



1 Yamaguchi University Graduate School of Medicine, Ube, 755-8505 Yamaguchi, Japan
Academic Editor: Gustavo Caetano-Anollés
Abstract
Recent studies provide evidence that similar to early-stage Parkinson’s disease, depression is a neurodegenerative disease characterized by the degeneration of monoamine axons. The major difference between the two disorders is that the symptoms of depression become evident without loss of monoamine neurons, while the motor symptoms of Parkinson’s disease appear after loss of the cell body. Given that the axonal degeneration of monoamine neurons underlies the pathophysiology of neurological (Parkinson’s disease) and neuropsychiatric (depression) diseases, axonal impairment of monoamine neurons is thought to also occur in schizophrenia and bipolar disorder and play a significant role in the pathophysiology of these mental illnesses. The positive symptoms of schizophrenia and manic symptoms of bipolar disorder are known to occur in hyper-monoaminergic states, opposite to depressive symptoms, negative/cognitive symptoms of schizophrenia, and motor disorders of Parkinson’s disease, all occurring in hypo-monoaminergic states. Since monoamine axons have the capacity to spontaneously regenerate or sprout in response to damage in the adult brain and sometimes show hyperinnervation due to excessive regeneration/sprouting beyond normal levels, it is possible that schizophrenia and bipolar disorder are disorders that include excessive regeneration/sprouting of monoamine axons leading to hyper-monoaminergic states. Together, based on accumulating data from animal and human studies, the pathophysiology of schizophrenia, major depression, and bipolar disorder is summarized as follows: The degeneration of monoamine axons is associated with the negative and cognitive symptoms of schizophrenia, major and bipolar depression, while hyper-regeneration/sprouting of monoamine axons underlies the positive symptoms of schizophrenia and bipolar mania. The integrated understanding of schizophrenia, major depression, and bipolar disorder as monoamine axon disorder will open the door to the development of new diagnosis and treatment methods for major mental illnesses as well as early-stage Parkinson’s disease.
Keywords
- Review
- Schizophrenia
- Depression
- Bipolar disorder
- Dopamine
- Noradrenaline: 5-HT
- Lithium
- Antidepressant
The three major brain monoamines, dopamine (DA), noradrenaline (NA) and serotonin (5-HT), have been extensively investigated in relation to neurological and psychiatric diseases, including Parkinson’s disease, schizophrenia, major depression, and bipolar disorder. Much evidence shows that the monoamine-related disorders occur in hypo-monoaminergic states (Parkinson’s disease, major depression, bipolar depression, and negative/cognitive symptoms of schizophrenia) and/or in hyper-monoaminergic states (bipolar mania and positive symptoms of schizophrenia). Parkinson’s disease is a neurodegenerative disease characterized by loss of DA neurons of the substantia nigra pars compacta (SNc) that project DA axons to the dorsal striatum, while the pathophysiology of schizophrenia, major depression and bipolar disorder remain elusive. Impairments of NA neurons of the locus coeruleus and 5-HT neurons of the raphe nucleus that each innervate almost the entire brain are thought to be involved in the pathophysiology of depression [1]. DA neurons of the ventral tegmental area (VTA) that send axons to the ventral striatum (nucleus accumbens) play a pivotal role in reward-related behavior and its disfunction is considered to be associated with anhedonia, one of the major symptoms of depression [2, 3, 4]. In addition, another group of DA neurons in the VTA sends axons to the medial prefrontal cortex (mPFC), and its functional impairment has been explored focusing, in particular, on schizophrenia.
Recent studies have shown that conventional antidepressant drugs possess the ability to induce the regeneration of NA and 5-HT axons [5, 6, 7, 8, 9, 10, 11], while in patients with depression as well as animal models of depression, depressive symptoms are associated with the degeneration of monoamine axons [8, 10, 11, 12, 13, 14, 15]. These findings support the view that depression is a neurodegenerative disease characterized by the degeneration of monoamine axons without loss of the cell bodies (cell death) of origin. In Parkinson’s disease, the degeneration of the distal axons of SNc DA neurons occurs first, and persistent retrograde axonal degeneration finally results in the degeneration of the cell body [16, 17]. Thus, axonal degeneration is thought to be a common phenomenon occurring in depression and early-stage Parkinson’s disease [12, 13, 14, 15, 16]. The major difference between the two disorders is that the symptoms of depression become evident without the degeneration of the cell bodies, while the movement disorders of Parkinson’s disease appear after loss of the cell body. Based on the view that axonal degeneration of monoamine neurons underlies the pathophysiology of neurological (early-stage Parkinson’s disease) and neuropsychiatric (depression) diseases, similarly to early-stage Parkinson’s disease and depression, axonal impairment of monoamine neurons is thought to also occur in schizophrenia and bipolar disorder and to play a significant role in their pathophysiology.
