









1 Milford Molecular Diagnostics Laboratory, Milford, CT 06460, USA
Abstract
The general assumption that spirochetemia does not occur at the early localized stage of Lyme disease is due to a lack of sensitive and specific methods for molecular diagnosis.
During a Lyme disease season in 2023, the platelet-rich plasma specimens of 145 people residing in Lyme disease-endemic areas in the United States were immediately separated from the blood cells following venous blood collection to prevent the spirochetes, if any, from invading the lymphocytes in the test tube. The entire DNA content was extracted from the platelet pellet and used for split sample polymerase chain reaction (PCR) amplification; Sanger sequencing was performed on the nested PCR products to detect the Borrelia burgdorferi flaB and 16S rRNA genes.
In 98 of the people who were clinically suspected of having early localized Lyme disease irrespective of the presence or absence of a skin lesion, 33 of their blood specimens (33.7%) were positive for Borrelia burgdorferi (B. burgdorferi), including 17 positive for flaB gene only, 15 positive for both the flaB and 16S rRNA genes, and one positive for 16S rRNA gene only. Eight (17.0%) of the 47 asymptomatic resident controls were positive for flaB PCR only.
The flaB gene is a more sensitive chromosomal target than the 16S rRNA gene for molecular detection of one to three B. burgdorferi cells due to spirochetes gaining or retaining flaB paralogs at the early localized stage of Lyme disease.
Keywords
- Sanger sequencing
- spirochetemia
- nested PCR
- early localized Lyme disease
- flaB paralogs
- SNP in flaB gene
- single Borrelia burgdorferi cell
- platelet-rich plasma
Lyme disease, a systemic bacterial infection caused by Borrelia burgdorferi (B. burgdorferi), is the most common tickborne disease in the United States. Released 2010–2018 insurance records suggest that each year approximately 476,000 Americans are diagnosed and treated for Lyme disease [1]. But the true number of Lyme disease patients is unknown due to the lack of reliable sensitive and specific routine laboratory diagnostic tools. It is believed that at the early stage of infection this disease can be treated successfully with a proper course of antibiotics, but the spirochete bacteria may disseminate from the site of the tick bite to other regions of the body if not timely and properly treated [2].
It has been assumed that the B. burgdorferi spirochetes begin to enter the blood stream at the stage 2 of Lyme disease [3, 4] about 3 to 12 weeks after the initial infection [5], and that during the early localized stage of infection, the number of B. burgdorferi cells simply increases in the dermal tissue in preparation for dissemination [6]. However, animal experiments showed that blood-borne dissemination of B. burgdorferi can be detected at day 2 after intradermal inoculation [7]. Like Leptospira interrogans, which enters the blood stream rapidly after invasion into the human body through the skin and mucosa [8], B. burgdorferi spirochetemia can occur in early infections based on epidemiological studies [9]. The key to diagnosing Lyme disease spirochetemia at the “early localized” stage of infection is to develop a highly sensitive, specific molecular blood test for the detection of Lyme disease spirochetes in low single-digit numbers.
As previously reported, nested polymerase chain reaction (PCR) amplification of the borrelial 16S rRNA gene DNA extracted from the spirochetes in the platelet pellet obtained by differential centrifugation followed by Sanger sequencing of the PCR products can diagnose spirochetemias with a bacterial density as low as 25 per mL of whole blood [10]. However, B. burgdorferi can actively attach to and invade human lymphocytes during co-incubation of the spirochetes and the blood cells in vitro [11]. If there were only a few B. burgdorferi spirochetes in the whole blood specimen, delayed exclusion of the blood cells from the platelet-rich plasma may lead to loss of all the spirochetes to the buffy coat by allowing the highly mobile spirochetes to attach to the lymphocytes prior to blood centrifugation.
This research project was designed to prove that immediate separation of the platelet-rich plasma after venous blood collection, using the entire DNA content extracted from the platelet pellet for nested PCR, and targeting the newly found duplicated paralogs of the flaB gene for PCR amplification can increase the chances of detecting a single B. burgdorferi spirochete in 1 mL of platelet-rich plasma. While plasmid gene duplication and loss by adaptation to multiple host species are well known phenomena in the life cycle of B. burgdorferi [12, 13], flaB gene duplication in the B. burgdorferi chromosome has not been reported.
Per agreement, DiaSorin, Inc., Stillwater, MN, USA, enrolled the patients and healthy donor controls and performed the initial specimen preparation for a research project after informed consent was obtained in accordance with applicable regulations in compliance with 21 CFR 812. A total of 98 symptomatic patients clinically suspected of having early localized Lyme disease and 47 asymptomatic residents serving as controls were included in this study. The patients clinically suspected of having early localized Lyme disease were defined as people residing in a known Lyme disease-endemic area in the United States who developed a recent onset of fatigue, skin rash, fever, muscle aches, neck pain, joint pain, or lymphadenopathy during the Lyme disease season in 2023. Their venous blood specimens were drawn for testing when any of these symptoms and signs first appeared. The asymptomatic resident controls, also referred to as healthy donors by DiaSorin, Inc., the specimen supplier, were people residing in the same communities with the symptomatic patients during the Lyme disease season in 2023.
Specimen collection was performed under the participating collection site’s
Institutional Review Board-approved protocols and under informed consent.
Preparation of platelet-rich plasma (PRP) followed a standard protocol [14] with
slight modification. Specifically, at the participating collection sites, about 3
mL of the patient’s venous blood was drawn into a lavender top tube containing
ethylenediaminetetraacetic acid (EDTA) as anticoagulant. After the whole blood
was mixed well for ~5 minutes, the rubber stopper was removed and
replaced with a plastic over cap. The whole blood specimen was centrifuged for 15
minutes at 400
This was a modified procedure from a previously published protocol [10]. At
Milford Molecular Diagnostics Laboratory, the frozen platelet-rich plasma was
defrosted and the entire thawed specimen was centrifuged in a 1.5 mL Eppendorf
tube at ~16,000
All Eppendorf tubes containing the 70% ethanol-washed pellets were put in an 85 °C heating block for 2 min with opened cap to evaporate the residual ethanol. Into each tube, 25 µL of complete “flaB primary PCR mixture” or 25 µL of complete “borrelial 16S rRNA gene primary PCR mixture” was added. The tubes were heated with closed caps at 85 °C for another 3 min, then vortexed to dissolve the nucleic acids into the PCR mixture.
After centrifugation at ~16,000
The 25 µL of complete “flaB primary PCR mixture” contained 20 µL of ready-to-use LoTemp® PCR mix (HiFi DNA Tech, LLC, Trumbull, CT, USA), 3 µL of molecular grade water, 1 µL of 10 µmolar flaB outer forward (FOF) primer (5′-GCATCACTTTCAGGGTCTCA-3′), and 1 µL of 10 µmolar flaB outer reverse (FOR) primer (5′-TGGGGAACTTGATTAGCCTG-3′) to amplify a 503-bp segment of the B. burgdorferi flaB gene [16].
The 25 µL of complete “borrelial 16S rRNA gene primary PCR mixture” contained 20 µL of ready-to-use LoTemp® PCR mix, 3 µL of molecular grade water, 1 µL of 10 µmolar M1 forward primer (5′-ACGATGCACACTTGGTGTTAA-3′), and 1 µL of 10 µmolar M2 reverse primer (5′-TCCGACTTATCACCGGCAGTC-3′) to amplify a 357/358-bp segment of the borrelial 16S rRNA gene [10]. DNA extracts from the cultured cells of Borrelia burgdorferi (ATCC 53210) and of Borrelia coriaceae (ATCC 43381) were used as the positive control for flaB amplification and as the positive control for borrelial 16S rRNA gene amplification, respectfully.
