

 , Laura Coimbra Teixeira 2, Luise Longo Angeloni 2, Julia Londero Heleno 2, Mariana Romano 2, Marcio Lopes Miranda 2, Tarsis Paiva Vieira 2, Mara Sanches Guaragna 2, Beatriz Amstaldem Barros 2, Andréa Trevas Maciel-Guerra 2, Gil Guerra-Junior 2,*
, Laura Coimbra Teixeira 2, Luise Longo Angeloni 2, Julia Londero Heleno 2, Mariana Romano 2, Marcio Lopes Miranda 2, Tarsis Paiva Vieira 2, Mara Sanches Guaragna 2, Beatriz Amstaldem Barros 2, Andréa Trevas Maciel-Guerra 2, Gil Guerra-Junior 2,*
1 Faculty of Medicine, Pontifical Catholic University of Campinas (PUCCAMP), 13083-887 Campinas, Sao Paulo, Brazil
2 Interdisciplinary Group for Studies of Sex Determination and Differentiation (GIEDDS), Faculty of Medical Sciences and Clinical Hospital, State University of Campinas (UNICAMP), 13083-887 Campinas, Sao Paulo, Brazil
Abstract
The 47,XYY syndrome is a genetic condition found in about 1 in 1000 male children. The expected phenotype is male but could vary greatly. Those with genitourinary abnormalities may also present with microphallus, hypoplastic scrotum, cryptorchidism, hypospadias and macroorchidism. This study reports a child with sex ambiguity who possesses an initial 47,XYY karyotype. We also conducted a narrative literature review of 47,XYY individuals and their respective genital phenotype and/or gender identity.
The narrative literature review was performed by searching for “47,XYY” in the PubMed database. All studies published in English, Spanish or Portuguese from January 1960 to January 2024 that contained the term “47,XYY” in the title or abstract were included. Studies that did not describe the genital phenotype and/or gender identity of cases were excluded. We also described the case of a 2-month-old patient with the 47,XYY karyotype and sex ambiguity.
Our patient underwent additional karyotype testing, resulting in 47,XYY [30] and another 45,X [2]/47,XYY [98] with mosaicism being confirmed by fluorescent in situ hybridization (FISH) on buccal smears (nuc ish (DXZ1 × 1, DYZ3 × 2)[64/100]/(DXZ1 × 1, DYZ3 × 0)[36/100]. A gonadal biopsy revealed an atrophic testis on the left and a streak gonad on the right, with a final diagnosis of mixed gonadal dysgenesis determined. The narrative review revealed 643 articles, of which 350 met the inclusion criteria. However, we excluded 132 articles because they presented no new cases. We included 138 articles, which presented a series containing less than 10 new cases with the 47,XYY karyotype (total of 327 cases), 58 articles presented 4001 cases and 22 articles presented 75 patients with the 47,XYY karyotype in mosaic with 45,X. For all 4403 analyzed cases, 4354 (98.90%) presented a male phenotype, of which 4322 had the 47,XYY karyotype and 32 had mosaicism with 45,X lineage. A further 23 (0.52%) presented a female phenotype, of which four had the 47,XYY karyotype and 19 had mosaicism with 45,X lineage. In addition, 23 (0.52%) cases presented ambiguous genitalia, of which two had the 47,XYY karyotype and 21 had mosaicism with 45,X lineage. Finally, three (0.06%) cases had undefined phenotypes, all with mosaicism with 45,X lineage. Of the six cases with the 47,XYY karyotype and no male phenotype, one had complete androgen insensitivity syndrome (CAIS), one had lipoid congenital adrenal hyperplasia, two had probable CAIS, and two presented an incomplete diagnostic investigation.
A female or ambiguous genital phenotype in an individual with 47,XYY karyotype is uncommon and should alert to the presence of the 45,X lineage or association with other causes of disorder/difference of sex development.
Keywords
- disorder of sex development
- 47,XYY
- turner syndrome
- mixed gonadal dysgenesis
The 47,XYY syndrome, also known as Jacob’s syndrome, is a genetic condition that occurs in about 1 in 1000 male children [1] and was first described in the 1960s [2].
The clinical presentation for this syndrome varies greatly, and many individuals have relatively few, if any, phenotypic abnormalities. It has been estimated that approximately 85% of 47,XYY men are undiagnosed [3]. Those who are diagnosed frequently exhibit tall stature, delayed speech and language development, low-set ears, malar flattening, and motor delay [3]. Other frequent features are asthma, autism spectrum disorder, atypical behavior, congenital night blindness, enuresis, finger clinodactyly, hyperactivity, hypertelorism, intellectual disability, macrocephaly, neonatal hypotonia and flat feet [3]. Other characteristics may also be present, including infertility, encopresis, macrodontism, seizures, tics and obsessive-compulsive behavior [3, 4, 5]. While some 47,XYY individuals are infertile, most are able to reproduce and often have offspring with a normal karyotype [3].