This review focuses on the role of monoamine axons in schizophrenia and bipolar disorder, and will provide an integrated view of the pathophysiology of major depression, schizophrenia, and bipolar disorder.
This review describes morphological changes of monoamine axons in relation to the pathophysiology of major mental disorders. The major morphological changes of monoamine axons include degeneration, regeneration, and sprouting: degeneration is defined as destruction of axons, regeneration as regrowth from damaged axons, and sprouting as new outgrowth from intact axons. Regarding measurements of axon density, immunohistochemistry using antibodies to monoamine synthesis enzymes or monoamine transporters is commonly used to visualize monoamine axons in animal studies and postmortem studies of human brains. In human studies, positron emission tomography (PET) imaging with radiolabeled monoamine transporters is also used to measure the density of monoamine axons. Since monoamine transporters are located on the membrane of monoamine axon terminals, the level of monoamine transporters is directly related to the density of monoamine axon terminals.
Schizophrenia is one of the major mental disorders, characterized by positive (e.g., delusion and hallucination), negative (e.g., social withdrawal and poor rapport), and cognitive symptoms (e.g., disorganized thinking and problem solving) [18]. There is clear and consistent evidence that positive symptoms are induced by hyperdopaminergic activity, whereas negative/cognitive symptoms are caused by hypodopaminergic activity [18, 19, 20, 21, 22]. Typical antipsychotics, such as chlorpromazine and haloperidol, improve the positive symptoms of schizophrenia by blockage of D2 receptors [23, 24], while they have rather worse effect on negative/cognitive symptoms [25]. It is also noted that psychostimulants, including amphetamine and cocaine, which increase extracellular DA levels, induce positive symptoms in normal subjects and make psychotic symptoms worse in schizophrenic patients [26, 27]. Moreover, L-dopa administration in Parkinson’s disease is found to produce positive symptoms in some cases [28]. In contrast, amphetamine and L-dopa have been reported to have beneficial effects on the negative/cognitive symptoms of schizophrenia [29, 30, 31, 32]. All these findings support the view that the positive symptoms of schizophrenia are related to excessive DA activity, while hypodopaminergic activity induces negative/cognitive symptoms.
The PFC, including the mPFC and dorsolateral PFC, is known to play a critical role in a variety of higher-order functions, such as working memory, decision-making, and interference control [33, 34], and patients with damage to the PFC show cognitive and memory impairments [35, 36, 37, 38, 39]. Given the function of the PFC, the negative/cognitive symptoms of schizophrenia are thought to occur due to a hypodopaminergic state in the PFC [20, 40]. However, the pathophysiology of hypodopaminergic states as well as hyperdopaminergic states remains unknown. In early-stage Parkinson’s disease, the degeneration of DA axons of SNc neurons projecting to the dorsal striatum leads to a hypodopaminergic state of the striatum. Similarly, it is possible that the degeneration of DA axons of VTA neurons projecting to the mPFC occurs in patients with schizophrenia, resulting in the hypodopaminergic state of the mPFC. Importantly, in a postmortem study, Akil et al. [41] reported direct evidence for degeneration of DA axons in the mPFC of schizophrenic patients. In this study, the density of DA axons, visualized by immunohistochemistry using antibodies to tyrosine hydroxylase (TH) or DA transporters, was examined in the dorsomedial PFC from 16 pairs of schizophrenic and matched control subjects. In the same subject, TH- and DA transporter-immunoreactive axons showed the same overall patterns of regional and laminar distribution. Compared to control subjects, the density of TH-immunoreactive axons was clearly reduced in 10 schizophrenic subjects, increased in two, and unchanged in four. In contrast, the density of cortical 5-HT axons labeled for the 5-HT transporter was not different between schizophrenic and control subjects. These findings strongly suggest the occurrence of degeneration of DA axons in the mPFC of schizophrenic patients.
Despite increasing evidence supporting the involvement of hyperdopaminergic activation in the expression of positive symptoms, the pathophysiology of hyperdopaminergic states in schizophrenia remains unclear. The majority of previous studies have focused on a hyperdopaminergic state in the striatum in relation to positive symptoms. Initially, the ventral striatum (nucleus accumbens) was considered a site where hyperdopaminergic states occur in schizophrenic patients with positive symptoms, and more recently, however, the focus appears to shift from the ventral striatum to the dorsal striatum, particularly to the associative striatum. For details, refer to the review by McCutcheon et al. [22].
