


Frontiers in Bioscience-Scholar (FBS) is published by IMR Press from Volume 13 Issue 1 (2021). Previous articles were published by another publisher on a subscription basis, and they are hosted by IMR Press on imrpress.com as a courtesy and upon agreement with Frontiers in Bioscience.

1 Department of Microbiology, Biochemistry and Immunology, Morehouse School of Medicine, Atlanta, GA, USA, 30310
Abstract
Breast cancer (BrCa) is the most commonly diagnosed cancer and the second leading cause of cancer-related death in women. Alarming increases in the cases quests for more effective treatment of BrCa. As most chemotherapeutic drugs are associated with drug resistance, cancer relapse, and side effects, scientists are turning to agents with more efficacy, such as natural compounds for treatment and prevention of BrCa. Selected natural compounds, substances derived from living organisms, promote apoptosis and inhibit metastasis, preventing cancer growth. As a result, these compounds have the potential to suppress BrCa progression, thus increasing patient survival rates and decreasing the number of BrCa-related deaths. In this review, we summarize natural compounds that have displayed, anti-cancer effects on BrCa cells in various studies. These natural compounds inhibit the development of BrCa, suppress the growth of cancer cells, and promote cell death. We conclude that natural compounds are efficient, effective and promising agents for treating BrCa other than therapeutic methods.
Keywords
- Natural compounds
- Breast Cancer
- Combination Therapy
- Review
Breast cancer (BrCa) is the most frequent type of cancer diagnosed in women worldwide (1). Approximately 1 in 8 women are diagnosed with BrCa sometime in their lives (2). In the United States, roughly 266,120 women are estimated to have been diagnosed with invasive BrCa in 2018 (3). In addition, it is the second most common cause of cancer-related death in females globally (4). Therefore, effective treatments for BrCa are needed to reduce the numbers of deaths from BrCa. Moreover, BrCa is more frequently diagnosed in certain races or ethnicities. African American women younger than age 40 are two times more likely to develop BrCa in comparison to white women of the same age. Another BrCa disparity is that African, American and Hispanic females are more at risk to be diagnosed with aggressive and advanced forms of BrCa (5). These BrCa disparities point to a need for more efficient treatments for BrCa.
Due to the high incidence of BrCa, effective agents are needed to treat this cancer (6). Surgery, chemotherapy, radiotherapy, and hormone therapy are the most frequently used methods of treatment for BrCa. Although BrCa is typically treated with chemotherapeutic drugs, the development of drug resistance, the occurrence of side effects, and reoccurrence of the disease indicate that these drugs have limited efficacy (4, 7). The well-studied drug efflux genes are MDR1, ABCG2, and BCRP (7). The side effects are unpredictable and depend upon the chemotherapeutics; some common effects include nausea and vomiting, neuropathy, constipation, diarrhea, and trouble breathing. Since clinicians need therapies that treat BrCa without extensive side effects and drug efflux, there is a need for replacing present chemotherapy drugs with natural compounds, which are organic chemical substances that are the products of living organisms (8) (Figure 1). Natural compounds are considered to be effective in the treatment of BrCa, as they affect several targets and have minimal or no side effects (Table 1). In addition, several studies have been identified in natural compound targeted therapies on subtypes of breast cancers (Basal-like, Luminal and HER-2) (9). In certain cases, natural compounds are safer, faster, cheaper, and less toxic in treatment of BrCa (10). Some of these compounds induce apoptosis and function in chemo-sensitization (11). The aim of this review is to evaluate natural compounds and show how they can be more effective than other therapeutic methods in treating BrCa.
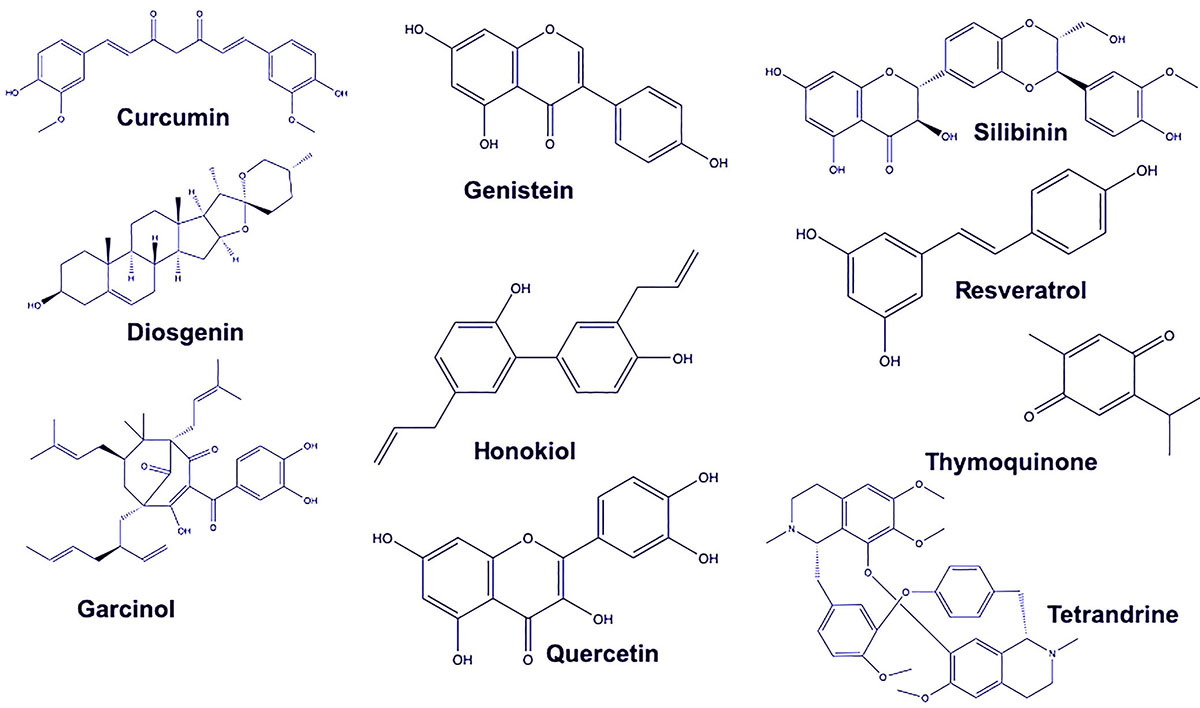 Figure 1
Figure 1Chemical structure of natural compound leads as therapeutics in treating breast cancer.