Borrelia coriaceae is rarely found in human specimens and its 16S rRNA gene has the same M1 and M2 primer sites as those of the pathogenic Borrelia species. Using B. coriaceae instead of B. burgdorferi as the positive control helps monitor bench work contamination. Contamination by B. coriaceae 16S rRNA gene DNA will be recognized easily at the stage of Sanger sequencing, thus avoiding false-positive results due to control contamination. However, the flaB gene sequences of various Borrelia species are heterogeneous. In the United States, it is necessary to use B. burgdorferi sensu stricto flaB DNA as the positive control for flaB PCR amplification.
Transferring of post-PCR materials from one test tube to another was accomplished by using a micro-glass rod in a 32″ PCR workstation (AirClean Systems, Raleigh, NC, USA) to avoid aerosols, which may occur in routine micropipetting. In order to minimize the potential for contamination, DNA extractions, PCR setup, and agarose gel electrophoresis were performed in three separate rooms.
A trace (about 0.2 µL) of each of the primary PCR products was transferred by a calibrated micro-glass rod to another 25 µL complete PCR mixture containing 20 µL of ready-to-use LoTemp® PCR mix, 3 µL of molecular grade water, 1 µL of 10 µmolar forward primer and 1 µL of 10 µmolar reverse primer for nested PCR or same-nested PCR amplification with identical thermocycling steps as for the primary PCR.
The primers for the flaB nested PCR amplification were the flaB inner forward (FIF) primer (5′-CTTTAAGAGTTCATGTTGGAG-3′) and the flaB inner reverse (FIR) primer (5′-TCATTGCCATTGCAGATTGT-3′) to amplify a 447-bp segment of the B. burgdorferi flaB gene [16].
The primers used for the 16S rRNA gene nested PCR amplification were the same M1 and M2 primers used in the borrelial 16S rRNA gene primary PCR mixture [10].
All nested PCR products, including those of the positive controls and negative controls, were tested by agarose gel electrophoresis. The nested PCR products showing a DNA band at agarose gel electrophoresis without further purification were subjected to bidirectional Sanger sequencing to verify the authenticity of the PCR-amplified products.
Based on a study reported by other investigators, the limit of detection of
16S rRNA gene in human blood samples by nested PCR assays was
approximately 2 (
Using one identical pair of primers for nested and primary amplification of borrelial 16S rRNA genes takes advantage of a unique 7-base discriminatory sequence immediately downstream of the M1 PCR primer between the Borrelia burgdorferi sensu lato complex and the relapsing fever borrelia group. The M1/M2 PCR primer pair can amplify all known pathogenic Borrelia 16S rRNA genes, including those of B. burgdorferi and B. miyamotoi. With these 2 PCR primers, all B. burgdorferi sensu lato complex members will generate a 357-bp amplicon while all members of the relapsing fever borrelia group will generate a 358-bp amplicon [18]. Further increasing the size of the amplicon will reduce detection sensitivity. Moving the primers inward for nested PCR will lose the discriminatory power of Sanger sequencing.
A trace (about 0.2 µL) of the nested PCR product showing a band at agarose
gel electrophoresis was transferred by a calibrated micro-glass rod from the
nested PCR tube into a Sanger reaction tube containing 1 µL of 10
µmolar sequencing primer, 1 µL of the BigDye®
Terminator (v 1.1/Sequencing Standard Kit, Applied Biosystems, Foster City, CA,
USA), 3.5 µL 5
To verify that Milford Molecular Diagnostics Laboratory is capable of using the described methods to detect borrelial spirochetes causing clinical Lyme disease in platelet-rich plasma specimens before launching this research project, DiaSorin, Inc. sent 20 blind-coded platelet-rich plasma specimens, some of which were spiked with cultured cells of Borrelia burgdorferi, Borrelia miyamotoi, a mixture of the two, and none of the two, to Milford Molecular Diagnostics Laboratory on June 29, 2023. Milford Molecular Diagnostics Laboratory was required to use the methods described above to identify all the blind-coded specimens correctly without cross contamination. However, the purpose of this work was to detect 1–3 Borrelia burgdorferi cells in 1 mL of patient platelet rich plasma at the early localized stage of infection. It is technically difficult to spike a sample with 1–3 spirochetes from a pure culture. As a result, these 20 blind-coded specimens did not mimic real clinical conditions.
The overall results of split sample testing on the venous blood specimens of 98 patients clinically suspected of having early localized Lyme disease showed that 65 specimens were negative for both flaB gene and 16S rRNA gene PCR amplifications and that 33 specimens (33.7%) were PCR-positive, including 17 specimens positive for the flaB gene only, and 15 specimens positive for both the flaB gene and 16S rRNA gene. One specimen was positive for the 16S rRNA gene only, indicating that this sample contained only two copies of B. burgdorferi chromosome and both copies were aliquoted for 16S rRNA gene PCR amplification by chance during sample splitting. Split sample testing on the venous blood specimens of 47 asymptomatic resident controls generated 8 positives (17.0%), all positive for flaB PCR only.
When tested in groups of 10, many specimens in each group only showed a flaB gene PCR product without a concomitant 16S rRNA gene amplification. The results of 20 selected consecutive nested PCRs from the symptomatic patients and 20 selected consecutive nested PCRs from the asymptomatic residents, respectively, are presented in Fig. 1. This decoupling of two chromosomal gene amplifications in split sample PCR testing suggested that there were more copies of the flaB gene than the 16S rRNA gene in one chromosome and this gene copy number difference became apparent only when split sample nested PCR testing of a specimen containing spirochetes in low single-digit numbers was carried out.
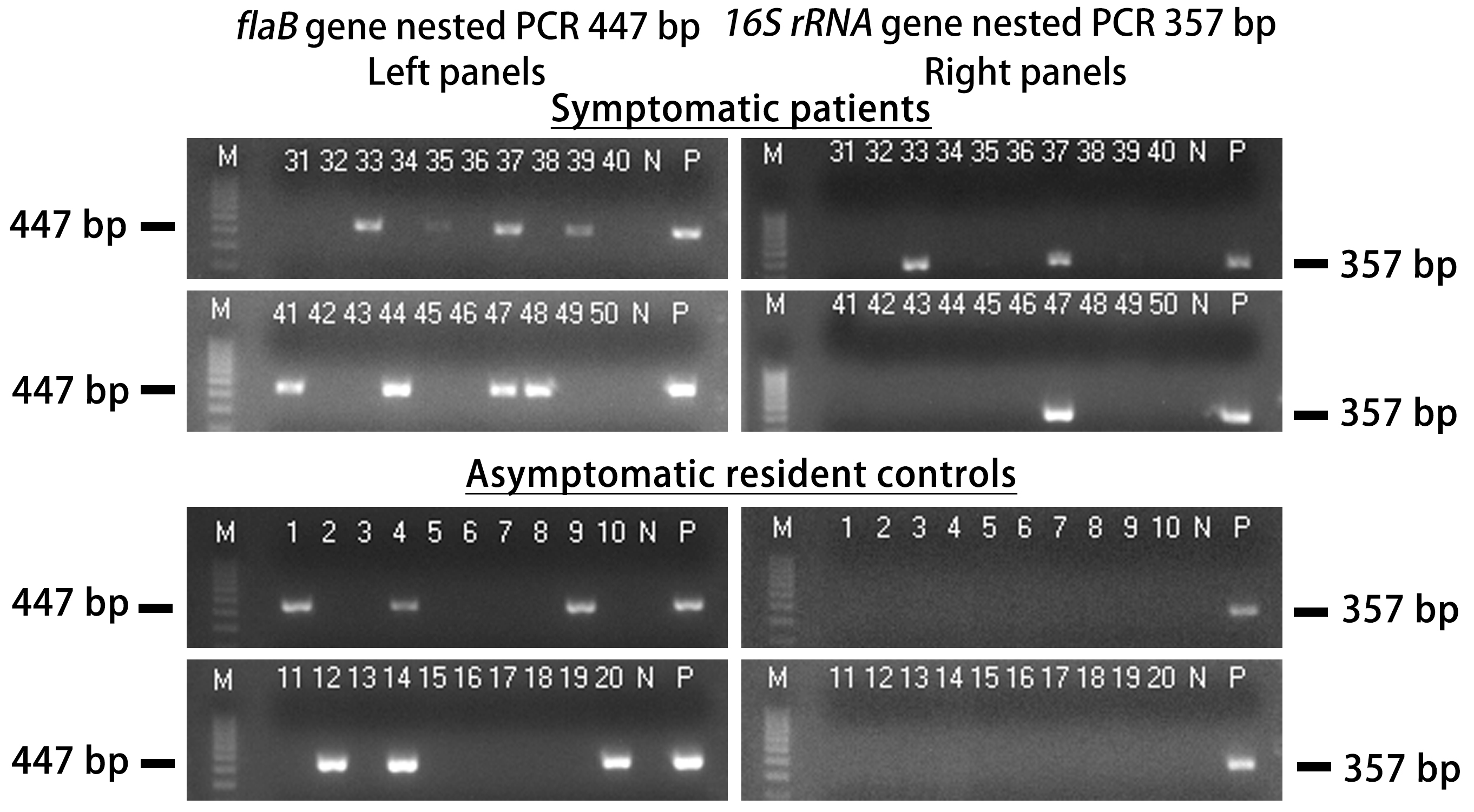 Fig. 1.