In addition to positive symptoms, high doses of amphetamine are known to induce behavioral alterations, such as euphoria and stereotyped behavior, in normal individuals. Animal and human studies have shown that amphetamine euphoria was associated with excessive DA release in the reward-related ventral striatum (nucleus accumbens) [42, 43, 44] that receives DA innervation from the VTA. Moreover, amphetamine-induced stereotyped behavior is reported to be due to a hyperdopaminergic state in the dorsal striatum [45, 46, 47] that is a site responsible for motor disorders of Parkinson’s disease with loss of DA innervation from the SNc. However, there is no direct evidence showing an association between amphetamine-induced positive symptoms and hyperdopaminergic activation in the striatum. In fact, it is difficult to identify the brain site responsible for amphetamine-induced positive symptoms such as hallucination and delusion, particularly in animal studies. On the other hand, although there is no doubt that high doses of amphetamine also release excessive DA in the mPFC as well as in the striatum, much attention has not been paid to a hyperdopaminergic state in the mPFC. It is possible that the mPFC is involved in at least some of positive symptoms due to hyperdopaminergic states related to amphetamine and schizophrenia.
The dopamine hypothesis of schizophrenia proposes that a hyperdopaminergic state in the striatum leads to the positive symptoms of patients with schizophrenia. This is based on the finding that excessive DA release and elevated DA synthesis capacity, which are associated with a hyperdopaminergic state, occur in the striatum of schizophrenics [18, 22]. In patients with schizophrenia, excessive DA release was indirectly estimated by a reduction in dopamine D2 receptor binding after amphetamine administration by PET imaging with specific D2 receptor radioligands [40]. However, the results of this measurement can be affected by the density of D2 receptors on striatal neurons and the number of DA transporters on striatal DA axon terminals. If these factors are altered in schizophrenia, the amount of DA release could not be accurately assessed. In fact, the density of D2 receptors and DA transporters is reported to be altered in the striatum of schizophrenia, though the results are inconsistent [18, 40]. Furthermore, elevated DA synthesis capacity in the striatum of schizophrenia was also indirectly assessed by PET imaging measuring the uptake of radiolabeled DA precursors. In a most recent study, Jauhar et al. [48] examined the effects of antipsychotic medication on striatal DA synthesis capacity in patients experiencing their first episode of psychotic symptoms and its relationship to symptom change. Striatal DA synthesis capacity was not altered by antipsychotic treatment, and there was no significant association between change in DA synthesis capacity and change in positive and negative symptoms. Notably, despite significant improvements in positive and negative symptoms, there was no significant change in DA synthesis capacity in all the striatal subdivisions including the associative, limbic and sensorimotor striatum. These findings suggest that the striatum may not play a major role in the occurrence of positive and negative symptoms in patients with schizophrenia.
Further evidence has been presented that there is a hyperdopaminergic state in the striatum and a hypodopaminergic state in the mPFC of patients with schizophrenia. In a postmortem study by Sekiguchi et al. [49], the level of DA transporters was found to be significantly lower in the dorsal striatum and higher in the mPFC (Brodmann’s area 10) in patients with schizophrenia. Since DA transporters, which are located on the membrane of DA axon terminals, regulate intra-synaptic DA levels by transporting DA from synaptic clefts into presynaptic DA terminals, lower and higher levels of DA transporters were interpreted as resulting in a hyper- and hypodopaminergic state, respectively. However, there is another possible interpretation for a change in the level of DA transporters. Because the level of DA transporters is directly related to the density of DA axon terminals, elevated levels of DA transporters due to an increase in the density of DA axon terminals (hyperinnervation) lead to a hyperdopaminergic rather than hypodopaminergic state. Thus, higher levels of DA transporters in the mPFC likely cause a hyperdopaminergic state in the mPFC, possibly due to excessive cortical DA axons. It is noted that in Parkinson’s disease, there is loss of DA transporters and a rise in the density of D1 and D2 receptors in the striatum [50], which is explained by the degeneration of striatal DA axons and the denervation supersensitivity of postsynaptic DA receptors, respectively.
As described before, the axonal degeneration of VTA DA neurons projecting to the mPFC may induce the negative/cognitive symptoms of schizophrenia. However, the pathophysiology of positive symptoms is unknown. One possible explanation is that a hyperdopaminergic state leading to positive symptoms is due to the hyperinnervation of the mPFC by DA axons. It is known that monoamine axons including DA axons have a great capacity to spontaneously regenerate or sprout in response to damage in the adult brain [51, 52, 53, 54, 55, 56, 57, 58, 59]. More importantly, monoamine axons sometimes show hyperinnervation due to excessive regeneration/sprouting following lesions, typically by treatment with monoaminergic neurotoxins such as DSP-4, 5,7-DHT and MPTP [53, 60, 61, 62, 63, 64]. Thus, monoamine axons are considered to have the ability to induce hyper-regeneration/sprouting far beyond normal levels after axonal degeneration, raising the possibility that a hyperdopaminergic state in patients with schizophrenia is induced by excessive regeneration/sprouting of DA axons. In schizophrenia, prodromal symptoms before the first episode of psychosis (hallucination and delusion) mainly comprise negative symptoms, such as reduced concentration, social withdrawal, and deterioration in role functioning [65]. This suggests that excessive regeneration/sprouting of DA axons leading to positive symptoms occurs following axonal degeneration leading to negative prodromal symptoms.
