as “Limited” or “Disputed” [6]. The renewed funding supported the phase 2 work of expanding to all PMD genes, which continuously updated clinical cases and functional evidence thus leading to GDR upgrading 16 of the 43 “Limited” GDRs to “Definitive” or “Moderate” (Shen L, personal communication, July 2025).
These 114 curated LSS GDRs represent one of the largest resources systematically evaluated by expert consensus and serves as a reference dataset through the Leigh Syndrome Spectrum portal on the Mitochondrial Disease Sequence Data Resource Consortium (MSeqDR) website ([51]; Portal URL: https://mseqdr.org/leigh.php). This GDR resource supports accurate variant interpretation, facilitates genetic diagnosis of LSS, and advances precision medicine efforts.
Because LSS is a rare disease, case-level, genetic, and mutational data are often sparse and dispersed across various database resources. This creates a challenge for the disease community due to a lack of comprehensive and centralized resources. This section describes several web-based, comprehensive resources that aggregate heterogeneous data for LSS genetics and molecular diagnosis.
ClinVar is a freely accessible public archive of interpretations of clinically relevant human genetic variations [52]. The U.S. FDA recognizes variant classifications submitted to ClinVar by ClinGen’s Variant Curation Expert Panels (VCEPs), which follow a standardized protocol. Mito-VCEP actively contributes expert-assessed variant-disease relationships for LSS and other PMDs. Other ClinGen expert panels and worldwide genetic testing providers also contribute pathogenicity assessments to ClinVar. LSS-related variant data can be searched and browsed on the ClinVar website and downloaded from its FTP site. As of July 20, 2025, there are 804 Leigh syndrome-associated entries at ClinVar, with ACMG classifications ranging from “Pathogenic” to “Uncertain Significance” or “Benign” (data not shown here).
The MSeqDR Consortium’s website resource (https://mseqdr.org) supports LSS gene and variant curation projects by the Mito-GCEP and Mito-VCEP expert panels and stores the curation results from these projects [51]. The MSeqDR Consortium is a global effort focused on collecting and sharing data for rare mitochondrial diseases, including rich data for LSS cases, genes, and variants. Three types of LSS data are available through the “Leigh Syndrome Spectrum Resource” Portal (URL: https://mseqdr.org/leigh.php).
6.2.2.1 LSS GDR Results from Mito-GCEP
These 114 curated LSS GDRs from section 6.1 serve as a reference dataset to facilitates genetic diagnosis of LSS, through the Leigh portal on the MSeqDR Consortium website.
6.2.2.2 LSS Case-Level Clinical and Genetic Data
The “LSS Virtual Registry” link directs visitors to a virtual registry of over 2000 published cases established through literature mining [53]. A demo subset includes 440 pseudo-cases that were expertly annotated for GDR classification by the Mito-GCEP expert panel and is accessible using a “guest” account without pre-registration.
6.2.2.3 Disease-to-Phenotype-to-Variant Associations
The MSeqDR platform provides integrated views of disease-phenotype-variant associations. Starting from the Leigh “Disease Ontology Tree” node, users can navigate to interconnected data from OMIM, the MONDO disease ontology, the Human Phenotype Ontology (HPO), and ClinVar. This data is integrated with analysis tools available on the MSeqDR website.
Leigh Syndrome Spectrum (LSS) research is moving rapidly from genetic discovery towards precise and highly personalized medicine including genetics-guided,
point mutation-targeted treatments such as the successful human MRT correction on
m.8993T
LS is the sole author, with contributions to all the manuscript work: the design, the literature and data collection, the data analysis, data summarization, manuscript drafting, editing, and submission. LS has prepared, read and approved the final manuscript, and agrees to be accountable for all aspects of the work.
Not applicable.
This work is supported by the Children’s Hospital Los Angeles as part of the author’s regular duty as a full-time employee, and the author is grateful to the support of the Children’s Hospital Los Angeles. The author thanks to Mr. Jitong Shen for helping with manuscript revision and proofreading, thanks to Mr. Dennis Maglinte and Dr. Srikar Chamala for administrative support and proofreading help, and thanks to all the peer reviewers for their opinions and suggestions.
This research received no external funding.
The author declares no conflict of interest.
Gemini-2.5 AI was used in grammar checking, spelling checking, making formatting, and expression suggestions, which are all human reviewed based on author’s domain knowledge relevant to this review article’s topic.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/FBS45427.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.