Fig. 1.
Decoupling of flaB gene from 16S rRNA gene in split sample polymerase chain reaction (PCR). Images of agarose gel electrophoresis of nested PCR products showing split sample testing results on 20 blood specimens taken from 98 symptomatic patients suspected of having early localized Lyme disease and 20 blood specimens taken from 47 asymptomatic residents living in the same communities during a Lyme disease season. The left panels show a 447-bp flaB nested PCR product band in 13 specimens. But only 3 of the 13 flaB-positive samples were associated with a concomitant 357-bp 16S rRNA gene same-nested PCR product band in the right panels, and all 3 were in the group of symptomatic patients. Lane numbers = specimen No. N = negative water control. P = DNA extract from cultured Borrelia burgdorferi (B. burgdorferi) cells (ATCC 53210) in the left panels, and DNA extract from cultured B. coriaceae cells (ATCC 43381) in the right panels.
All nested PCR products showing a band at agarose gel electrophoresis were verified by Sanger sequencing without purification. A typical specimen positive for B. burgdorferi (case No. M23-167) containing a single-nucleotide silent mutation in the flaB gene (Figs. 2,3) and a positive B. burgdorferi 16S rRNA gene (Supplementary Fig. 1) was chosen for illustration.
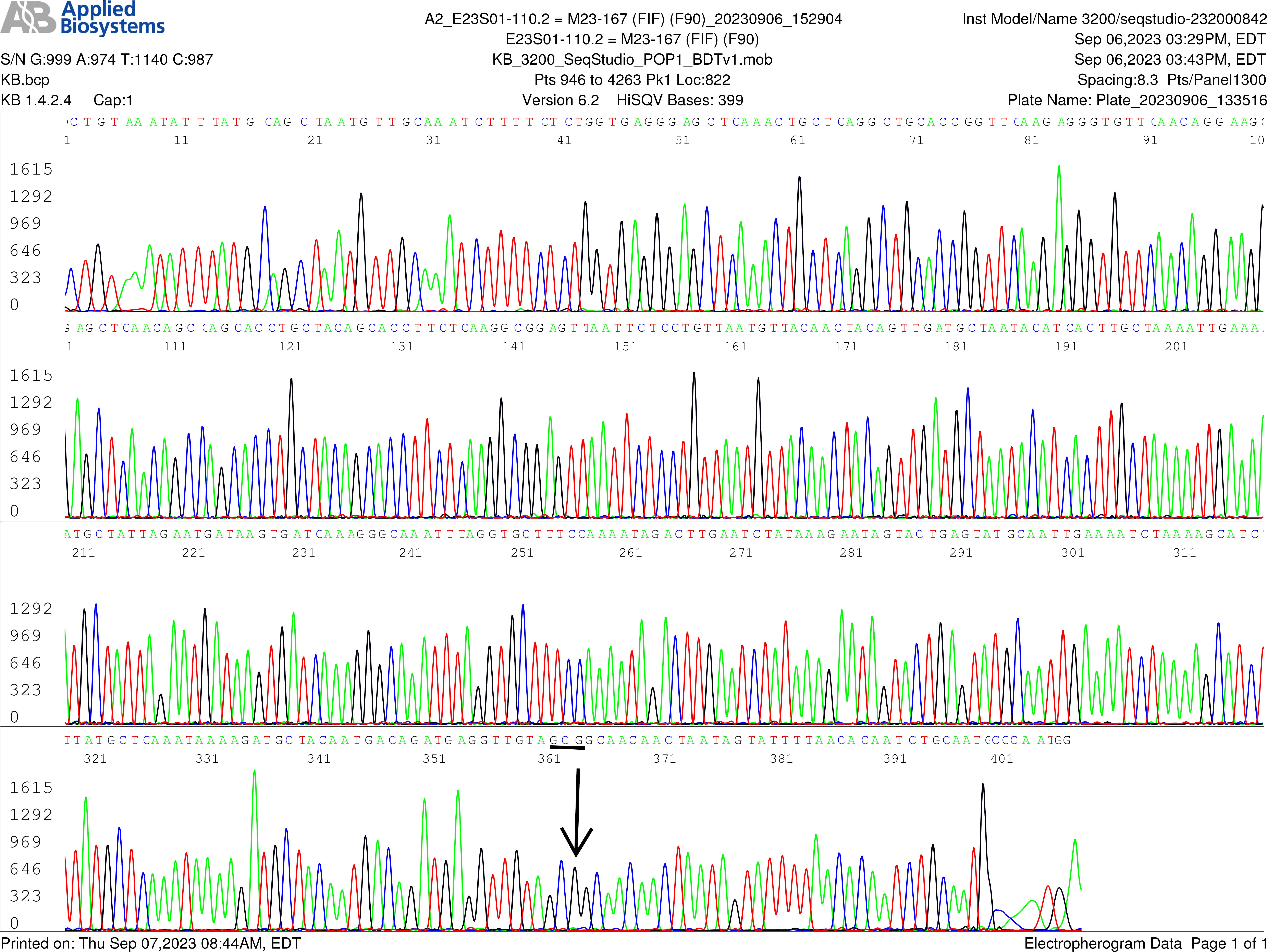 Fig. 2.
Fig. 2.
Single-nucleotide silent mutation in the flaB gene forward sequence. Image of a flaB gene forward sequencing electropherogram, using the flaB inner forward (FIF) nested PCR primer as the sequencing primer, shows an A-to-G (indicated by an arrow) single-nucleotide silent mutation in the 447-bp flaB gene PCR amplicon. In this FlaB protein, the 306th amino acid alanine was encoded by codon GCG instead of GCA as annotated in the GenBank database for all wild-type B. burgdorferi isolates.