According to Bardsley et al. [3], macroorchidism is a common finding in 47,XYY individuals. In their study, 42% of individuals aged 11–13 years had a testicular volume of 10 mL or more. Most individuals (78%) had a greater than average testicular volume for their age, and in 50% of cases it was significantly above average [3]. However, hypospadias and cryptorchidism were both noted to be very rare, with only one and two cases, respectively [3]. Moreover, there was no increased incidence of precocious puberty or precocious adrenarche [3].
Although the 47,XYY karyotype is usually associated with a normal male phenotype, in the present study we had the opportunity to evaluate a child with sex ambiguity whose chromosome constitution was initially diagnosed as 47,XYY. The aim of our study was to report on this case and to conduct a comprehensive literature search of 47,XYY individuals who were reported with information on their genital phenotype and/or gender identity.
A narrative review of the literature was performed in the PubMed database using the terms “47,XYY” OR “Jacob syndrome”. All studies published in English, Spanish or Portuguese from January 1960 to January 2024 with the term “47,XYY” in the title or abstract were included. Studies that did not report the genital phenotype or the gender identity of the patients were excluded. Additional data was obtained by examining the reference list of articles and then reviewing any relevant papers.
We also describe a 2-month-old patient with sex ambiguity and an initial diagnosis of 47,XYY karyotype. This study was carried out in accordance with the Declaration of Helsinki. The child’s parents authorized the disclosure of data after signing the consent form approved by the Research Ethics Committee of the UNICAMP (No. 434/2006 – CAAE: 0340.0.146.000-06).
Only descriptive statistical analysis was used, with data shown in Table and Figure format.
An 87-day-old infant with a female sex assignment was referred to us due to genital ambiguity. The infant was born at term by cesarean section, had no neonatal complications, and had a birth weight of 3630 g (p100), length of 48 cm (p22), head circumference of 35 cm (p88), and Apgar 8/8. The infant was the second child of healthy unrelated parents (39-year-old mother and 41-year-old father), and had a healthy 7-year-old brother and a 21-year-old maternal half-brother.
Physical examination revealed a healthy infant with normal growth (weight of 5100 g; length of 57 cm, head circumference of 38.5 cm) and no dysmorphic features for the external genitalia. These comprised a 2.8 cm phallus with chordee, penoscrotal hypospadias, unpalpable right gonad and left gonad palpable in the inguinal region. The features of the external genitalia were compatible with a Prader grade III/IV, and the external masculinization score [6] was 4.0.
Karyotype analysis for the infant was performed by another service on cells cultivated from peripheral blood. This revealed a 47,XYY karyotype in 20 analyzed cells. No uterus was visualized on pelvic ultrasound.
Basal levels of gonadotropins (Follicle Stimulating Hormone (FSH): 1.54 IU/L; Luteinizing Hormone (LH): 2.95 IU/L), androstenedione (1.07 mg/dL) and 17-hydroxyprogesterone (512.0 ng/dL) were normal for the reference values at minipuberty, while total testosterone (96 ng/dL) was slightly below the level for age.
A second karyotype analysis on peripheral blood cells also showed the 47,XYY in
30 analyzed cells. Fluorescent in situ hybridization (FISH) with X and Y
centromeric probes conducted oin buccal smear cells revealed a cell line without
Y chromosome signals in 36% of the analyzed cells, nuc ish (DXZ1
Videolaparoscopy and cystoscopy performed at the age of 5 months showed a 3.5-cm vagina, no uterus, an atrophic testis in the left inguinal region and a streak gonad on the right. Diagnosis was therefore given as Mixed Gonadal Dysgenesis (MGD).
After discussing the case with the multidisciplinary team and the family, it was decided to maintain the female sex and to perform feminizing genitoplasty and bilateral gonadectomy in the future. Abdominal ultrasound showed no urinary tract malformations, and echocardiography results were also normal.