| Natural Compound | Source | Action | Reference |
|---|---|---|---|
| Curcumin | Curcuma longa | Promotes apoptosis; blocks angiogenesis and tumor metastasis; regulates the NF-κB signaling pathway; downregulates expression of human epidermal growth factor 2 and the phosphorylation of Akt; prevents the activation of NF-kB; downregulates vascular endothelial growth factor and intercellular adhesion molecule-1; promotes cell cycle arrest and beta-catenin nuclear translocation; and inhibits EMT markers. | (25, 26) |
| Diosgenin | Dioscorea villosa and Trigonellafoenum graecum | Suppresses cancer growth and progression; promotes apoptosis; inhibits actin polymerization, inhibits ER-α protein and mRNA expression; induces the intrinsic antioxidant defense system; affects CSCs via the Wnt β-catenin pathway (enhances β-catenin expression); reduces mTOR and Akt phosphorylation; induces JNK phosphorylation; suppresses the Raf/MEK/ERK pathway; induces cell cycle arrest; regulates DNA methylation; activates GATA3; downregulates MMP9. | (85-91) |
| Garcinol | Garcinia indica | Regulates the NF-κB signaling pathway; suppresses histone acetyltransferases and ROS; induces cell cycle arrest; reverses EMT markers; regulates β-catenin; and Wnt signaling pathway. | (102, 104) |
| Genistein | Fabaceae | Reduces tumorigenesis; induces cell differentiation; inactivates the epidermal growth factor signaling pathway, modulates gene transcription by regulating epigenetic activities; suppresses topoisomerase I and II and DNA polymerase; suppresses tyrosine kinases; regulates Hedgehog-Gli1 signaling; cell cycle arrest; and PI3K/Akt and MEK/ERK signaling pathways | (92, 93, 95, 96, 98) |
| Honokiol | Magnolia grandiflora | Inhibits angiogenesis; prevents tumor cell proliferation, induces apoptosis; regulates immunoresistance through PI3K/mTOR pathway; induces autophosphorylation; suppresses leptin-induced Wnt1-MTA1-β-catenin signaling; reduces phosphorylation of STAT3; inhibits PLD activity; suppresses mammosphere formation, ALDH activity, and expression of iPSC inducers; induces cell cycle arrest; inhibits EGFR; suppresses phosphorylation of c-Src. | (75, 76) |
| Quercetin | Allagopappus viscosissimus, Opuntia ficus-indica var. saboten, Lychnophora staavioides, and Rhamnus species | Promotes apoptosis, cell cycle; prevents invasive activity of BCSCs | (13, 15, 17, 109) |
| Resveratrol | Polygonum cuspidatum | Prevents tumorigenesis, DNA damage, and cancer metastasis; induces cell cycle arrest and apoptosis; modifies genetic and epigenetic profiles of cells; hinders COX activity; diminishes DNA binding activity of NF-κB; reduces cell viability, glucose consumption, and ATP content; downregulates expression of TGFβ1; reduces survival of BCSCs; induces autophagy by inhibiting the Wnt/β-catenin signaling pathway; diminishes PI3K/Akt/mTOR signaling. | (61-64, 68) |
| Silibinin | Silybum marianum | Induces apoptosis through extrinsic and intrinsic pathways; initiates autophagy; diminishes ROS production; lowers EGF-induced FN expression; inhibits STAT3 phosphorylation; inhibits the EGF/STAT3 signaling pathway; downregulates the expression of MMP-9; blocks MEK and ERK phosphorylation; downregulates the expression of TGF-β2, basal FN, and MMP-2. | (39, 40, 44, 46, 48) |
| Tetrandrine | Stephania tetrandra | Contributes to blockade of positive ion channels; overcomes multiple drug resistance; induces autophagy; promotes apoptosis. | (18, 19, 22) |
| Thymoquinone | Nigella Sativa | Promotes apoptosis by p53-dependent and p53-independent pathways; initiates p38 and ROS signaling; cell cycle arrest; hinders tumor growth by targeting NF-κB; enhances the PPAR-γ activation pathway/ PPAR-γ activity; diminishes Akt,4E-BP1, eIF4E, S6R and p70S6K phosphorylation. | (49, 51, 53, 54, 56, 58) |
Quercetin (QC), a flavonol from the flavonoid group of polyphenols, is produced by plants, including Allagopappus viscosissimus, Opuntia ficus-indica var. saboten, Lychnophora staavioides, and Rhamnus species (12, 13). The geographical distribution determines, it is found in more than twenty pants material (14). This compound present in various vegetables and fruits and in wine, tea, and coffee (15), has anticancer, antioxidant, antitumor, and anti-inflammatory properties (16). QC promotes apoptosis in a wide variety of cells, including those of prostate, lung, breast, colon, and cervical cancers (15). In addition, nanoparticles containing QC reduce the growth of cancer cells and cause them to undergo apoptosis, (12). To promote apoptosis of cancer cells, QC decreases the expression of anti-apoptotic proteins, such as survivin, Bcl-xL, and Bcl-2, and increases the expression of the pro-apoptotic proteins, such as Bad and Bax, (15). Since it targets cancer cells and causes them to undergo apoptosis, this compound is a potential therapeutic for treating various cancers (17).
QC is a promising agent for prevention and therapy of BrCa (12). For BrCa cells (BT-20 and MCF-7), QC promotes apoptosis by inducing the inactivation of c-FLIPL and upregulating DR5. In addition, QC arrests proliferation of BrCa cells (MCF-7) by inhibiting the cell cycle (15). Moreover, it inhibits the growth and invasive capacity of BrCa stem cells (MDA-MB-231) and downregulates various proteins, including aldehyde dehydrogenase 1A1, epithelial cell adhesion proteins, and others that are associated with the growth of BrCa cells. Therefore, by downregulating these proteins, the proliferation of BrCa cells is inhibited, showing that QC has anticancer properties (17). Thus, since QC promotes apoptosis of BrCa cells, it is likely to be effective in treating BrCa.
Tetrandrine, which demonstrates anti-proliferation and antitumor properties (18), is a dibenzyl tetrahydro isoquinoline alkaloid present in Stephania tetrandra, an Asian herb (Chinese plant) used for medicinal purposes (18, 19). This natural compound has proapoptotic effects on cancers, including leukemias, melanomas, and prostate and breast cancers (18). Tetrandrine has pharmacological properties in that it contributes to the blockade of positive ion channels and blocks various drug resistance proteins (19). Used to treat various cancers, tetrandrine affects the resistance of tumor cells (20), and it reverses drug resistance in human BrCa cells (21). Further, tetrandrine induces autophagy. As a result, cells that are resistant to apoptosis and thus are resistant to cell death in general, undergo autophagic cell death (22). Therefore, since tetrandrine decreases the growth of cancer cells, it is a promising agent for treating various cancers.