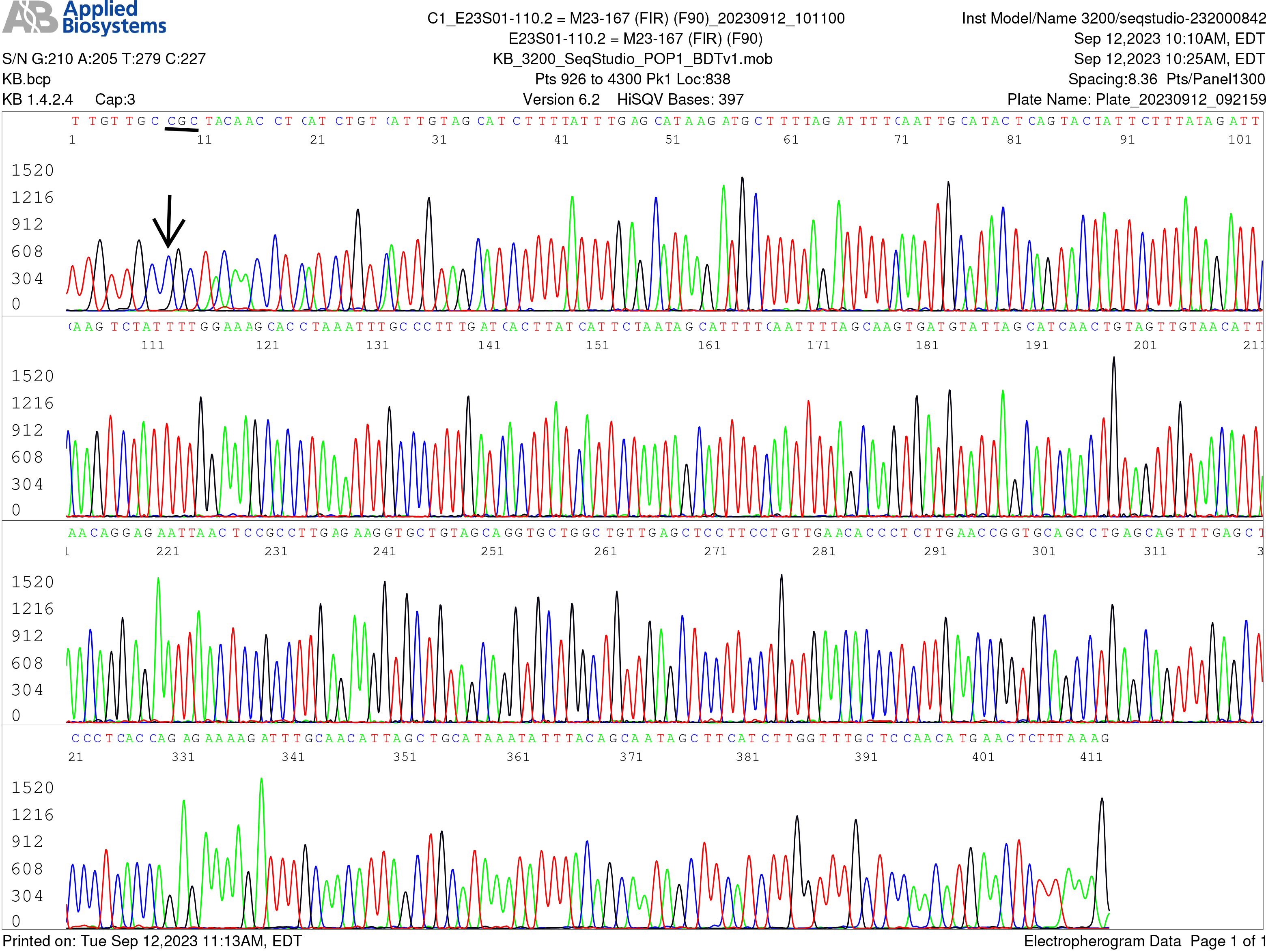 Fig. 3.
Fig. 3.
Single-nucleotide silent mutation in the flaB gene reverse sequence. Image of a flaB gene reverse sequencing, using the flaB inner reverse (FIR) nested PCR primer as the sequencing primer, of the same nested PCR product used to generate the electropherogram shown in Fig. 2, confirming the A-to-G mutation in the flaB gene (the reverse complementary base C is indicated by an arrow).
Nucleotide substitutions in flaB were found in 5 of the 32 (15%) flaB-positive specimens derived from symptomatic patients, and in 1 of 8 (12%) flaB-positive specimens derived from asymptomatic residents.
All the single nucleotide polymorphisms (SNPs) and nucleotide substitutions were verified by bidirectional Sanger sequencing in Figs. 2,3,4,5,6 and Supplementary Fig. 2.
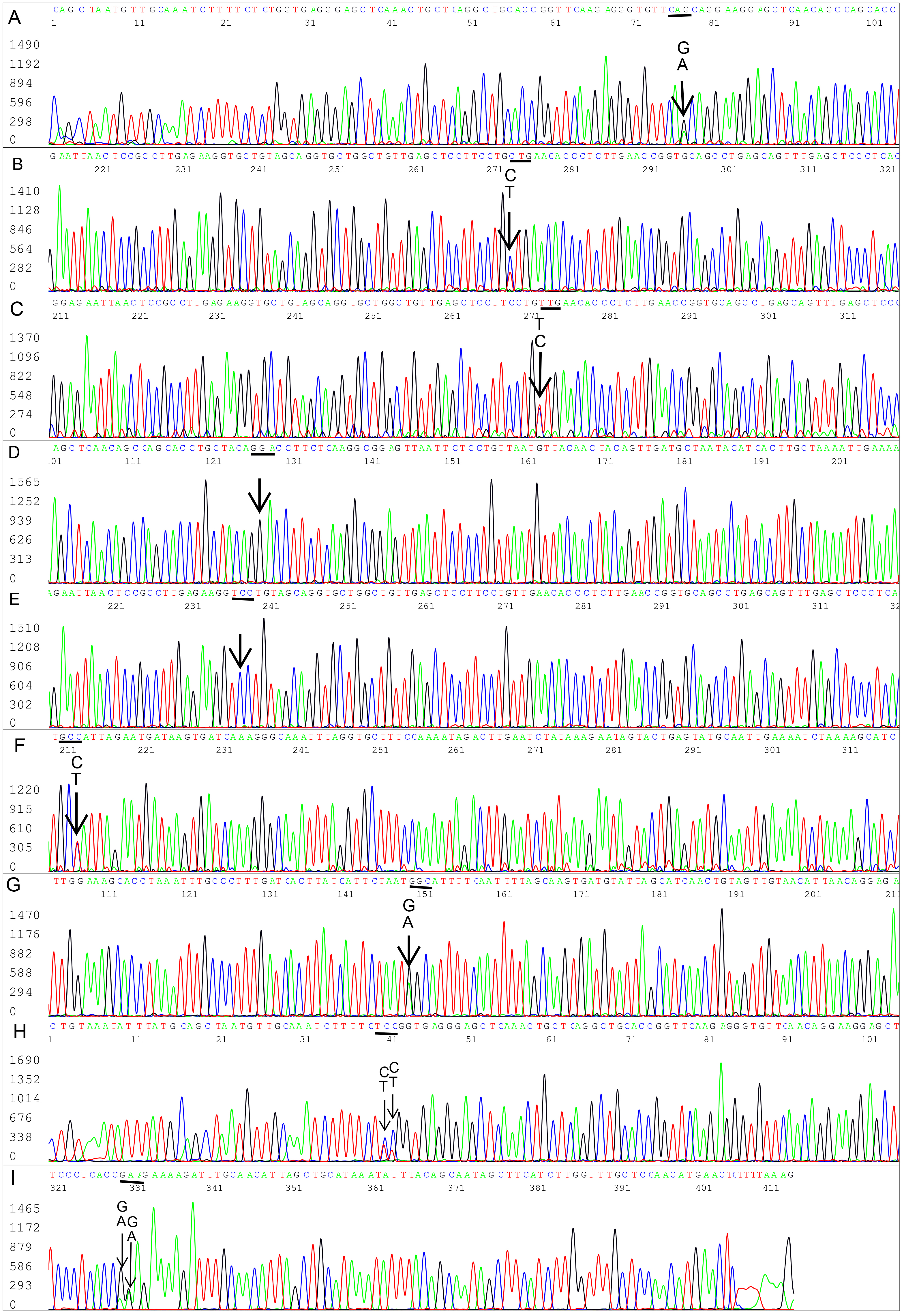 Fig. 4.