The search in the PubMed database revealed 643 published articles with the terms “47,XYY” OR “Jacob syndrome”, of which the full text was available for 432. Of these, 390 were related to humans and 350 were published in English, Spanish or Portuguese. All titles and abstracts from the 350 selected articles were read. Among these 22 articles, a total of 75 cases presented the 47,XYY karyotype in mosaic with 45,X or variants with female genitals (Turner syndrome—TS) or ambiguous genitals (MGD or Ovotesticular Disorder/Difference of Sex Development—OT DSD) (Supplementary Table 1 and Fig. 1). Of these 328 articles, 132 were not included because they did not present any new cases (Fig. 1). Among 196 articles, we included 58 [3, 5, 7, 8, 9, 10, 11, 12, 13, 14, 15, 16, 17, 18, 19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52, 53, 54, 55, 56, 57, 58, 59, 60, 61, 62] comprising 4001 cases with the 47,XYY karyotype (Supplementary Table 2 and Fig. 1) and 327 cases from 138 articles (Fig. 1).
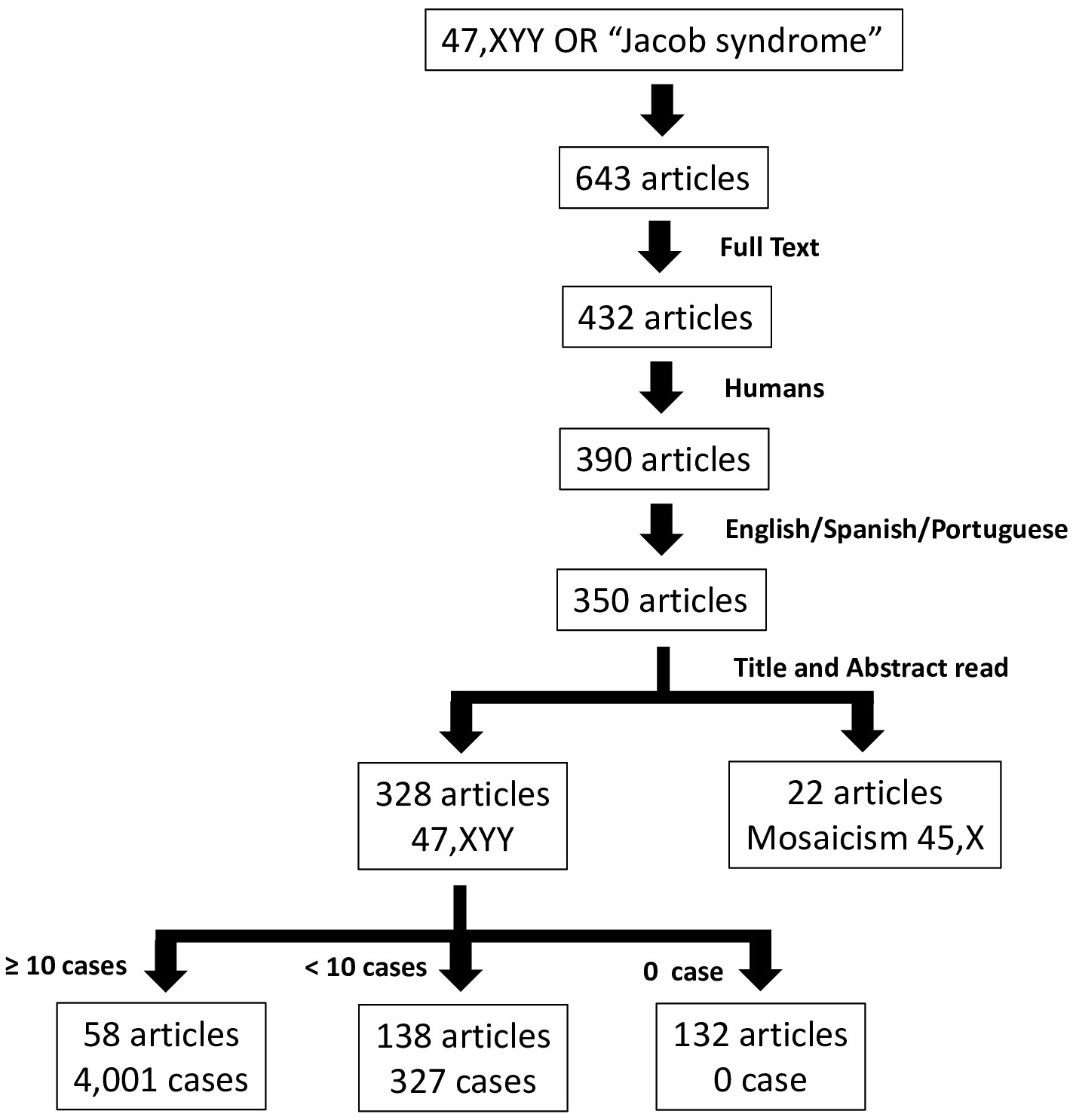 Fig. 1.
