

 , Andrei V. Stepanov 2,3, Galina A. Sakuta 4, Mikhail L. Vorobev 5, Ekaterina V. Baidyuk 6,7,*
, Andrei V. Stepanov 2,3, Galina A. Sakuta 4, Mikhail L. Vorobev 5, Ekaterina V. Baidyuk 6,7,*
1 Laboratory of Toxinology and Molecular Systematics, L.A. Orbeli Institute of Physiology, National Academy of Sciences, 0028 Yerevan, Armenia
2 Group of Neuroregulation of Muscle Function, Sechenov Institute of Evolutionary Physiology and Biochemistry, Russian Academy of Sciences, 194223 St. Petersburg, Russia
3 Department of Histology and Embryology named after Prof. A.G. Knorre, St. Petersburg State Pediatric Medical University, 194100 St. Petersburg, Russia
4 Department of Physiology and Cell Biology, College of Medicine, The Ohio State University, Columbus, OH 43210, USA
5 Confocal Microscopy and Image Analysis Group, Institute of Cytology, Russian Academy of Sciences, 194064 St. Petersburg, Russia
6 Laboratory of Hyperspectral Imaging of Surgical Targets, Center of Excellence, L.A. Orbeli Institute of Physiology, National Academy of Sciences, 0028 Yerevan, Armenia
7 Laboratory of Comparative Biochemistry of Enzymes, Sechenov Institute of Evolutionary Physiology and Biochemistry, Russian Academy of Sciences, 194223 St. Petersburg, Russia
Abstract
Cardiovascular diseases remain a leading cause of mortality worldwide, with myocardial infarction (MI) being the most severe form. Despite advances in treatment, MI is still associated with an estimated mortality rate of approximately 35%, and survivors frequently develop heart failure and arrhythmias, underscoring the need for new therapeutic strategies. Growing evidence indicates that mitochondrial dysfunction in cardiomyocytes (CMCs) is a major driver of post-MI remodeling. Consequently, targeting mitochondrial dynamics and subpopulation-specific responses has emerged as a promising cardioprotective approach. While acute-phase mitochondrial changes after MI have been extensively studied, remodeling during the subacute and chronic stages remains less understood, despite its critical role in scar expansion and the progression of heart failure. In this study, we investigated functional and morphological alterations in distinct mitochondrial subpopulations of left ventricular CMCs two weeks after MI.
MI was induced in adult rats by permanent ligation of the left anterior descending coronary artery. Mitochondrial morphology was analyzed by transmission electron microscopy. Mitochondrial function and oxidative stress were assessed in live isolated CMCs using fluorescence and confocal microscopy.
Two weeks after MI, CMCs exhibited a reduction in total mitochondrial membrane potential (MMP) and an increase in reactive oxygen species levels. Herewith, mitochondrial activity differed among mitochondrial subpopulations. The MMP of perinuclear (PNM) and subsarcolemmal mitochondria (SSM) decreased by ~30% more than that of intermyofibrillar mitochondria (IFM). These functional impairments were accompanied by reductions in mitochondrial size: IFM area decreased by 22%, whereas PNM and SSM decreased by 32% and 29%, respectively. At the same time, mitochondrial volume density decreased in SSM and IFM regions but remained unchanged in PNM regions. Consequently, the overall functional alterations in the PNM regions were comparable to those observed in IFM regions.
Our data demonstrate a decrease in the activity of CMC mitochondria associated with their fragmentation and reduced volume density two weeks after MI, with the most pronounced changes in SSM. These findings underscore the importance of subpopulation-specific mitochondrial analysis for understanding subacute post-infarction remodeling and for identifying novel therapeutic targets.