Fig. 4.
Four cases of single nucleotide polymorphisms (SNPs) or nucleotide substitutions verified with bidirectional sequencing. (A–I) Bidirectional Sanger sequences excised from 9 electropherograms show 5 B. burgdorferi flaB gene single-nucleotide mutations (all indicated by arrows) in 4 patient blood specimens. (A) There were two copies of flaB gene with a silent mutation. In one copy, the codon for the 216th amino acid glutamine was CAA as annotated in the GenBank sequence database. In the other copy, the 216th amino acid glutamine codon was CAG (not annotated in the GenBank database). (B,C) Reverse complement sequencing of the same nested PCR product used for (A), confirming the 216th amino acid glutamine was encoded by 2 codons CAG (B) and CAA (C), called by the computer base-caller as CTG and TTG in reverse complement when the sequencing was repeated. (D) There was a C-to-G single-nucleotide missense mutation. The 228th amino acid alanine encoded by codon GCA has been converted to glycine encoded by codon GGA (not annotated in the GenBank database). (E) Reverse complement sequencing of the same nested PCR product used for (D), confirming the 228th amino acid codon was GGA (TCC in reverse complement). (F) There were two copies of flaB gene with a silent nutation. In one copy, the codon for the 208th amino acid alanine was GCT. In the other copy, the codon was GCC (not annotated in the GenBank database). (G) Reverse complement sequencing of the same nested PCR product used for (F), confirming 2 copies of flaB in one of which the 208th amino acid alanine was encoded by GCT while the other by GCC. (H) There were two C/T SNPs in the underlined codon TCT, encoding the 199th amino acid serine. When the third nucleotide of the codon changed from T to C, the result was a silent mutation. When the second nucleotide changed from C to T, it caused a missense mutation, turning the amino acid serine into phenylalanine encoded by codon TTT or TTC (a phenylalanine in this amino acid position has not been annotated in the GenBank database). (I) Reverse complement sequencing of the same nested PCR product used for (H), confirming the two C/T SNPs in the codon encoding the 199th amino acid. The existence of two SNPs in one codon verified by Sanger sequencing raised the possibility of more than one duplicated flaB paralog in a chromosome.
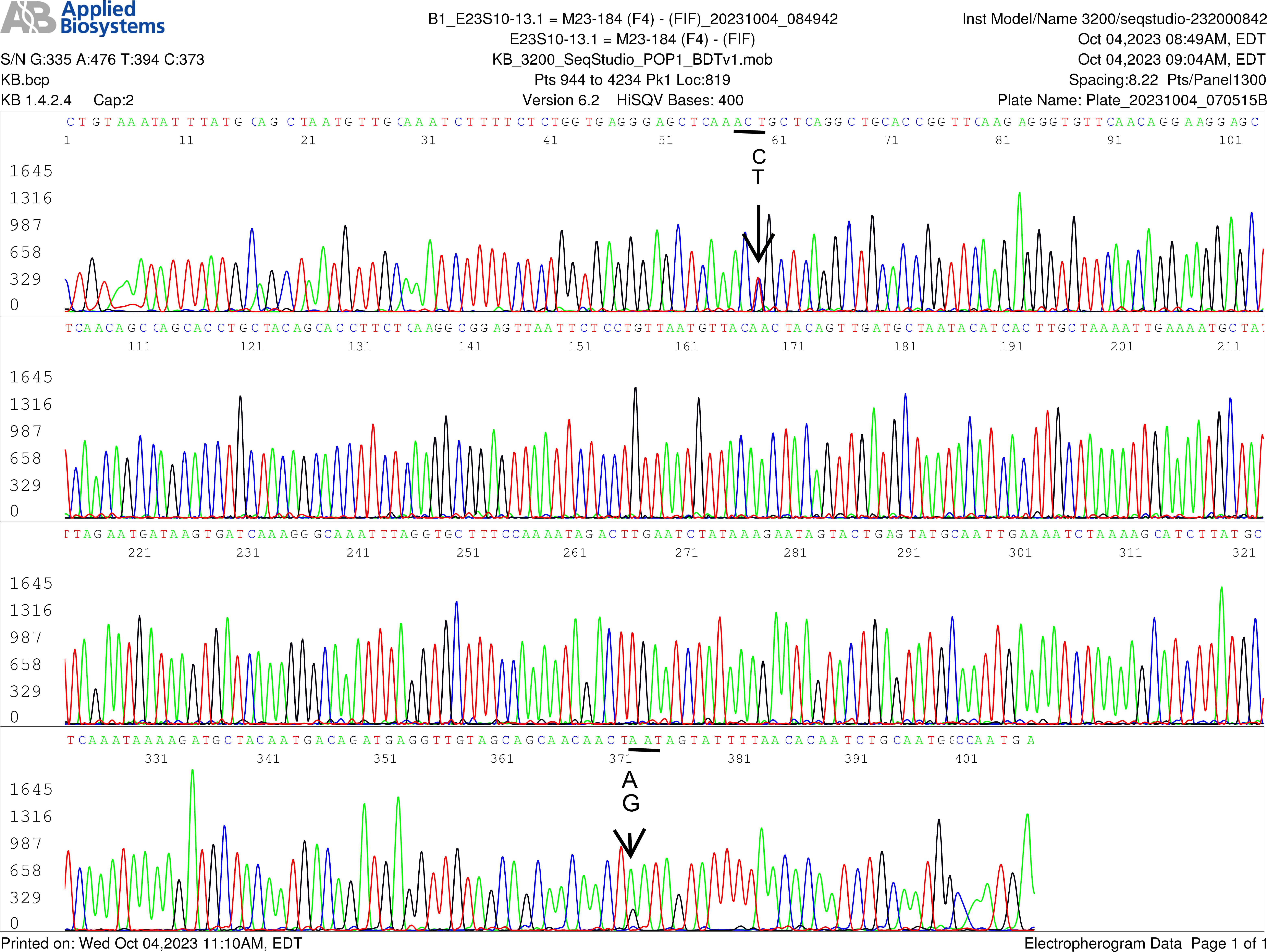 Fig. 5.
Fig. 5.
SNPs in flaB gene of B. burgdorferi in blood of an asymptomatic resident-forward sequencing. This electropherogram shows a forward sequencing of the flaB nested PCR product illustrated in Fig. 1, Left Panel, Lane 4 of an asymptomatic resident control. In this specimen, there were more than one paralogous flaB gene in the B. burgdorferi chromosome. The first T/C SNP in this electropherogram (pointed by an arrow) indicates that there were at least two copies of flaB gene in one of which the 205th amino acid threonine was encoded by codon ACT, and in the other the 205th amino acid threonine was encoded by codon ACC, a silent mutation, which has not been annotated in the GenBank sequence database. In addition, there was an A/G SNP at position 371 (also pointed by an arrow), indicating that there was another duplicate paralog in which the 310th amino acid asparagine encoded by codon AAT was changed to aspartate encoded by codon GAT, a missense mutation. The co-existence of AAT and GAT coding the 310th amino acid was confirmed by a repeated forward sequencing electropherogram (Fig. 6) and a reverve complement sequencing electropherogram presented in Supplementary Fig. 2. A strain of Borrelia burgdorferi containing a flagellin B protein with aspartate in its 310th amino acid position has not been annotated in the GenBank database.