The neurodevelopment hypothesis of schizophrenia proposes that a brain lesion during pre- and postnatal periods leads to the late appearance of clinical symptoms in adolescence or early adulthood without evidence of a neurodegenerative process [66, 67, 68]. An early study reported that neonatal lesions of 5-HT neurons by the injection of the neurotoxin 5,7-DHT into the dorsal raphe nucleus produced a hyperinnervation of DA axons in the mPFC of adult rats [63]. More recently, based on the finding that the expression of the neurotrophic factor neuregulin-1 (NRG1) is increased by neonatal hypoxia [69], Kato et al. [70] demonstrated that neonatal mice, transiently exposed to the NRG1 during postnatal days 2–10, showed hyperinnervation of the mPFC, but not the striatum, by DA axons in adults. The NRG1-treated mice exhibited behavioral impairments, such as lack of prepulse inhibition and latent inhibition, reduced social interactions, and hypersensitivity to amphetamine, most of which are included as positive symptoms occurring in hyperdopaminergic states in patients of schizophrenia [71]. These findings support the possible association between dopaminergic hyperinnervation of the mPFC and the positive symptoms of schizophrenia. NRG1 was identified as a schizophrenia susceptibility gene [72, 73], and in a postmortem study, NRG1 protein levels in the PFC were elevated in schizophrenic patients compared to normal, bipolar and major depressed subjects [74]. Thus, it is possible that perinatal brain insults, such as neonatal hypoxia, affect the development of DA axons projecting to the mPFC, and result in the expression of psychosis due to hyper-regeneration/sprouting of DA axons of the mPFC in early adulthood, possibly by inducing neurotrophic factors such as NRG1. Further studies are needed to confirm this possibility.
Bipolar disorder, formerly called manic depression, is a mental disorder that causes extreme shifts in mood, energy, concentration, and behavior. Bipolar depression is essentially the same as unipolar depression (major depression), and therefore, the depressive symptoms of bipolar disorder are thought to result from the degeneration of monoamine axons. On the other hand, mania that is characterized by symptoms including euphoric mood, psychomotor agitation, and increased energy is the opposite state of depression, suggesting the occurrence of manic symptoms in hyper-monoaminergic states. As described before, since monoamine axons including DA axons are found to have the capacity to regenerate beyond normal levels, it is proposed that hyperinnervation of the mPFC by DA axons may play a critical role in the positive symptoms of schizophrenia. Similar to DA axons, there may be hyper-noradrenergic and hyper-serotonergic states due to excessive regeneration/sprouting of NA and 5-HT axons, respectively, and hyper-monoaminergic states due to these monoamine axons are considered to contribute to some of neuropsychiatric and neurological symptoms. In fact, hyper-noradrenergic and hyper-serotonergic states, as well as hyperdopaminergic states, have been reported to be associated with manic symptoms, although most studies are concerned, in particular, with the association between DA and mania [75, 76, 77, 78, 79]. Notably, many symptoms overlap between mania and the positive symptoms of schizophrenia [80], supporting that some of manic symptoms occur in hyperdopaminergic states. It is also noted that mood switches from depression to mania can occur spontaneously or after antidepressant treatment for bipolar depression, as well as after administration of amphetamine and L-dopa [75, 77, 78, 79, 81]. Thus, considering the similarity to the positive symptoms of schizophrenia possibly due to hyper-regeneration/sprouting of DA axons, mania is likely to result from excessive regeneration/sprouting of NA and 5-HT axons as well as DA axons.
Since NA and 5-HT axons innervate almost the entire brain and are thought to play a pivotal role in the regulation of mood, hyperinnervation by these monoamine axons may contribute to symptoms such as elevated mood, increased energy, and reduced need for sleep. On the other hand, hyperinnervation of the nucleus accumbens by DA neurons of the VTA may play a role in euphoria, and hyperinnervation of the dorsal striatum by DA neurons of the SNc is likely associated with stereotyped behavior, which occurs in mania as well as schizophrenia. It is noteworthy that a rat model of depression, which was developed by repeated exposure to forced walking stress for two weeks, exhibited degeneration of NA axons in the cerebral cortex. However, a subset of animals, which revealed no depressive behaviors even after exposure to the same chronic stress, rather showed a marked increase in the density of cortical NA axons compared with control animals not exposed to the stress [8]. This study provides evidence that hyper-regeneration/sprouting of NA axons occurs in response to insults, such as repeated stress, although it remains to be known whether this group of animals revealed manic behaviors.