Tetrandrine displays a preventive effect against the growth of inflammatory and breast tumor-initiating cells by killing these cells. This compound reduces mammosphere formation, which is a surrogate for the proliferation of cancer cells, and it reduces the protein expression of aldehyde dehydrogenase. For SUM-149 and SUM-159 BrCa cells, tetrandrine exhibits anti-proliferative properties. In addition, by downregulating aldehyde dehydrogenase proteins, it demonstrates anti-proliferation characteristics because these proteins are related to the growth of BrCa cells (19). For BrCa MCF-7/TAM cells, tetrandrine reverses drug resistance of tamoxifen. As an activator of autophagy, tetrandrine has pro-autophagic effects on numerous BrCa cell lines, and it promotes cell death in cells that are resistant to apoptosis. Due to autophagy, apoptosis-resistant cell lines that have low expression of caspase 3, caspase 7, and Bax, Bak undergo cell death when treated with tetrandrine. Thus, tetrandrine is useful in activating cell death amongst cancer cells (22), and it shows properties that make it a promising treatment for the prevention and treatment of BrCa (21).
Curcumin (CUR),a component of the spice plant Curcuma longa turmeric, probably originated from South and South-East Asia, is commonly used for medicinal purposes and is effective in the treatment of a variety of cancers (23-25). It has anti-inflammatory, anti-tumor, anti-microbial, anti-oxidative, and anticancer properties, and it has anti-carcinogenic effects on squamous cell carcinomas and on lung, breast, pancreatic, brain, head and neck, and colorectal cancers (24-27). In addition, CUR displays anti-proliferative effects on cancer cells, including those of melanoma and mantle cell lymphoma and hepatic, ovarian, and prostatic carcinomas (28, 29). CUR regulates the expressions of several proteins, including inflammatory cytokines and enzymes, transcription factors, and gene products associated with cell survival and growth (26). This polyphenol derivative also promotes apoptosis and hinders angiogenesis and tumor metastasis (30). By disrupting a variety of signaling pathways, CUR inhibits the survival, growth, and invasive migration of cancer cells (31). Furthermore, unlike most chemotherapeutic drugs, CUR has minimal side effects (24). Therefore, CUR displays characteristics of a promising agent for treatment of various cancers.
For BrCa cells, CUR has a wide range of effects. CUR, either alone or in combination with other natural compounds or chemotherapeutics, hinders tumorigenesis and cancer cell growth. For instance, CUR reduces proliferation of human BrCa cells by preventing the activation of nuclear factor kappaB (32), which is associated with cancer cell survival, cell growth, and metastasis. Furthermore, in BT-474 and SK-BR-3 cells, CUR downregulates MAPK, and NF-κB(32).
CUR reduces paclitaxel-induced NF-κB by inhibiting the activation of IκBα kinase and through IκBα phosphorylation and degradation. This compound also downregulates the expression of anti-apoptotic proteins, such as BCL-xL and BCL-2; proliferative proteins, such as cyclin-D1 and c-Myc; and metastatic proteins, such as vascular endothelial growth factor and intercellular adhesion molecule-1 (33). It decreases the number of viable cells in the BrCa cell line, MDA-MB-231. CUR inhibits growth of tumors, showing that it has therapeutic potential for treatment of BrCa (25).
For MCF-7 BrCa cells, CUR reduces the proliferative effects of bisphenol A, which is associated with the development of BrCa (34). It also reverses the upregulation of oncogenic miR-19a and miR-19b and the downregulation of miR-19-related downstream proteins (PTEN, p-AKT, p-MDM2, p53) and proliferating cell nuclear antigen, which is elevated by bisphenol A. CUR modulates the miR-19/PTEN/AKT/p53 axis to reduce the growth of bisphenol A-promoted MCF-7 cells (34). Furthermore, in BrCa cells, the compound promotes cell cycle arrest (24). For instance, CUR decreases the expression of CD44 and CD24 in MCF-10F cells, and CD24 in MCF-7 cells. In addition, CUR downregulates the expression of CD44 in MDA-MB-231 cells. Thus, by enhancing the amount of CD44+/CD24+ cells and reducing the amount of CD44+/CD24-cells, CUR decreases the growth of BrCa cells (35).
CUR reduces expression of the inflammatory cytokines, CXCL1 and -2, which results in a decrease in BrCa metastasis. Moreover, in BrCa metastasis is regulated by the expression of a variety of miRNAs, including miR181b, which diminishes the expression of CXCL1 and -2 by binding to their 3’-UTRs.This is relevant to its anticancer activity against BrCa cells (36).
CUR enhances the expression of Nrf2, a regulator of antioxidant defense systems in BrCa cells and decreases expression of the Flap endonuclease 1 (Fen1) protein, a DNA-repair nuclease (37). Additionally, CUR causes Nrf2 translocation from the cytoplasm to the nucleus and suppresses Fen1-induced activity through reduced recruitment of Nrf2 to the Fen1 promoter. CUR reduces the proliferation of BrCa cells, providing a new strategy for the inhibition of tumor growth (37). In the BrCa cell lines, MDA-MB-231 and MCF-7, CUR downregulates the expression of Bcl-2, an anti-apoptotic protein, and upregulates the expression of Bax, a pro-apoptotic protein (29).
CUR targets cancer stem cells, in which it diminishes the formation of the E-cadherin/beta-catenin complex (38) and enhances expression of genes associated with the epithelial-mesenchymal transition (EMT), such as Slug. By downregulation of E-cadherin and promotion of the EMT, the migration of the BrCa stem cells is enhanced. However, CUR reduces nuclear translocation of beta-catenin. Therefore, CUR activates EMT-promoting target genes, and upregulates the expression of E-cadherin. By enhancing formation of the E-cadherin/beta-catenin complex and retention of beta-catenin, the EMT is inhibited, as is the migration of the BrCa stem cells. In sum, CUR suppresses the migration and growth of BrCa cells (38).
Silibinin (INN), from the plant Silybum marianum, a milk thistle, possesses anti-carcinogenic and anti-proliferative properties (39). In Europe and Asia, INN is a commonly used medicine with hepatoprotective effects (40). This compound has inhibitory effects on various cancers, including those of the breast, colon, prostate, skin, brain, and lung (41). The hepatoprotective, anti-inflammatory, anti-fibrotic, and anti-tumor effects of INN show that it has potential as a therapeutic agent in the treatment of various cancers (42).
INN is effective in treating BrCa. Against MCF-7 BrCa cells, it induces apoptosis and displays anti-proliferative properties. In these cells, INN activates the conversion of light chain 3 (LC3)-I to LC3-II, upregulates Atg12-Atg5 formation, raises beclin-1 expression, and diminish es Bcl-2 expression (39). These effects show that, for MCF-7 BrCa cells, INN initiates autophagy, and demonstrate that it promotes autophagic cell death. INN promotes cell death by reducing the expression of Bcl-2 adenovirus E1B 19-kDa-interacting protein 3 (BNIP3), increasing ROS production, and regulating ΔΨm and ATP levels (39).