Fig. 1.
Flowchart used for the screening and selection articles reporting 47,XYY karyotype cases.
The Table 1 shows the association of karyotype with genital phenotype for the
4403 individuals analyzed. Of 4328 cases with 47,XYY karyotype, 4322 (98.3%)
presented with male genitalia, four with female genitalia [63, 64, 65, 66], and two with
ambiguous genitalia [63]. Of 75 cases with 47,XYY in mosaicism with the 45,X
lineage (Supplementary Table 1), 32 had male genitalia, 19 had female
genitalia (probably TS), 21 had ambiguous genital (probably MGD or OT DSD), and
three had an undescribed genital phenotype. von Westarp et al. [63]
described a 47,XYY patient with associated Complete Androgen Insensitivity
Syndrome (CAIS), thus explaining the female genitalia. These authors [63] also
described two other 47,XYY cases with female phenotype and enlargement of the
clitoris, but did not carry out further hormonal and cytogenetic investigations.
Therefore, it is unclear whether they had a 45,X/46,XYY mosaicism characterizing
MGD or OT DSD or another DSD etiology such as disorder in testosterone synthesis
or action [67, 68]. Katsumata et al. [64] described a 47,XYY case with
associated Lipoid Congenital Adrenal Hyperplasia (LCAH) which could also explain
the female genitalia. Boczkowski [65] described a case that was similar to that
of von Westarp et al. [63], but without genetic molecular investigation.
Their patient had breast development, absence of pubic hair and uterus, tall
stature and increased levels of gonadotropins, suggestive of CAIS. Benasayag
et al. [66] described a similar case of a 47,XYY woman with breasts,
sparse hair, absence of uterus, presence of two testes, LH
| Genital phenotype | Karyotype | % | |
| 47,XYY | Mosaicism with 45,X lineage | ||
| Male (n = 4354) | 4322 | 32 | 98.90 |
| Female (n = 23) | 4 | 19 | 0.52 |
| Ambiguous (n = 23) | 2 | 21 | 0.52 |
| Not described (n = 3) | 0 | 3 | 0.06 |
| % | 98.30 | 1.70 | 100.00 |
Following referral of the 47,XYY infant described above to our service, we conducted a narrative review of thousands of cases with the 47,XYY karyotype. The vast majority (98.9%) were found to have a male phenotype and/or male gender identity. However, six cases with a homogeneous 47,XYY karyotype had a female phenotype (four cases) [63, 64, 65, 66] or ambiguous genital phenotype (two cases) [63]. One of these had a confirmed association with CAIS [63], two had suspected associations with CAIS [65, 66], one had a confirmed association with LCAH [64], and two cases from the 1960’s had an ambiguous genital phenotype without diagnostic confirmation [63]. These latter two cases potentially involved unidentified 45,X/47,XYY mosaicism indicating MGD or OT DSD, or association with another DSD etiology, due to a disorder in testosterone synthesis or action. In addition, 22 articles were identified that presented cases of 45,X/47,XYY mosaicism or variants with a female phenotype and a diagnosis of TS, or an ambiguous phenotype and a possible diagnosis of MGD or OT DSD.
A diagnosis of TS may be established in cases of 45,X/47,XYY mosaicism or variants with a female phenotype. The management of such cases should follow the guidelines of the Turner Syndrome Consensus. This includes bilateral prophylactic gonadectomy due to the high risk (10–15%) of gonadal tumor development [69]. In our experience, 6% of TS cases present an intact Y chromosome or a structural change in its chromosomal constitution [70]. Furthermore, a diagnosis of MGD is more frequent in 45,X/47,XYY mosaicism individuals who present genital ambiguity. A diagnosis of OT DSD is also possible [67, 68, 71], and differs from TS exclusively by genital ambiguity and gonadal constitution. There are two streak gonads in TS, there may be a streak gonad and a dysgenetic testis or two dysgenetic testes in MGD, while testicular and ovarian tissue are present in the same gonad or in different gonads in OT DSD [72]. However, after deciding upon the sex of rearing and the maintenance of gonads [67, 68], these cases must be followed up in same way of TS due to the presence of the 45,X lineage [69]. Therefore, whenever a female or ambiguous phenotype is present in a 47,XYY individual, FISH with X and Y centromere probes should be performed to search a 45,X lineage. This approach was carried out for the case we described, thereby allowing the diagnosis of MGD.