Lithium (Li) is one of the most commonly used medicines for treating bipolar disorder. Although the mechanism of therapeutic action of Li remains unclear, Li has been reported to exhibit inhibitory and facilitatory effects on growth and regeneration of central and peripheral axons, including 5-HT axons in adult rats [82, 83, 84, 85]. The dual effect of Li on axonal growth and regeneration is concentration-dependent, i.e., facilitatory at low concentrations and inhibitory at high concentrations, and the facilitatory effect of Li at low concentrations and the inhibitory effect at high concentrations are reported to occur by inducing partial and strong inactivation of glycogen synthase kinase-3, respectively [83, 84, 85]. These findings suggest that Li may treat bipolar disorder by exerting enhancing effects on regeneration of monoamine axons in a depressive phase and suppressive effects in a manic phase, leading to stabilizing moods.
The dissociative anesthetics and N-methyl-D-aspartate (NMDA) receptor antagonists, phencyclidine and ketamine, have been known to induce schizophrenia-like symptoms, including both positive and negative/cognitive symptoms, while the psychostimulants amphetamine and cocaine produce positive symptoms alone [33, 86, 87].
Although positive symptoms in schizophrenic patients and psychostimulant users are mediated by hyperactivation of dopamine D2 receptors, NMDA-related psychosis can be induced by blocking NMDA receptor activity without any association with DA transmission. Moreover, NMDA receptor inhibition by phencyclidine and ketamine leads to positive symptoms simultaneously with negative/cognitive symptoms, suggesting the involvement of NMDA hypofunction in positive symptoms as well as negative/cognitive symptoms through different neuronal pathways [33, 87]. Importantly, Zheng et al. [88] reported DA concentration-dependent modulation of NMDA receptor activity of pyramidal neurons of the mPFC using a slice preparation: DA at low concentrations promotes NMDA activity via D1 receptors and DA at high concentrations inhibits it via D2 receptors. Since neuroanatomical studies have shown that separate populations of pyramidal neurons of the mPFC reveal expression of either D1 or D2 receptors and few neurons express both [89, 90, 91], the D1 mediated activation and D2 mediated