Fibronectin (FN) is associated with cell adhesion, migration, and oncogenic transformation, and its expression correlates with a poor prognosis for various types of cancer, including BrCa (40). A study focused on the effect of INN on the expression of the epidermal growth factor (EGF)-induced FN in triple negative BrCa (TNBC) cells found that a STAT3 inhibitor reduced the expression of EGF-induced FN (40). In MDA-MB-468 and BT20 BrCa cells, EGF increases the expression of FN mRNA. As a result, EGF-induced FN expression is diminished by AG1478 and gefitinib, which are EGFR inhibitors. In addition, MEK1/2, PI3K, and STAT3 inhibitors downregulate EGF-induced FN expression. INN diminishes the EGF-induced FN expression, which inhibits STAT3 phosphorylation. Thus, INN inhibits the EGF/STAT3 signaling pathway, which leads to the inhibition of FN expression in TNBC cells. Consequently, INN has anti-proliferative and anti-migratory effects on BrCa cells, which show that it is a promising agent for treatment of TNBCs (40). Moreover, for MDA-MB-231 cells, INN suppresses EGFR phosphorylation; reduces the expressions of COX-2, VEGF, and MMP-9; and reduces tumor sizes in mice, showing that it has anti-tumor and anti-cancer activities (43).
Matrix metalloproteinases (MMPs) contribute to cell migration, cell invasion, and cancer metastasis. INN downregulates the expression of MMP-9 and reduces 12-O-tetradecanoylphorbol-13-acetate-induced cell migration. In addition, in MCF-7 BrCa cells, INN lowers MEK and ERK phosphorylation (44). Moreover, the expressions of miR-21 and miR-155 are low in INN-treated T47D BrCa cells, and the expressions of potential targets, CASP-9 and APAF-1, are upregulated. In cells, INN downregulates miR-21 and miR-155, which are frequently over-expressed in cancers (45). INN has a similar effect on MCF-7 cells by promoting apoptosis and by decreasing the expression miR-21 and miR-155 while increasing the expression of potential targets. Additionally, the increase in expression of CASP-9 and BID in MCF-7 cells reveals that INN promotes apoptosis through the extrinsic and intrinsic pathways (46).
INN, acting as a CXCR4 antagonist, affects CXCR4 signaling. It reduces chemokine ligand 12 (CXCL12)-induced CXCR4 internalization; as a consequence, downstream intracellular signaling is suppressed. By suppressing CXCL12-induced migration in MDA-MB-231 cells, INN demonstrates its capacity to regulate cancer proliferation and metastasis (47). High levels of TGF-β2 correlate with a poor prognosis. In TNBC cells, INN downregulates the expression of TGF-β2, basal FN, and MMP-2 (48). Thus, INN is a promising agent to prevent BrCa metastasis and a promising treatment for BrCa.
Thymoquinone (TQ) is present in the seeds of Nigella sativa, which is cultivated in the Mediterranean region and in Western Asian countries (49, 50). This compound has activity against varying types of cancers, including myeloblastic leukemia, osteosarcoma, and pancreatic adenocarcinoma and breast, liver, ovarian, larynx, prostate, and colorectal cancers(49-53). The antitumor activity of TQ involves several targets, including p53, p73, STAT3, NF-κB, PPAR-γ, and ROS (53). In MCF7, HCT-116, and HL-60 cancer cells, TQ increases the ratio between Bax and BCL-2, raising amounts of the pro-apoptotic protein and diminishing amounts of the anti-apoptotic protein and demonstrating its anti-proliferative properties (51). Thus, TQ is a promising compound for treatment of cancers.
TQ has anticancer effects on BrCa cells. It exhibits anti-migratory and pro-apoptotic properties against BrCa cells by elevating phosphorylation of p38 and ROS signaling. In addition, TQ suppresses the expression of anti-apoptotic proteins, including survivin, Bcl-xL, and Bcl-2, displaying its anti-proliferation activity (54). The downregulation of anti-apoptotic proteins, promotion of p38 phosphorylation, and reduction of the size of breast tumors indicates that TQ is an effective treatment for BrCa (53). In addition, TQ inhibits molecules associated with the S phase and, in cells, induces sub-G1 arrest, which relates to its capacity to inhibit cell proliferation. TQ promotes apoptosis by p53-dependent and p53-independent pathways through modulation of various targets. TQ hinders cell proliferation and tumor growth by targeting NF-κB and affecting the cell cycle. With long-term treatment, even low concentrations suppress BrCa cells (54).
TQ causes cleavage of poly (ADP-ribose) polymerase, enhancement of γH2AX, a decline in phosphorylation of Akt, and downregulation of the expression of X-linked inhibitor of apoptosis (55). In addition, it serves as a ligand of PPAR-γ and inhibits proliferation of BrCa MCF-7/DOX cells (56). Consequently, TQ enhances PTEN protein expression, and reduces Akt phosphorylation. Since Akt phosphorylation maintains cell survival, by decreasing it, TQ inhibits cell growth. Further, TQ blocks MCF-7/DOX cells at the G2/M phase (57, 58). TQ also reduces the expression of cyclin D1 and cyclin E and reduces Akt by diminishing the phosphorylation of 4E-BP1, eIF4E, S6R and p70S6K (58). Thus, TQ is an effective compound for the treatment of BrCa, as it induces apoptosis among BrCa cells.
Resveratrol (trans-3,5,4′-trihydroxystilbene, RES), a polyphenolic compound with anti-carcinogenic activity, is present in plant foods and dietary sources, including grapes, peanuts, soybeans, pomegranates, and berries (59, 60). A plant that contains considerable amounts of resveratrol is Polygonum cuspidatum, or Japanese knotweed, which has beneficial effects against inflammation. For hundreds of years in Asian countries, this plant has been used to prevent and treat several diseases, including cancer. RES induces cell cycle arrest and causes apoptosis of tumor cells. RES also downregulates the expression of tumor-derived nitric oxide synthase, functions as an antioxidant, and prevents DNA damage; it also reduces tumor growth (61). This compound modifies genetic and epigenetic profiles of cells within tumors, demonstrating its antitumor properties (62). In addition, it diminishes the DNA-binding activity of NF-κB. Since this factor promotes the transcription of genes that induce tumor cell proliferation, RES prevents tumor growth by diminishing its binding activity (61). RES reduces intracellular ROS, mitochondrial membrane potential, and phosphorylation of mTOR, RP-S6, and 4EBP1. It prevents inflammation and leukemia and inhibits viruses. RES has neuroprotective and apoptotic-inducing properties (63). RES causes apoptosis through several pathways, such as by targeting p53, Rb, and cell cycle kinases (64). RES hinders cancer cell proliferation and promotes apoptosis of various cancer cells (65). It has antitumor effects on colorectal, liver, pancreatic, prostate, and breast cancers (61, 66). Because RES displays multi-target effectiveness, medical safety, convenience, and cost efficacy, it provides an effective method of treatment for various cancers (67).