Keywords
- myocardial infarction
- heart failure
- cardiomyocytes
- left anterior descending coronary artery
- mitochondrial dynamics
- mitochondrial membrane potential
- reactive oxygen species
- transmission electron microscopy
Cardiovascular diseases rank first among the causes of death worldwide (32%), according to the WHO [1], with 85% of these deaths related to heart attacks and strokes. Despite modern treatment methods, including primary percutaneous coronary intervention and coronary artery bypass grafting, chronic total occlusion (CTO) of the left coronary artery remains a common form of coronary artery disease (15–30%) [2, 3]. Studying myocardial cellular changes under CTO conditions is therefore of fundamental importance, as it is key to assessing long-term myocardial viability and developing therapeutic strategies aimed at preventing the progression of heart failure. Increasing evidence indicates that mitochondrial dysfunction in cardiomyocytes (CMCs) plays a central role in remodeling following myocardial infarction (MI) [4, 5, 6].
Mitochondria are essential for CMC survival and function, supplying most of the cellular adenosine triphosphate (ATP) through oxidative phosphorylation and regulating key cell death pathways [7, 8]. Importantly, mitochondria in CMCs are not homogeneous but consist of distinct subpopulations with specific localizations and functional roles [9, 10]. Intermyofibrillar mitochondria (IFM) are located between the myofibrils and are thought to primarily supply ATP for the contractile activity of CMCs. Subsarcolemmal mitochondria (SSM), located beneath the sarcolemma, regulate ion transport and membrane-related processes. Perinuclear mitochondria (PNM), clustered around the nucleus, contribute to transcriptional regulation, signaling, and mitochondrial biogenesis. Numerous studies have demonstrated that these mitochondrial subpopulations respond differently to myocardial injury [11, 12, 13, 14, 15, 16]. Studying these differences is essential because the heterogeneous responses of mitochondrial subpopulations help explain how MI affects multiple aspects of CMC physiology, from contractile function to ion homeostasis and gene regulation. Moreover, therapeutic strategies may need to be tailored to target specific subpopulations. For example, inhibitors of mitochondrial permeability transition pore opening or mitochondria-targeted antioxidants may preferentially protect IFM and SSM, whereas activators of mitophagy and biogenesis may be particularly beneficial for PNM [7, 17]. Thus, investigating morpho-functional changes in distinct mitochondrial subpopulations after MI is critical not only for understanding the mechanisms of post-infarction remodeling but also for identifying precise therapeutic targets that may enhance cardioprotection and improve clinical outcomes.
Most existing research has focused on mitochondrial changes during the acute phase of MI or acute ischemia-reperfusion injury [18, 19, 20]. Far fewer studies have examined alterations in mitochondrial morphology and function in CMCs following CTO of the left coronary artery [21, 22], and virtually no studies have specifically addressed changes across the distinct mitochondrial subpopulations under this condition. The limited available data, derived from different experimental models, are inconsistent. For example, in a rat model of heart failure induced by transverse aortic constriction, IFM exhibited reduced mitochondrial content and respiratory capacity, whereas SSM function was only modestly affected [23]. In contrast, a microembolization-induced heart failure dog model showed no differences between SSM and IFM in oxidative phosphorylation or electron transport chain organization [14]. In a mouse model of pressure overload, PNM were more susceptible than IFM to mitochondrial membrane depolarization, reactive oxygen species (ROS) generation, and impaired Ca2+ uptake [16]. Notably, no previous study has compared all three CMC mitochondrial subpopulations simultaneously, leaving a significant gap in understanding subacute and chronic remodeling.
In the present study, we focused on the 2-week time point after MI, which corresponds to the subacute phase of post-infarction remodeling, when granulation tissue transitions into a forming scar and CMCs undergo pronounced yet still reversible structural and metabolic adaptations [24, 25, 26, 27]. We aimed to provide a detailed analysis of mitochondrial subpopulation remodeling at this biologically important stage. Focusing on this time window enables the identification of subpopulation-specific mitochondrial vulnerabilities during a period in which remodeling is substantial but remains potentially reversible, thereby offering insights into mechanisms that may shape the trajectory toward chronic heart failure. Accordingly, we investigated both morphological and functional alterations in all three mitochondrial subpopulations (IFM, SSM, PNM) two weeks after MI induced by CTO of the left coronary artery in a rat model.