inhibition of the NMDA function are thought to occur independently in two distinct (D1- or D2-expressing) neuronal systems. Thus, hyperactivation of D2 receptors in D2-exprssing neurons in the PFC results in supernormal levels of NMDA hypofunction leading to positive symptoms, while negative/cognitive symptoms may be associated, at least in part, with reduced NMDA activity due to hypoactivation of D1-expressing neurons in the PFC. Therefore, the co-existence of positive symptoms and negative/cognitive symptoms seen in users of the NMDA antagonists ketamine and phencyclidine can be explained by their direct inhibitory effects on NMDA activity of separate (D1- or D2-exprssing) neuron populations without dopaminergic modulation (Fig. 1).
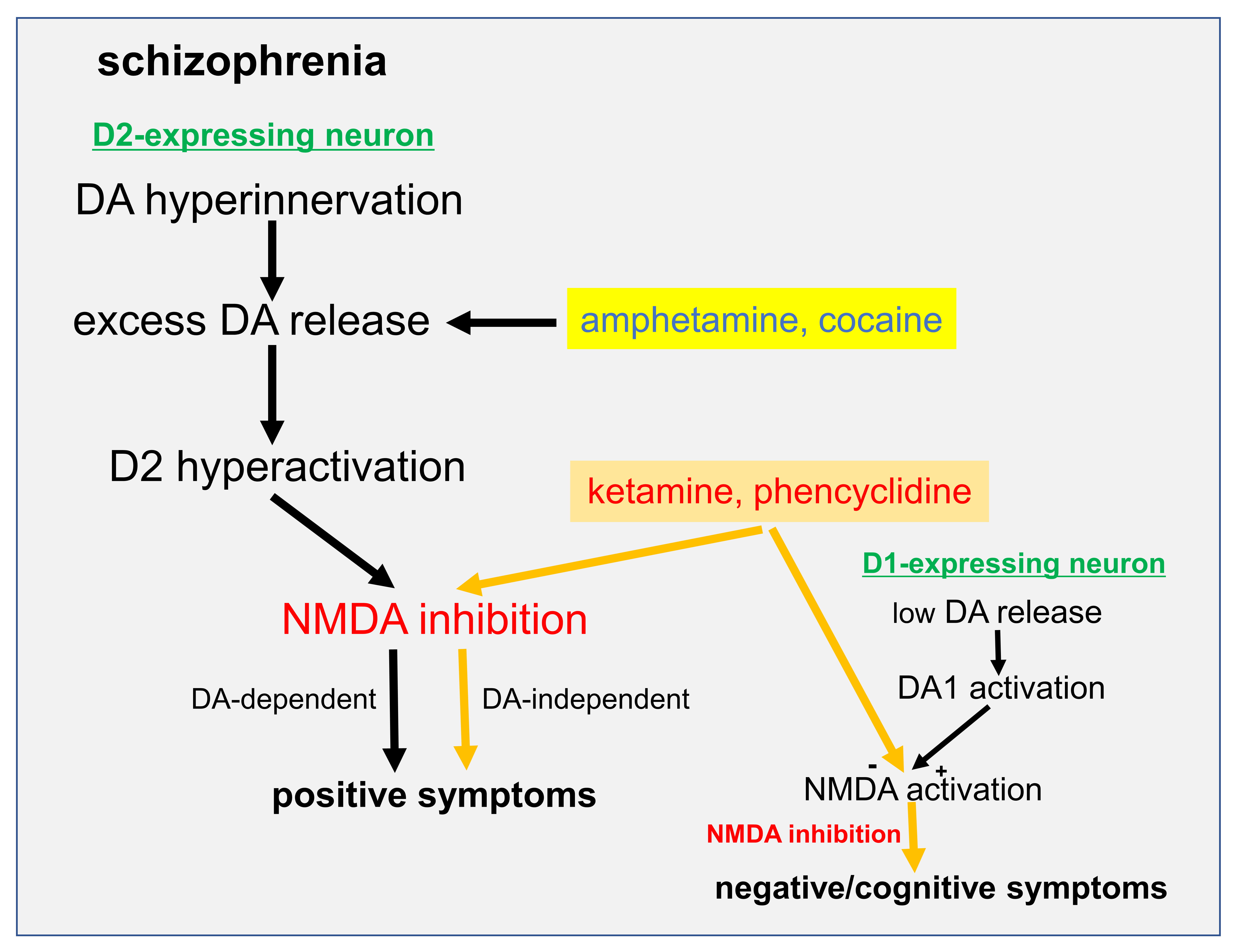 Fig. 1.
Fig. 1.A possible mechanism of the simultaneous occurrence of positive symptoms and negative/cognitive symptoms. DA at low concentrations promotes NMDA activity via D1 receptors and DA at high concentrations inhibits it via D2 receptors. The D1 mediated activation and D2 mediated inhibition of the NMDA function occur independently in two distinct (D1- or D2-expressing) neuronal systems. In schizophrenic patients and psychostimulant (e.g., amphetamine and cocaine) users, hyperactivation of D2 receptors due to excessive DA release in the PFC leads to NMDA hypofunction resulting in positive symptoms, while NMDA antagonists (e.g., phencyclidine and ketamine) lead to positive symptoms simultaneously with negative/cognitive symptoms by inducing NMDA hypofunction in separate (D1- or D2-exprssing) neuron populations in the PFC without any association with DA transmission.
The present review proposes that the major mental disorders, schizophrenia, major depression, and bipolar disorder, all are disorders of monoamine neurons characterized by the degeneration and/or excessive regeneration/sprouting of monoamine axons. Based on this view, the pathophysiology of depression, schizophrenia, and bipolar disorder is summarized in Fig. 2: The degeneration of monoamine axons is associated with the negative/cognitive symptoms of schizophrenia, major and bipolar depression as well as early-stage Parkinson’s disease, while hyper-regeneration/sprouting of monoamine axons plays a role in the positive symptoms of schizophrenia and bipolar mania.