RES has anticancer effects on BrCa cells. Against triple-negative BrCa cell lines, MDA-MB-231 and MDA-MB-231/PacR, RES prevents cell growth, promotes senescence, downregulates the expression of survivin, and initiates apoptosis. In promoting apoptosis, it activates caspase 7 (65). Moreover, RES reduces cell viability, glucose consumption, and the ATP content in MCF-7 cells; it also suppresses PFK. In this manner, RES reduces the survival and proliferation of these cells (68).
RES inhibits tumor growth in various animal models. For instance, it reduces the incidence of tumor formation in female rats. In rats, RES decreases the expression of COX2 and the binding of NF-κB to DNA. It reduces the expression of single-strand DNA; decreases DNA damage; and downregulates the expression of 5-LOX, TGFβ1, and NF-κB. Additionally, RES decreases BrCa tumor growth and metastasis(61).
In BrCa cells, RES modulates apoptotic and cell cycle machinery by regulating tumor-suppressive miRNAs, including miR-125b-5p, miR-200c-3p, miR-409-3p, miR-122-5p, and miR-542-3p. miR-542-3p is involved in inhibition of apoptosis in MCF-7 cells and miR-122-5p in MDA-MB-231 cells. By modulating miRNAs, RES demonstrates its anticancer and anti-proliferative properties against BrCa (69). Furthermore, in MCF-7 cells, RES enhances the expression of ASPP1, a protein activator of p53 that stimulates apoptosis. RES also upregulates BAX and p21 (70). Its modulation of Bcl-2 inhibits cancer progression. RES increases p53 expression, reduces procaspase 8, and activates caspases 7 and 9. In addition, RES induces cell cycle arrest in the S phase and raises p-Chk2 levels. RES reduces the active form of CDK2 and blocks CDK7 activity (71). It promotes p53-dependent apoptosis through plasma membrane integrin αvβ3, demonstrating its anti-proliferative activity(72). In HER2-positive BrCa cells, RES modifies cell cycle progression and promotes apoptosis by blocking FASN (73). Therefore, RES is a promising agent for treating BrCa.
Honokiol (HNK), a natural compound derived from the plant, Magnolia grandiflora, native to the Southeastern United States and many other regions of the globe (74), demonstrates antimicrobial, anti-oxidative, and anti-inflammatory properties (75, 76). It also inhibits angiogenesis, which is associated with tumor metastasis (75) and suppresses vascular endothelium growth, leading to anti-tumor effects (77). It reduces proliferation of tumor cells in culture and inhibits growth of tumor xenografts in mice. For B-cell chronic lymphocytic leukemia cells, HNK promotes caspase-dependent apoptosis (75). This is accomplished through p53-independent pathways (76). Similarly, HNK suppresses bone metastasis of prostate cancer cells (75). Furthermore, in glioma, breast, and prostate cancer cells, it diminishes PI3K/mTOR pathway-mediated immunoresistance (78). Moreover, HNK induces differentiation of human HL-60 cells. The compound reduces VEGF-induced KDR autophosphorylation in HUVECs and angiosarcoma proliferation in mice. Additionally, it suppresses growth of RKO colon cancer cells and, in mice, growth of RKO solid tumors. HNK extends the lifespan of the mice with solid tumors (76). Therefore, HNK is a promising as a cancer therapeutic agent.
HNK shows potential as an agent for treating BrCa. The compound suppresses the growth of the BrCa cells and enhances the efficacy of other drugs against these cells. In mice, HNK causes cell cycle arrest of BrCa cells (79). For MDA-MB-231 cells, it promotes the activation of caspase 3 and induces pro-apoptotic traits. In HNK-treated mice, tumor cell proliferation is suppressed (80). In addition, HNK suppresses leptin-induced Wnt1-MTA1-β-catenin signaling. It reduces the phosphorylation of STAT3, and, because of the release of repressor-STAT3, activates miRNAs (81).
The survival of cancer cells is greater when phospholipase D (PLD) activity is enhanced. Thus, an agent that suppresses PLD activity is likely to be effective in inhibiting proliferation of BrCa cells. HNK inhibits PLD activity, hence preventing the proliferation of cancer cells. In MDA-MB-231 cells, enhancement in PLD activity correlates with Ras activation; HNK inhibits PLD activity as well as Ras activation. By suppressing Ras and PLD activity, which promotes cell survival, HNK shows potential as a therapeutic agent for BrCa (75).
Furthermore, HNK demonstrates anticancer effects on BrCa cells through suppression of mammosphere formation, ALDH activity, and expression of iPSC inducers. By downregulating iPSC inducers through STAT3 inactivation, HNK promotes LKB1 and suppresses the stem-like phenotype of BrCa cells (82). Additionally, HNK suppresses the growth of MDA-MB-231 BrCa cells by inducing G0/G1 phase cell cycle arrest. This is associated with the increase of a CDK inhibitor, p27Kip-1, and with increases of CDK4, cyclin D1, CDK2, cyclin A, and cyclin E. Further, HNK promotes the cleavage of PARP and DNA fragmentation by activating a caspase cascade. Hence, apoptosis of these BrCa cells is enhanced. Moreover, HNK shows an antigrowth effect by regulating cell signal transduction pathways. This compound inhibits EGFR, the receptor tyrosine kinase ErbB, and c-Src, which stimulates EGFR through phosphorylation of the receptor at itsTyr845 position. The phosphorylation of c-Src is also suppressed by HNK. For MDA-MB-231 cells, inhibition of factors that contribute to BrCa cell growth and angiogenesis relate to the anti-proliferative traits of HNK. In addition to diminishing the expression of c-Src, HNK reduces the expression of Akt, which promotes cell survival and suppresses apoptosis. The inhibition of Akt and c-Src is regulated by modulation of Hsp90; HNK also inhibits Hsp90. Thus, by modulating c-Src/EGFR-mediated signaling and inhibiting the expression of c-Src and Akt, HNK is a treatment option for BrCa (83).
Diosgenin (DG) is a steroid saponin derived from the plants Dioscorea villosa and Trigonellafoenum graecum, mianly occur in China, India, Thailand, and South-East Asian countries, Mediterranean region, and Northern Africa (84). This compound is recognized for its contribution to synthetic steroidal drugs produced in the pharmaceutical field. DG, a constituent of traditional medicines, has anti-hypercholesterolemia, anti-hyperglycemia, antifungal, antiviral, and anti-diabetes properties (85, 86). For various cancer cells, this compound suppresses growth and progression, and promotes apoptosis. The cancers beneficially affected by DG include osteosarcoma, colon carcinoma, leukemia, hepatoma, and BrCa. DG inhibits cancer growth by regulating various cell-signaling events related to cancer proliferation, differentiation, apoptosis, and growth (85). There are no reports indicating a toxic effect on non-cancerous cells (87). Thus, DG, which acts on numerous targets in various types of cancer, is a promising agent for treating cancer (85).