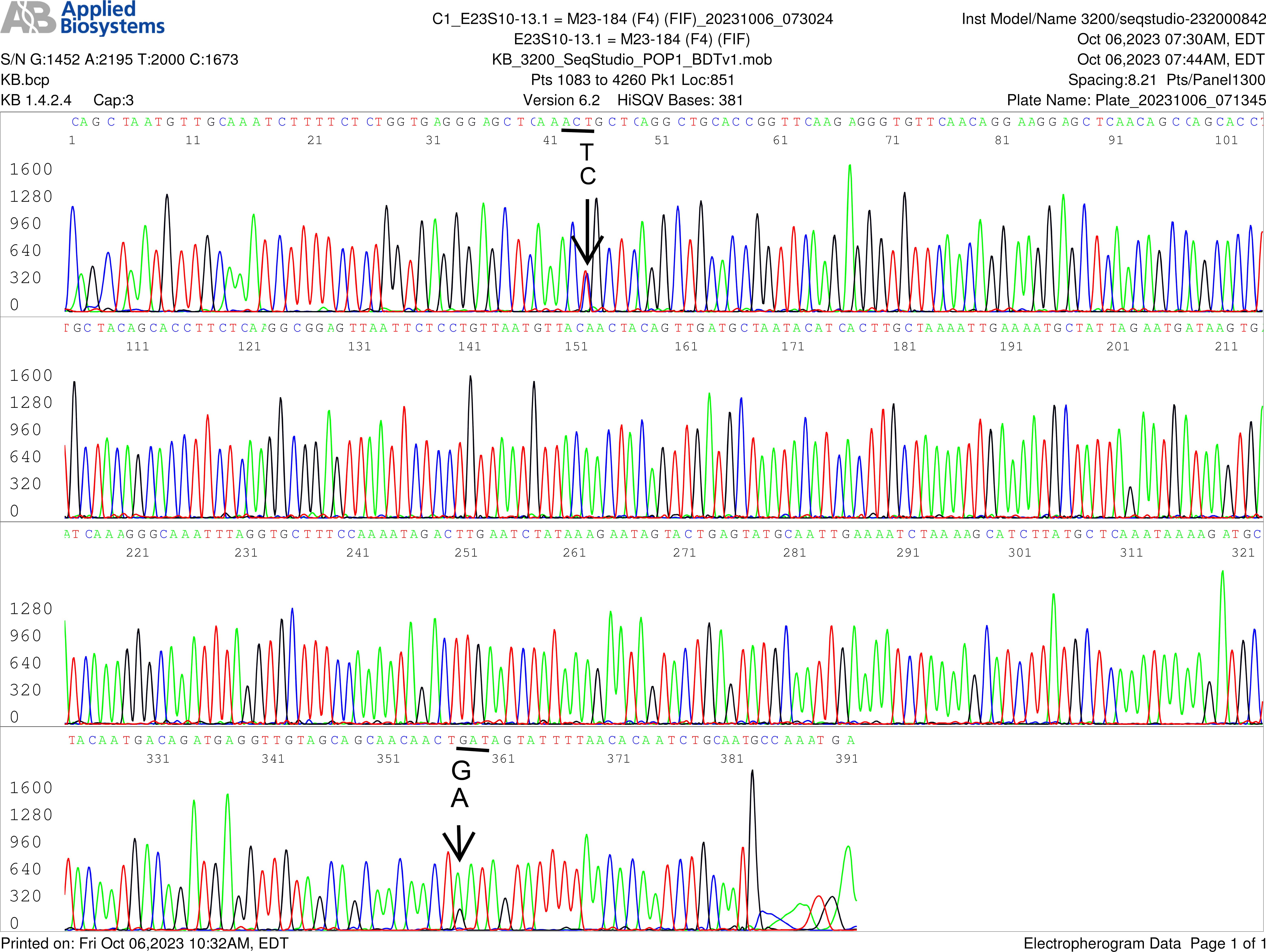 Fig. 6.
Fig. 6.
SNPs in flaB gene of B. burgdorferi in blood of an asymptomatic resident-repeated sequencing. Electropherogram of re-sequencing of the nested PCR product used to generate the sequence shown in Fig. 5, confirming an A/G SNP in position 358, pointed by a GA arrow. The computer base-caller called the codon as GAT in this repeated sequencing instead of AAT. The computer base-caller failed to consistently determine whether “A” or “G” was the dominant base in this position even though the “G” peak appears lower than the “A” peak in the A/G SNP in the electropherograms. Sanger sequencing is designed for qualitative analysis, peak heights in Sanger sequencing analysis are not necessarily quantitative and may not indicate the amount of DNA.
Of the 41 positive specimens, 6 (one in Fig. 2, four in Fig. 4 and one in Fig. 5) showed 8 single- nucleotide mutations or substitutions (summarized in Fig. 7) all of which have not been annotated in the GenBank sequence database in the 447-base segment of the flaB DNA selected for PCR amplification and sequencing. Six of these 8 single-nucleotide mutations occurred as SNPs with two overlapping peaks due to the presence of duplicated flaB paralogs pointed by arrows (four in Fig. 4A,F,H, and two in Fig. 5), and two as single-nucleotide substitutions that resulted in one silent mutation (Fig. 2) and one missense mutation (Fig. 4D). Two of the SNPs (one in Fig. 4H and one in Fig. 5) also resulted in missense mutations.
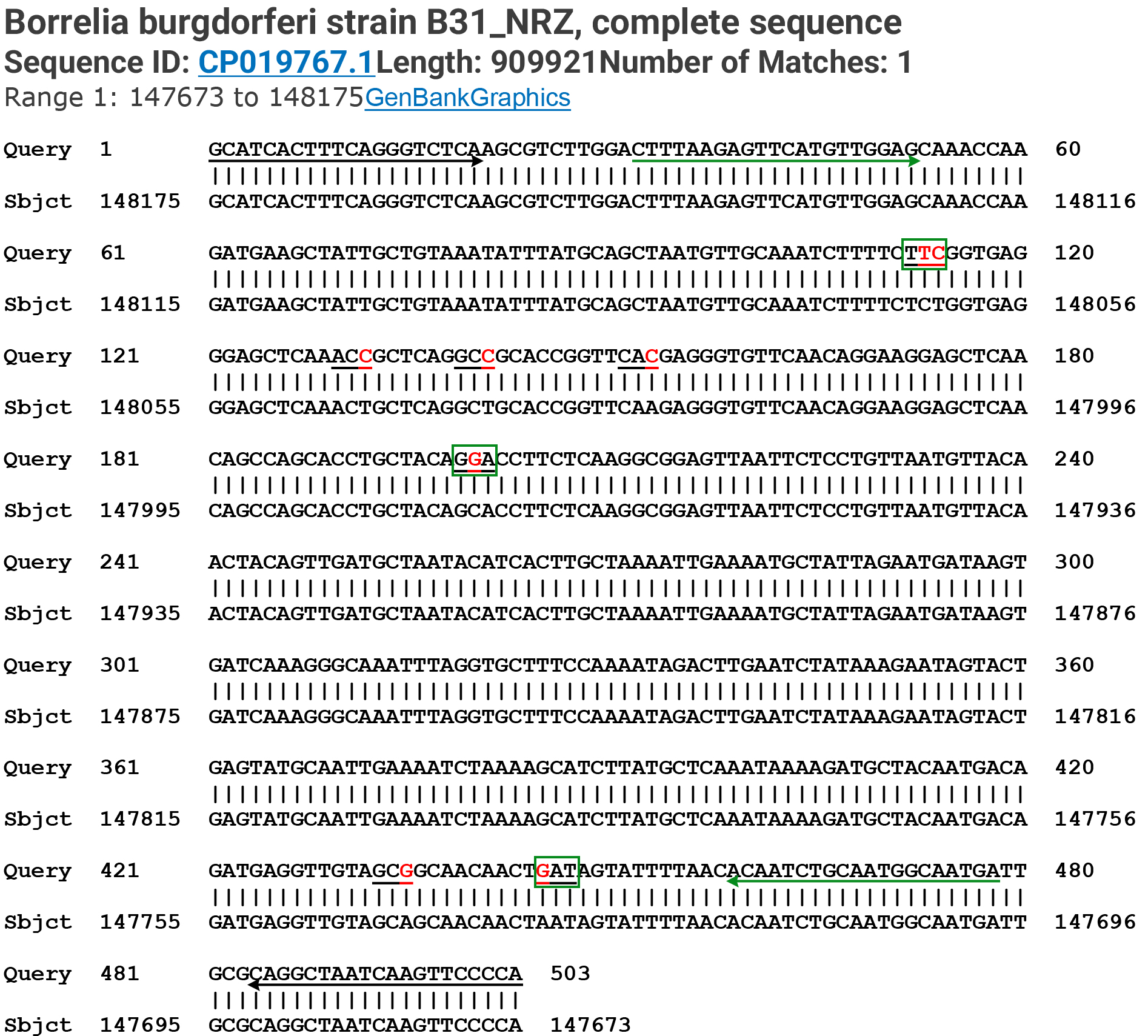 Fig. 7.