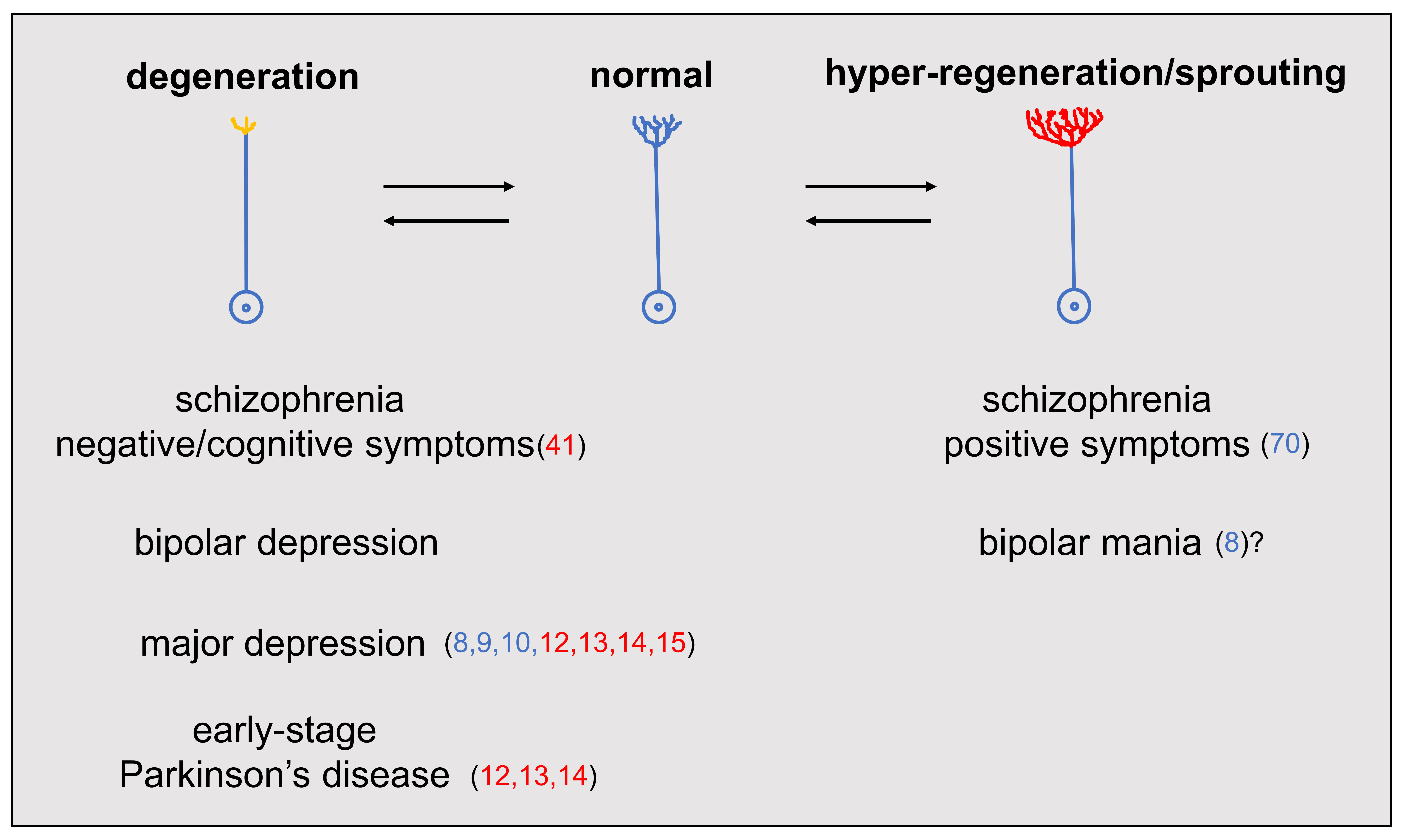 Fig. 2.
Fig. 2.Schizophrenia, major depression, and bipolar disorder as monoamine axon disorder. The degeneration of monoamine axons leads to the negative/cognitive symptoms of schizophrenia, major and bipolar depression as well as early-stage Parkinson’s disease, while the hyper-regeneration/sprouting of monoamine axons induces the positive symptoms of schizophrenia and bipolar mania. Supporting references are indicated by red (human study) and blue (animal model) numbers in the parentheses. Reference 8 (with a question mark) reported that while chronic stress produced a rat model of depression with degeneration of cortical NA axons, a subset of stressed animals showed excessive regeneration/sprouting of cortical NA axons. However, behavioral tests were not performed to assess manic behavior.