For BrCa MDA-MB-231 cells, DG suppresses migration and reduces actin polymerization, Vav2 phosphorylation, and Cdc42 activation, which are associated with cancer cell migration and invasion and with BrCa progression. Suppression of these factors may relate to the capacity of DG to inhibit cancer metastasis (85). Furthermore, in MDA-MB-231, MDA-MB-453, and T47D BrCa cells, DG downregulates the expressions of the anti-apoptotic proteins, Bcl-2 and cIAP-1 (86). It decreases the expression of myeloid cell leukemia-1 (Mcl-1), which is associated with cell survival. Moreover, DG promotes apoptosis by causing apoptosis inducing factor (AIF) to be released from mitochondria and translocated to the nucleus. Thus, the AIF-facilitating, caspase-independent pathway regulates DG-induced apoptosis. By decreasing the expression of inhibitors of apoptosis, such as Bcl-2 and clap-1, and inducers of cell survival, DG demonstrates its anti-proliferative effects on various BrCa cell lines (86).
For ER-positive MCF-7 BrCa cells, DG suppresses growth and promotes apoptosis. For these cells, DG downregulates the expression of procaspase-3, procaspase-8, and survivin and upregulates the expression of Fas ligand and cleaved PARP1, demonstrating that DG-promoted apoptosis is regulated by the extrinsic pathway (88). In addition, DG suppresses ER binding to the estrogen response element. In MCF-7 cells exposed to DG, C-Myc and cyclin D1, which are ERα-mediated genes, are downregulated. Additionally, DG also downregulates activation of p38 and ERK1/2. N-methyl-N-nitrosourea (NMU), a mammary carcinogen, promotes the development, in female Sprague Dawley rats, of BrCas that resemble those in humans. For these rats, DG reduces the occurrence of cancers by downregulating the peroxidation reaction and marker enzymes and by inducing the intrinsic antioxidant defense system. Therefore, DG demonstrates anticancer effects on NMU-induced BrCas through the reduction of lipid peroxidation by inducing the antioxidant defense system (89). Additionally, in MCF7, T47D, and MDA-MB-231cells, DG reduces the numbers of cancer stem cells (CSCs) by acting on the Wnt β-catenin pathway. DG enhances β-catenin expression and reduces GSK3β expression. In addition, it reduces the expression of epithelial markers of CSC and thereby suppresses the growth of BrCas (87).
In HER2-overexpressing BrCa cells, DG inhibits the expression of fatty acid synthase. It also reduces cell growth, promotes apoptosis, and reduces mTOR and Akt phosphorylation. In ER+ and ER- cells, DG suppresses pAkt expression and Akt kinase. This suppression occurs without effects on PI3 kinase levels, which lead to the suppression of its downstream targets. Targets that are downregulated include kappaB, Bcl-2, survivin, and XIAP. In ER+ cells, DG also suppresses the Raf/MEK/ERK pathway, another downstream target of Akt. Moreover, in ER+ and ER- BrCa cells, there is G1 cell cycle arrest as a result of DG decreasing the expression of cyclin D1, cdk-2, and cdk-4. In nude mice, it reduces cancer cell growth and promotes apoptosis. Thus, DG is a promising therapeutic for BrCa as it demonstrates antitumor effects on MCF-7 and MDA-MB-231 cells by suppressing their proliferation (90).
In MDA-MB-231 and MCF-7 cells, DG activates GATA3 by targeting the epigenome. Since low GATA3 expression correlates with a poor prognosis for BrCa patients, DG can be beneficial, as it induces the expression of GATA3. In addition, DG regulates DNA methylation and decreases cancer cell survival of both ER+ and ER- cells (91). These cells, exposed to DG, have morphological traits of epithelial cells. For instance, mRNA expression of DNMT3A, TET2, TET3, ZFPM2, and E-cad are upregulated; and TET1, VIM, and MMP9 are downregulated. In addition, DG enhances the expression of TET2, TET3, ZFPM2, and DNMT3A transcripts. TET2 contributes to DNA demethylation, and downregulation of TET2 correlates with enhancement of cell growth. Thus, by increasing the expression of this enzyme, DG displays its anti-proliferative and anti-growth characteristics. Additionally, DG decreases MMP9, and, in MCF-7 and MDA-MB-231 cells, VIM mRNA levels are downregulated. The modulation of TET and GATA3 by DG is associated with the suppression of cell migration and invasion (91). Thus, DG is promising as an agent for treating BrCa.
Genistein, an isoflavone phytoestrogen present in Leguminosae (Fabaceae), possesses anti-tumor effects for various cancers (92). It is present notably in soybeans, and native of South-East Asia (93, 94). The compound overcomes cancer drug resistance and suppresses the recurrence of cancers (92). It also reduces tumorigenesis of cancers that require estrogen (93). Administered to rats, genistein prevents tumors, cardiovascular disease, and osteoporosis, and it is a preventive agent for chemically induced mammary tumors. Genistein induces cell differentiation and inactivation of the epidermal growth factor signaling pathway (93). In addition, it has anti-oxidation, anti-proliferation, anti-cancer activities; it also promotes apoptosis and suppresses angiogenesis and metastasis (95, 96). Genistein is thought to modulate gene transcription by regulating epigenetic activities (95). Furthermore, it suppresses topoisomerase I and II and DNA polymerase II, and it downregulates genes encoding cyclins, such as B1, D1, CDK-1, and Wee1. Genistein inhibits expression of Bcl-2, IAP, XIAP, and survivin, which are inhibitors of apoptosis. Moreover, for cancer cells, it increases expression of p53, p21, p27, and p16. Tyrosine kinases are factors involved in signaling pathways regulating cell growth and viability. Genistein suppresses tyrosine kinases, thus inhibiting cell survival and cancer progression. Additionally, it inhibits angiogenesis by modulating genes encoding VEGF, PTK, and MAPK, and it reduces proteolysis of cancer-related tissue (96). Thus, it a potential therapeutic agent for treating various types of cancers.