Fig. 7.
Summary of the amino acid codons affected by the SNPs verified by Sanger sequencing. A modified Basic Local Alignment Search Tool (BLAST) report from the GenBank including a 503-base Query sequence defined by the flaB outer forward (FOF) PCR primer (marked by a left-to-right black arrow) and the flaB outer reverse (FOR) PCR primer (marked by a right-to-left black arrow) showing the positions of the 8 newly discovered single-nucleotide mutations or SNPs (8 red letters in the 7 underlined codons) in the 447-base segment of the B. burgdorferi flaB gene defined by the flaB inner forward (FIF) nested PCR primer (marked by a left-to-right green arrow) and the flaB inner reverse (FIR) nested PCR primer (marked by a right-to-left green arrow). A single-nucleotide change in the 3 codons framed by green rectangles resulted in missense mutations. The single-nucleotide changes in the 4 unframed codons are silent mutations. The first green-framed codon encoding the 199th amino acid of the FlaB protein had two SNPs one of which also resulted in a silent mutation (see Fig. 4H).
The amino acid codons in the 447-base flaB gene segment affected by the single-nucleotide mutations described above and their relationship to the outer and inner PCR primers were summarized in Fig. 7.
In bacterial genetics, the DNA sequence of a gene in Borrelia burgdorferi strain B31 is considered the “wild type”. Any base changes in a gene sequence from the wild type are mutations. Since the flaB gene in B. burgdorferi strain 31 cultured cells (ATCC 53210) is well characterized, the sequence of the amplicon generated by the PCR primers listed in Fig. 7 was used as the wild type comparator sequence for investigation of the flaB gene complex found in the spirochetes in the positive samples. The bidirectional sequencing electropherograms of the 503-base wild type B. burgdorferi flaB gene sequence defined by both outer PCR primers are presented in Supplementary Figs. 3,4 as reference. Concatenation of the two bidirectional sequences shown in Supplementary Figs. 3,4 generated one contiguous 503-base DNA sequence identical to that illustrated in Fig. 7 (range 147673 to 148175, Sequence ID: CP019767).
To confirm that decoupling of flaB gene from 16S rRNA gene does not occur in cultured B. burgdorferi PCRs, two-fold serial dilutions of a DNA extract from cultured B. burgdorferi cells (ATCC 53210) were made and used as the templates for flaB gene and 16S rRNA gene nested PCRs in parallel. Since both the flaB gene and 16S rRNA gene nested PCRs invariably ended at the same endpoint dilution (Supplementary Fig. 5), the data indicated that the chromosome of cultured B. burgdorferi indeed contains a single 16S rRNA gene and a single flaB gene as reported in literature.
The results of this study confirm that if the platelet-rich plasma of the patients is separated immediately from the red cells and the white cells after venous blood collection before the spirochetes can attach to the lymphocytes in the test tube [11] and the entire DNA content extracted from the platelet pellet obtained by differential centrifugation is used to initiate a primary PCR, Sanger sequencing of the nested PCR products can accurately diagnose B. burgdorferi spirochetemia at the early localized stage of Lyme disease when the density of spirochetes is in the low single-digit numbers (1–3) per mL of platelet-rich plasma. A highly sensitive reliable nucleic acid-based blood test is also needed for identification of the post-treatment Lyme disease syndrome patients whose infected tissues continue releasing a small number of spirochetes into the circulating blood [10]. In contrast to metagenomic (next-generation) sequencing detection of B. burgdorferi cell-free DNAs [19] that can be continuously detected even after the B. burgdorferi cells have been eliminated by antibiotics [20, 21], Sanger sequencing of a segment of the chromosomal DNA of the B. burgdorferi cells spun down in the pellet of the platelet fraction can diagnose active infections in patients with persistent Lyme disease. Dead bacteria in the circulating blood are quickly removed and digested by the reticuloendothelial system (RES), particularly in the liver and the spleen [22]. In practice, next-generation sequencing is unable to detect B. burgdorferi cell-free DNA in the plasma of patients with active erythema migrans rashes and is considered unlikely to be helpful in diagnosing early localized Lyme disease [23].
The finding of 33 spirochetemic cases (33.7%) among 98 symptomatic patients clinically suspected of having early localized Lyme disease was a surprise because it is generally assumed that spirochetemia only begins at the “early disseminated stage” [3, 4]. Given the fact that spirochetemia can occur at the early localized stage of Lyme disease even without an erythema migrans, a highly sensitive and specific diagnostic laboratory test for detecting very low-density B. burgdorferi cells in the blood specimen is needed to diagnose Lyme disease patients for early, proper treatment to prevent deep tissue damage.
Another unexpected finding was that 25 of the 41 split samples tested positive for flaB gene without a concomitant 16S rRNA gene PCR amplification. Since the chromosome of the cultured “wild type” B. burgdorferi is known to contain a single copy of flaB gene [24] and a single copy of 16S rRNA gene [25], a 1:1 gene ratio, which is also supported by simultaneously testing these two genes on double serial dilutions of the DNA extract from cultured B. burgdorferi cells (Supplementary Fig. 5), the B. burgdorferi spirochetes invading the blood stream of the patients at the early localized stage of Lyme disease must have more than one copy of flaB gene in the chromosome through gene duplication to account for the split sample discordance between these two gene PCR test results (Fig. 1). Other investigators also reported that when the synovial fluid DNA of Lyme disease patients was tested for both the 16S rRNA gene and flagellin gene, the PCR targeting the 16S rRNA gene yielded amplification in only 8 cases while the PCR targeting the flagellin gene yielded amplification in 9 cases, a phenomenon referred to as “target imbalance”. Such target imbalance could not be demonstrated when serial dilutions of B. burgdorferi DNA extracted from cultured organisms were tested [26], as observed in the current study.