Since the 47,XYY chromosome constitution is not rare, the presence of a female or ambiguous phenotype and the absence of a 45,X lineage by FISH analysis does not rule out a possible association with other diseases. This was observed in four cases described in the literature, one of which had CAIS confirmed by a pathogenic variant in the androgen receptor gene [63] and two of which showed strong clinical suspicion of this condition [65, 66]. CAIS is an X-linked genetic disease and is the most common cause of DSD in 46,XY individuals [73]. The phenotype ranges from normal female external genitalia in CAIS to normal male external genitalia associated with infertility and/or gynecomastia in the mild form (Mild Androgen Insensitivity Syndrome, MAIS) [73]. Moreover, a large spectrum of undervirilized male external genitalia is observed in the partial form (Partial Androgen Insensitivity Syndrome, PAIS) [73]. Pathogenic variants in the androgen receptor gene (AR – OMIM (On line Mendelian Inheritance in Man) *313700) are found in 65% to 95% of individuals with CAIS but in only 40% to 45% of individuals with PAIS [73].
Another case described in the literature was a Japanese patient with a 47,XYY karyotype who exhibited a female phenotype attributed to LCAH [64]. LCAH (OMIM #201710) is caused by loss-of-function pathogenic variants in the STAR gene (OMIM *600617) which encodes steroidogenic acute regulatory protein [74]. This disorder is characterized clinically by impaired steroidogenesis in the adrenal gland, resulting in severe deficiencies of glucocorticoids and mineralocorticoids, as well as gonadal dysfunction manifesting as female genitalia in both 46,XY and 46,XX karyotypes with hypergonadotropic hypogonadism. Pathologically, LCAH it is characterized by the accumulation of cholesterol ester in the cytosol of the steroidogenic cells in the adrenal and gonadal tissues [74]. LCAH demonstrates ethnic variations in prevalence that have been attributed to a founder effect of STAR-Gln258*, notably in Korea and potentially extending to others East Asian countries [74]. There is limited epidemiological and clinical data regarding his disorder, even in East Asia, where LCAH has a higher prevalence compared to other regions [74].
Another two cases with the 47,XYY karyotype and genital ambiguity (female phenotype with enlarged clitoris) were cited by von Westarp et al. [63]. These two cases were originally reported in the 1960s, but unfortunately both articles were unavailable. It is therefore uncertain whether they involved a 45,X/47,XYY mosaicism characterizing MGD or OT DSD or another etiology of DSD with a disorder in testosterone synthesis or action [67, 68].
Among the 75 cases with 47,XYY in mosaicism with the 45,X lineage, 32 had a male phenotype, 19 had a female phenotype (probably TS), 21 had ambiguous genitalia (probably MGD or OT DSD), and in three cases no genital features were described. These results highlight the great diversity of genital phenotype when a 45,X lineage is present in mosaicism with the 47,XYY lineage.
The current review and case presentation confirm that the expected genital and identity phenotype in the vast majority of cases with a homogeneous 47,XYY karyotype is male, except when there is mosaicism. These data are of great importance for pre- and postnatal evaluation of cases with the 47,XYY karyotype.
An important limitation of this work is that the review included articles from 1960 to 2024, and the chromosomal analysis has been improved throughout the years, especially for the detection of structural chromosomal rearrangements and low-level mosaicisms. Considering that this review focused on a numerical abnormality, the lack of banding techniques in the 60s would not impact the results of this review. However, we cannot rule out that cases of a homogeneous 47,XYY karyotype and a female phenotype or ambiguous genitalia had a hidden mosaicism with a 45,X cell lineage. Appropriately, these cases should be investigated by FISH technique, which only became available at the end of 80s. On the other hand, the main goal and a strong point of the current study was that it analyzed a large number of cases with the 47,XYY karyotype in relation to their genital phenotype or gender identity.
The expected genital phenotype and/or gender identity in an individual with a 47,XYY karyotype is male. However, in the presence of a female or ambiguous genital phenotype, it is crucial to investigate for the presence of a 45,X lineage, and also to consider possible associations with other causes of DSD.
Data from this study can be requested from the corresponding author at any time.
MJP, ATMG and GGJ designed the study; MJP and GGJ performed the review of the literature; LCT, LLA, JLH, MR and MLM cared the patient involved in this study; TPV performed karyotype and FISH analyses. MSG and BAB contributed with critical analysis of the study. All authors helped in writing this manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work agreed to be accountable for all aspects of the work.
This study was carried out in accordance with the Declaration of Helsinki. The child’s parents authorized the disclosure of data after signing the consent form approved by the Research Ethics Committee of the UNICAMP (No. 434/2006 – CAAE: 0340.0.146.000-06).
Not applicable.
This research received no external funding.
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/FBS25251.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.