Genistein is an effective preventive and therapeutic agent for BrCas. For MCF-7 and MDA-MB-231 BrCa cells, it downregulates global DNA methylation levels, DNA methyltransferase (DNMT) activity, and DNMT1 expression levels (97). Genistein interacts with the catalytic domain of DNMT1 and thereby suppresses the binding of hemi-methylated DNA to the catalytic domain of DNMT1. In addition, genistein reduces DNA methylation in the promoter area of various tumor suppressor genes (TSGs), including ataxia telangiectasia mutated (ATM), adenomatous polyposis coli (APC), phosphatase and tensin homolog (PTEN), and mammary serpin peptidase inhibitor (SERPINB5). Genistein upregulates the mRNA expressions of these TSGs. It demethylates methylation-silenced TSGs by interacting with the catalytic domain of DNMT1 and reducing the expression of DNMT1(97). For MCF-7 cells, genistein decreases their survival and growth and induces apoptosis. It also reduces breast cancer stem cells (BCSCs) and BrCa stem-like cells by downregulating the Hedgehog-Gli1 signaling pathway. Blocking of this pathway reduces CSC survival by lowering of the proteins, SMO and/or Gli1. Exposure of BrCa cells to genistein downregulates SMO and Gli1 expression. ALDH1 is a marker for BCSCs. For mice, dosing with genistein decreases ALDH protein and mRNA levels, making it a promising therapeutic agent for BrCa. Downregulation of the Hedgehog-Gli1 signaling pathway and ALDH1 is associated with a decrease of the stemness of BCSCs. Thus, by decreasing the expression of these BCSCs, which are involved in drug resistance, cancer relapse, and metastasis, genistein prevents a primary cause of cancer, making it an effective preventive agent for BrCa (92).
For MCF-7 BrCa cells, high concentrations of genistein promote changes in the expression of differentially expressed genes (DEGs) in the cell cycle. The key function of the DEGs is in the cell cycle, as 47 of these genes are involved in the cell cycle pathway. These DEGs include CDC20, BUB1, MCM2, and cyclin B1. Exposure to genistein results in cell cycle arrest, which happens at various phases in the cell cycle, including the G2/M, G0/G1, and G1/S phases (93). Genistein suppresses BrCa cell growth and prevents the development of cancers, suggesting that it is an effective treatment for BrCa. In addition, genistein restores ERα-dependent cellular responses to the activator, 17β-estradiol (E2). Thus, by targeting ERα reactivation, it is a promising therapeutic for BrCa (95). For T47D cells, genistein causes an increase in ERβ and enhances cytochrome c oxidase. As a result, the ATP synthase/cytochrome c oxidase ratio is lowered. Thus, genistein causes cell cycle arrest and improves the mitochondrial functionality of T47D BrCa cells (98).
Genistein suppresses BrCa cell growth and stimulates apoptosis by promoting the inactivation of IGF-1R and p-Akt. Moreover, genistein decreases the Bcl-2/Bax ratio, suggesting that it can prevent BrCa progression (99). Furthermore, genistein induces morphological alterations of mammospheres that correlate with PI3K/Akt and MEK/ERK signaling pathways. The release of amphiregulin from ER+ BrCa cells activates these pathways. For mammospheres, genistein decreases the ratio of a subset of CD44+/CD24-/ESA+ cells and enhances the expression of differentiated cell markers. It promotes differentiation of BCSCs by interacting with ER+ cancer cells (99). In addition, for T47D cells, genistein decreases the expression of MMP-2, MMP-3, MMP-13, MMP-15, TIMP-1, TIMP-2, and TIMP-3. Thus, it prevents BrCa angiogenesis and metastasis. In clinical practice, this compound has the potential to increase survival rates of patients with BrCa (96).
Garcinol, a polyisoprenylated benzophenone extracted from the plant Garcinia, popularly valued in the Indian subcontinent, Africa, and China (100), possesses anti-oxidative, anti-bacterial, anti-fungal, anti-inflammatory, anti-glycative, and anticancer characteristics (101, 102). This acetyltransferase inhibitor is present in plants that are in tropical areas (103). Traditionally used for its antioxidant properties, garcinol is now being utilized for its anticancer characteristics. Garcinol suppresses histone acetyltransferases and induces ROS. It down-regulates the NF-κB signaling pathway by suppressing constitutive NF-κB and by decreasing the expression of NF-κB-associated genes (102). Therefore, garcinol is a promising therapeutic agent for treating cancer.
By modulating the NF-κB signaling pathway in BrCa cells, it suppresses cell growth and promotes apoptosis. For MCF-7 BrCa cells, garcinol suppresses E2-promoted proliferation and enhances apoptosis. Cell cycle arrest occurs at the G0/G1 phase. For these cells, garcinol decreases the expression of ac-H3, ac-H4, and NF-κB/ac-p65 proteins. It also suppresses the nuclear translocation of NF-κB/p65, as well as the mRNA and protein levels of cyclin D1, Bcl-2, and Bcl-xL Thus, garcinol reduces the progression of MCF-7 BrCa cells by lowering ac-p65 expression in the NF-κB pathway and by modulating expressions of various genes(103). For MDA-MB-231 and BT-549 BrCa cells, garcinol affects EMT markers, with an increase in E-cadherin, an epithelial marker, and decreases in vimentin, ZEB-1, and ZEB-2, which are mesenchymal markers. Garcinol enhances the expression of miR-200 and let-7 family miRNAs. It also enhances phosphorylation of β-catenin concomitant with its decreased nuclear localization. In addition to promoting apoptosis and suppressing cancer cell invasion, garcinol suppresses the Wnt signaling pathway. Administered to mice, garcinol suppresses NF-κB, miRNAs, vimentin, and nuclear β-catenin. Therefore, the anti-carcinogenic effects of garcinol against BrCa are associated with reversal of the EMT phenotype (102).
The promotion of apoptosis by garcinol is associated with downregulation of the NF-κB signaling pathway. For MCF-7 and MDA-MB-231 BrCa cells, garcinol suppresses constitutive NF-kappa events, correlating with the downregulation of genes related to NF-κB. Furthermore, this compound suppresses BrCa growth promoted by nicotine. Garcinol prevents the migration of these MDA-MB-231 BrCa cells by decreasing the expression of α9-nAChR and cyclin D3, which are associated with breast tumorigenesis promoted by nicotine. Thus, by suppressing the factors that contribute to the growth of breast tumors, garcinol is an anticancer agent (101) with potential to be useful in treating BrCa (104).
The combination of drugs has been proven to play an important role in treating breast cancer. As the synthetic drugs have side effects and expensive, scientists/ researchers are turning to methods such as combination treatment for treating BrCa. The combination therapy is expected to reduce the dosage regimen, fewer side effects, and cost-effective. In addition, using combination treatment can increase the quality of life of BrCa patients. Natural compounds and herbal medicine have been evidenced that the use of these compounds in adjunct of chemotherapeutic is not only enhanced therapeutic efficacy but reduces toxicity and inhibits resistance associated with multiple drugs (105).
Furthermore, we showed in Table 2, the use of a natural compound in combination with other such compounds or other therapeutic agents, and their mode of action in BrCa therapy that has proven to be efficient in a low dosage of the carcinogenic compounds. These substances are potential therapeutic agents for BrCa (Figure 2). This combination approach is a new therapeutic option, as it is generally more beneficial than single therapeutics alone. Combination therapy triggers cytotoxicity among cancer cells, affects the tumor environment, and alters the immune response to tumors (10). By combining therapeutics, the benefits of various mechanisms for inhibition of cancer cells are likely to be shown. This concept demonstrates the evolution of medical treatments for cancer. As cancer treatment progresses, improvements will allow for efficient treatment options for patients (106).