The findings of 6 SNPs in the form of 2 overlapping nucleotide peaks in 4 of the 40 specimens positive for flaB gene PCR (Fig. 4A,F,H, and Fig. 5) provided evidence for gene duplication in the chromosomal flaB gene of the B. burgdorferi cells invading the blood stream of the patients during the early localized stage of Lyme disease. Since duplicate genes initially have identical sequences but diverge in coding and regulatory regions during subsequent evolution [27], the spirochetes in the other 36 flaB-positive specimens without a SNP in their flaB gene sequence probably also had more than one identical flaB gene copy in the form of paralogs in their chromosome at the early stage of duplication. When duplicated paralogs with an identical sequence are amplified by a pair of PCR primers, only one PCR amplicon will be generated for Sanger sequencing.
Using PCR amplification of chromosomal genes for laboratory diagnosis of B. burgdorferi spirochetemia at the early localized stage of Lyme disease has been hampered by its low sensitivity [28]. When agarose gel electrophoresis is used as the tool for detection of PCR products, approximately 104 copies of target DNA are required as the template to generate a visible PCR product band after 25–30 cycles of PCR amplification [29] because in testing for a pathogen’s genomic DNA in human specimens PCR amplification never reaches its theoretical 100% efficiency due to the presence of PCR inhibitors among many other interfering factors [30, 31]. In the current nested PCR study, the primary PCR mixture contained all the PCR inhibitors, especially the mitochondrial DNA, derived from the platelets isolated from about 2 mL of whole blood. The primary PCR needed at least two copies of target DNA [17] to initiate an exponential amplification to deliver the required copy number of target DNA in about 0.2 µL for the nested PCR to generate a visible product band at agarose gel electrophoresis after 30 cycles of nested PCR amplification.
Using the same pair of primers for nested PCR, referred to as same-nested PCR in this report, is generally not advisable because one pair of PCR primers can generate many unwanted PCR products after 40 cycles of DNA amplification, including exponential amplification of sequences with “Taq errors”. However, when a high-fidelity DNA polymerase is chosen for the same-nested PCR, a segment of borrelial 16S rRNA gene and a segment of flaB gene of Borrelia burgdorferi can be amplified after 60 cycles of PCR for Sanger sequencing without ambiguous base-calling sequences as reported previously [10, 18] and illustrated in Supplementary Figs. 3,4, respectively. Comparing the results of the conventional nested PCR amplification of the flaB gene and those of the same-nested PCR amplification of the 16S rRNA gene of cultured Borrelia burgdorferi cells, there is no observed difference in sensitivity between the two nested PCRs (Supplementary Fig. 5).
When each of the two split sample primary PCR mixtures contained only one copy of B. burgdorferi chromosome, the split sample nested PCR testing would generate a negative 16S rRNA gene amplification and a positive flaB gene amplification because each chromosome contained only one copy of 16S rRNA gene and at least two copies of flaB gene, one in the form of paralog. It required at least 3 copies of chromosome, one copy aliquoted to the flaB gene amplification and two copies aliquoted to the 16S rRNA gene amplification, to generate a positive flaB gene PCR and a positive 16S rRNA gene PCR. When there was only one copy of chromosome in the entire platelet-rich plasma specimen, the specimen would test positive for B. burgdorferi if this single copy of chromosome was aliquoted for the flaB primary PCR during sample splitting. However, if this single copy of chromosome was aliquoted for the 16S rRNA gene primary PCR by chance during sample splitting, the same specimen would generate a false-negative result.
If the entire platelet pellet DNA extract containing a single copy of B. burgdorferi chromosome were used to initiate a primary flaB PCR without sample splitting, the false-negative result would be avoided. However, the flaB gene PCR primers are designed specifically for B. burgdorferi assays, and will fail to amplify the flaB genes of the relapsing fever Borrelia species, including the flaB gene of Borrelia miyamotoi. Since the 16S rRNA gene PCR primers used in this study are designed to amplify a highly conserved segment of the 16S rRNA gene of all known pathogenic Borrelia species [18], a split sample testing to include the 16S rRNA gene PCR can detect spirochetemia caused by a relapsing fever Borrelia species, a B. burgdorferi sensu lato species, or a mixture of the two [10, 32]. The trade-off for ensuring detection of a single B. burgdorferi cell by allocating the entire DNA extract to a flaB gene PCR without a concomitant 16S rRNA gene PCR is to risk missing spirochetemic cases caused by a tick-borne relapsing fever Borrelia species.
The presence of SNPs in a sequencing electropherogram may indicate mixed infections with two species of Borrelia as reported previously [10] rather than duplicate paralogs. However, in the current study SNPs were present in 3 specimens that were positive for flaB gene PCR and negative for 16S rRNA gene PCR in split sample testing. One of these 3 cases is shown in Fig. 1 lanes 4, Figs. 5,6 and Supplementary Fig. 2. Since the 16S rRNA nested PCR was negative, the number of chromosome copies aliquoted to each split sample PCR in these 3 cases must be less than 2, which practically rules out mixed Borrelia infections being the cause for the detected SNPs.
The finding of one to two (1–2) B. burgdorferi cells, detected by flaB sequencing only, in 1 mL of platelet-rich plasma among 17.0% of asymptomatic people residing in Lyme disease-endemic areas in the United States is not a surprise since spirochetemia among asymptomatic patients in early Lyme disease has been reported to be 22.9% by blood cultures [33]. Blood culture seems to be more effective for the diagnosis of spirochetemia in asymptomatic Lyme disease patients than the flaB gene nested PCR amplification method (22.9% versus 17.0%). However, this difference might be due to different criteria being used for the selection of asymptomatic patients. Since B. burgdorferi does not have lipopolysaccharides, B. burgdorferi spirochetemia, unlike other Gram-negative bacteremias, may generate very mild nonspecific symptoms that can be ignored by some patients. To what extent the human innate immunity can clear the B. burgdorferi cells from early Lyme disease patients’ blood needs further study. The biological significance of the duplicate flaB gene paralogs is totally unknown. The B. burgdorferi spirochetes may gain these flaB gene paralogs at certain stage of their life cycle for a specific function and lose the paralogs when the spirochetes are cultured in artificial media. Since almost all DNA sequencing works on B. burgdorferi were carried out on pure cultures, no flaB gene paralog sequences have been published.
This study shows that B. burgdorferi cells causing spirochetemia at the early localized stage of Lyme disease have more than one copy of flaB gene due to the presence of flaB paralogs. Targeting a 447-base segment of the flaB gene for nested PCR amplification followed by Sanger sequencing of the PCR product for verification can detect a single B. burgdorferi cell spun down in the platelet pellet derived from about 2 mL of whole blood with ~100% specificity. Since the highly mobile B. burgdorferi spirochetes can invade co-incubated lymphocytes in the test tube at ambient temperature, the spirochetes along with the platelets must be separated from the whole blood by differential centrifugation as soon as possible after blood collection to prevent losing the limited number of spirochetes to the buffy coat.
Data is available upon written request sent to the author.
SHL developed the methodology, supervised the testing, analyzed the PCR and Sanger sequencing results, wrote the initial manuscript and revised the manuscript after peer reviews. The author has participated sufficiently in the work and agreed to be accountable for all aspects of the work.
DiaSorin, Inc., Stillwater, MN, USA enrolled the patients and healthy donor controls and performed the initial specimen preparation for a research project after informed consent was obtained in accordance with applicable regulations in compliance with 21 CFR 812. Written document is available upon request. The study was carried out in accordance with the guidelines of the Declaration of Helsinki.
The author thanks Ashli Rode and her colleagues in DiaSorin, Inc. for supplying the platelet-rich plasma specimens used in this study. The author also thanks Wilda Garayua for her technical assistance in performing the nested PCR and Sanger sequencing.
This research received no external funding.
The author declares no conflict of interest despite the author’s affiliation is a commercial entity performing tests for fees. The author had full access to all of the data in this study and takes complete responsibility for the integrity of the data and the accuracy of the data analysis.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/FBS31280.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.