Moreover, as shown in Table 1, the degeneration of NA and 5-HT axons contributes to depressed mood, and the degeneration of VTA DA axons projecting to the ventral striatum (nucleus accumbens) is considered to induce anhedonia. The degeneration of VTA DA axons projecting to the mPFC plays a critical role in the negative/cognitive symptoms of schizophrenia. On the other hand, hyperinnervation of the mPFC by DA axons of VTA neurons induces the positive symptoms (delusion and hallucination) of schizophrenia, while hyperinnervation of the striatum by DA neurons of the VTA and SNc contributes to manic symptoms of bipolar disorder and some positive symptoms of schizophrenia. In addition, hyperinnervation of many brain regions, including the cerebral cortex, by NA neurons of the locus coeruleus and 5-HT neurons of the raphe nucleus is attributable mainly to elevated mood. Importantly, since patients with mental illness often have overlapping symptoms of different mental disorders (e.g., psychotic symptoms of schizophrenia and bipolar disorder), it is most likely that in these patients, more than two monoaminergic systems are simultaneously impaired. Notably, in MPTP-treated monkeys with Parkinsonian symptoms, a marked hyperinnervation of DA and 5HT axons occurs in the globus pallidus, despite the massive degeneration of DA axons in the striatum [64]. This finding indicates the possibility of simultaneous occurrence of hyper- and hypodopaminergic states as well as hyper-serotonergic states in the same brain, supporting the possible co-occurrence of symptoms due to hypo- and hyper-monoaminergic states. Furthermore, it is noteworthy that the monoaminergic state in one brain region can influence the monoaminergic state in another brain region. Pycock et al. [92] reported that 6-hydroxydopamine lesions of the mPFC enhanced DA turnover and utilization in the striatum, indicating that a hypodopaminergic state in the mPFC leads to a hyperdopaminergic state in the striatum.
| monoamine system | major symptom | |
| cell body/innervation | degeneration | hyper-regeneration/sprouting |
| 5-HT | depressed mood | elevated mood |
| raphe nucleus/cerebral cortex | ||
| NA | ||
| locus coeruleus/cerebral cortex | ||
| DA | ||
| VTA/ventral striatum | anhedonia | euphoria |
| VTA/mPFC | negative/cognitive symptoms | positive symptoms |
| SNc/dorsal striatum | impaired movement | stereotyped behavior |
| 5-HT, serotonin; NA, noradrenaline; DA, dopamine; VTA, ventral tegmental area; mPFC, medial prefrontal cortex; SNc, substantia nigra compacta. | ||
Recent studies of PET imaging with radiotracers of presynaptic monoamine transporters have provided direct evidence for the involvement of degeneration of monoamine axons in depressive symptoms [12, 13, 14]. Similarly, to ascertain the degeneration and excessive regeneration/sprouting of monoamine axon terminals in patients with schizophrenia and bipolar disorder, the density of monoamine axon terminals must be assessed by imaging techniques with radiolabeled monoamine transporters. However, there is no imaging study that shows degeneration and excessive regeneration/sprouting of monoamine axons in relation to the symptoms of schizophrenia and bipolar disorder. Future imaging (and anatomical) studies in patients with mental illness will be needed to identify impaired monoamine axons and involved brain regions, leading to further improvement of diagnosis and treatment of mental disorders.
Based on the view that depression, bipolar disorder, and schizophrenia are disorders caused by impairment of monoamine axons, the development of drugs for these mental diseases will be needed. Regarding depression that is thought to be caused by the degeneration of monoamine axons, NA- and 5-HT-specific antidepressant drugs are clinically effective possibly by their regenerative action on NA and 5-HT axons, respectively, but so far there is no report on drugs that show regenerative effects on DA axons. If such drugs are developed, they will contribute to treatment of hypodopaminergic disorders, including not only major and bipolar depression and the negative/cognitive symptoms of schizophrenia, but also early-stage Parkinson’s disease. On the other hand, although positive symptoms, such as delusion and hallucination, are treated by administration of D2 receptor blockers, there are no drugs to suppress excessive regeneration/sprouting of monoamine axons, possibly leading to the positive symptoms of schizophrenia as well as manic symptoms of bipolar disorder.
The mechanism of hyper-regeneration/sprouting of monoamine axons remains unknown. It is likely that neurotrophic factors, such as NRG1, play a role in hyper-monoaminergic states due to excessive regeneration/sprouting of monoamine axons. Furthermore, recent studies have demonstrated that the regeneration of NA axons induced by NA-specific antidepressants is mediated by activation of phospholipase A2 (PLA2) [7] and subsequently that calcium-independent PLA2 (iPLA2, a PLA2 family member) and its products (ecosapentaenoic acid and docosahexaenoic acid) are involved in the regeneration/sprouting of axons in the periphery and brain, including DA axons [93, 94, 95, 96, 97, 98]. It is thus worth to see whether the PLA2 (particularly iPLA2) signaling pathway plays a significant role in hyper-regeneration/sprouting of monoamine axons leading to hyper-monoaminergic states in patients with mania and schizophrenia. Finally, since molecular and genetic studies provide clear evidence that genes are involved in the etiology of schizophrenia and bipolar disorder [99], future studies of these mental disorders will need to pay more attention to genes associated with the regeneration/sprouting of monoamine axons.
SN is a single author responsible for all contents of the manuscript.
Not applicable.
I would like to deeply thank James M Tepper, Distinguished Professor of Rutgers University-Newark, for reviewing the manuscript and improving the English.
This research received no external funding.
The author declares no conflict of interest.