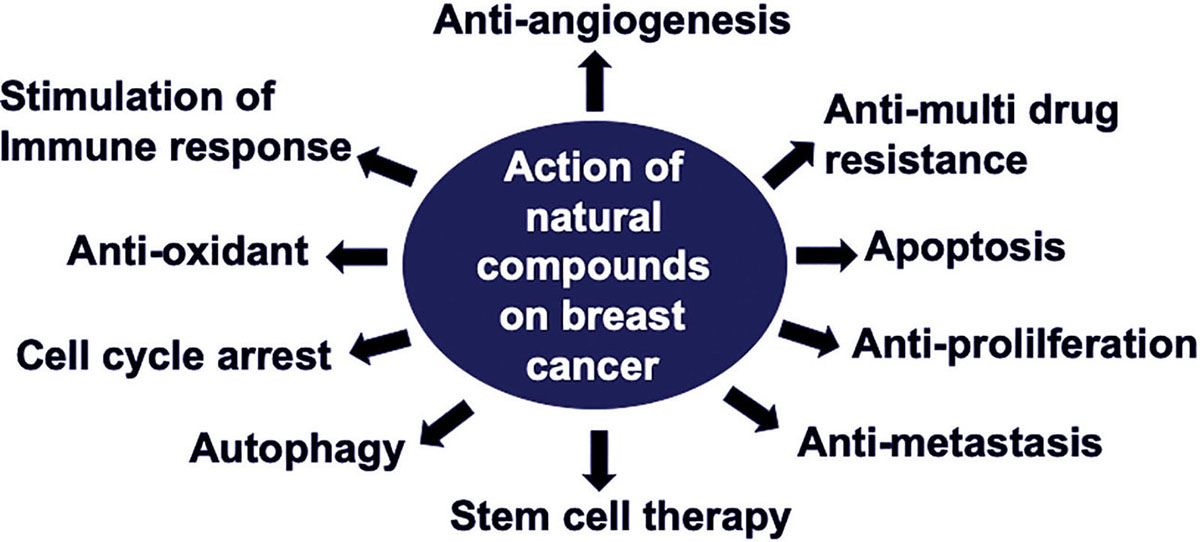 Figure 2
Figure 2Schematic representation of action of natural compounds on breast cancer.
| Combination | Action | REFERENCES |
|---|---|---|
| Tetrandrine and Arsenic | Increases expression of FOXO3a, p21, and p27; downregulates expression of cyclin D1; induces G0/G1 phase arrest; promotes autophagy; suppresses expression of survivin. | (110, 111) |
| Curcumin and Berberine | Promotes caspase-dependent apoptosis by activating ERK pathways; initiates autophagy; increases phosphorylation of JNK and beclin1; downregulates phosphorylation of Bcl-2. | (112) |
| Thymoquinone and Tamoxifen | Diminishes relapse rates, TNF-α, IL-6 and TGF-β1; upregulates caspase-3 expression; downregulates Bcl-2 expression; decreases cell viability via the PI3-K/Akt pathway through suppression of Akt phosphorylation; induces XIAP degradation; activates caspase-9; promotes apoptosis. | (113) |
| Silibinin and Chrysin | Suppresses BrCa cell proliferation; decreases mRNA expression of hTERT and cyclin D1. | (114) |
| Resveratrol and Salinomycin | Regulates cell cycle ; induces caspase activation; initiates apoptosis; decreases expression of protein components of Wnt signaling; decreases vimentin and increases E-cadherin; suppresses cell migration and invasion; induces caspase-8 and -9 activity; downregulates Wnt/EMT signaling. | (115) |
| Garcinol and Paclitaxel | Promotes cell cycle arrest; suppresses the caspase-3/cytosolic Ca2+-independent phospholipase A2 (iPLA2) signaling pathway; hinders nuclear factor-κB (NF-κB)/Twist-related protein 1 (Twist1) signaling pathway; inhibits cell viability, inflammation, angiogenesis, and cell migration. | (116) |
| Honokiol and Lapatinib | Suppresses tumor cell growth; promotes apoptosis in cells over-expressing HER-2; inhibits HER-2 expression. | (117) |
A promising approach for treating BrCa involves natural compounds, which are chemical substances derived from living organisms (8). Various natural compounds reverse the effects of drug resistance and affect various targets, demonstrating that they have therapeutic benefits (10). An analysis was organized to identify which areas of BrCa research, if targeted, could result in the greatest impact on BrCa patients (107). They recognized a gap of current knowledge on BrCa treatment, i.e. natural compound absorption, bioavailability, initiation, progression, knowledge of genetic changes, targets and diagnostic markers (107). However, several reports showed that natural compounds can suppress the promotion of carcinogenesis and reverse the progression of cancers by promoting apoptosis and cell cycle arrest. They act on tumor cells by modulating cell death pathways, including extrinsic and intrinsic apoptotic and autophagic pathways (11). In these processes, these substances inhibit the growth of cancer cells without displaying extensive toxic effects on normal cells (108). Because they demonstrate various anticancer and apoptotic effects and have little toxicity, natural compounds are now being considered for use in clinical practice. As the effects of additional natural compounds against BrCa are shown, many of these substances will likely be used to treat this disease (106).
In conclusion, the natural compounds described are only a few of the various substances that show therapeutic effects against BrCa. These substances are bringing scientists a step closer to effective treatment of BrCa. They have the potential to reduce the numbers of BrCa-related deaths and to prolong the lives of patients around the world. Thus, the use of natural compounds as a strategy for treatment of BrCa is being widely considered.
Authors declare no potential conflicts of interest. This study was supported in part by the National Cancer Institute of the National Institutes of Health under Award Numbers SC1CA193758 and U54CA118638, and by the Department of Defense under Award Number W81XWH1810429. RS designed the concept and drafted the manuscript. JWL critically reviewed the manuscript. BN and SKS prepared the figures. BN and SKS contributed equally to the writing of the final version of the manuscript. All authors read and approved the final manuscript.
BrCa
Breast cancer
Multi-drug resistance
Quercetin
Curcumin
Flap endonuclease 1
Epithelial-mesenchymal transition
Silibinin
Light chain 3
Fibronectin
Triple negative breast cancer
Epidermal growth factor
Matrix metalloproteinases
Thymoquinone
Resveratrol
phospholipase D
Honokiol
Diosgenin
Myeloid cell leukemia-1
Breast cancer stem cells
N-methyl-N-nitrosourea
Phosphatase and tensin homolog
Tumor suppressor genes
Serpin peptidase inhibitor B5
DNA methyltransferase
Ataxia telangiectasia mutated
Differentially expressed genes
Nuclear factor- κ B