The study was conducted on 4-month-old male Wistar rats (approximately 250 g), divided into two groups (10 animals per group): MI-induced rats and intact controls. Two weeks after coronary occlusion, the hearts of rats from both groups were excised under general anesthesia. Anesthesia was induced with isoflurane inhalation (4–5% in oxygen, 1–2 L/min) in a closed chamber. After the onset of anesthesia (1–2 min), animals were placed on a surgical platform and maintained under anesthesia via a nose cone, with the isoflurane concentration reduced to 3%. The surgical area was shaved, after which the thoracic cavity was rapidly opened, and the heart was excised. For analgesia, meloxicam (2 mg/kg) was administered subcutaneously 30 min before the procedure. Left ventricular (LV) samples from three animals per group were fixed for histological and electron microscopy analyses, while the hearts of the other animals were used for enzymatic CMC isolation for functional assessments.
All animal procedures were carried out in accordance with the Treaty of Lisbon amending the Treaty on European Union and the Treaty establishing the European Community (effective from 1 December 2009). The study was approved by the Ethical Committee for Animal Research of the Institute of Cytology of the Russian Academy of Science (Protocol No. 3; approval date: 28 June 2016) and the Animal Care and Use Committee at the L.A. Orbeli Institute of Physiology NAS RA (Protocol No. 07.02.2025/1; approval date: 19 April 2025).
MI was induced in rats under anesthesia (intramuscular Zoletil, Virbac, France, 40 mg/kg, and Xylazine, Nita-Pharm, Russia, 10 mg/kg) by permanent ligation of the left anterior descending coronary artery. Artificial ventilation was provided via tracheal intubation using a TOPOTM dual-mode ventilator (Kent Scientific, Torrington, CT, USA) at a frequency of 50/min and a volume of 2 mL/100 g. Before intubation, the throat was treated with 2% lidocaine. Thoracotomy was performed in the fourth intercostal space, left of the sternum, extending to the anterior axillary line. After opening the pericardium, the left anterior descending artery was ligated using a 6-0 atraumatic needle. The wound was then closed in layers. Throughout the entire surgical procedure, electrocardiographic monitoring was performed. Correct ligature placement and the onset of myocardial ischemia were confirmed by the ST-segment elevation 30 min after the procedure. Electrocardiograms (ECG) were recorded in three standard leads using disposable needle electrodes connected to a PolySpectrum 8/V (Ivanovo, Russia) veterinary electrocardiograph, and Lead II was used for analysis. To improve signal quality, cardiac complexes were averaged in MATLAB based on R-wave peaks after five minutes of recording. Additionally, ECG recordings were obtained two weeks after surgery, immediately before tissue collection. The extent of ischemic injury was additionally assessed post-mortem by staining 5–6 transverse heart sections with 1% 2,3,5-triphenyl-tetrazolium chloride (TTC) solution prepared in 0.2 M Tris buffer (pH 7.8) [28].
Adult live ventricular CMCs were isolated using the Langendorff perfusion method. Ten minutes before anesthesia, rats received an intraperitoneal injection of heparin (1000 IU/kg body weight) [29]. The animals were then anesthetized with 5% isoflurane, after which the hearts were excised, cannulated, and perfused with a calcium-free Krebs-Henseleit buffer, followed by enzymatic digestion with collagenase II (Worthington, Lakewood, NJ, USA, 0.6–1.0 mg/mL) for approximately 30 min at 37 °C [30]. The LV was subsequently dissected, gently dispersed, and subjected to gradual reintroduction of Ca2+, resulting in viable rod-shaped CMCs with well-defined striation (Fig. 1A). Cell viability was assessed using Trypan blue staining (0.2%).
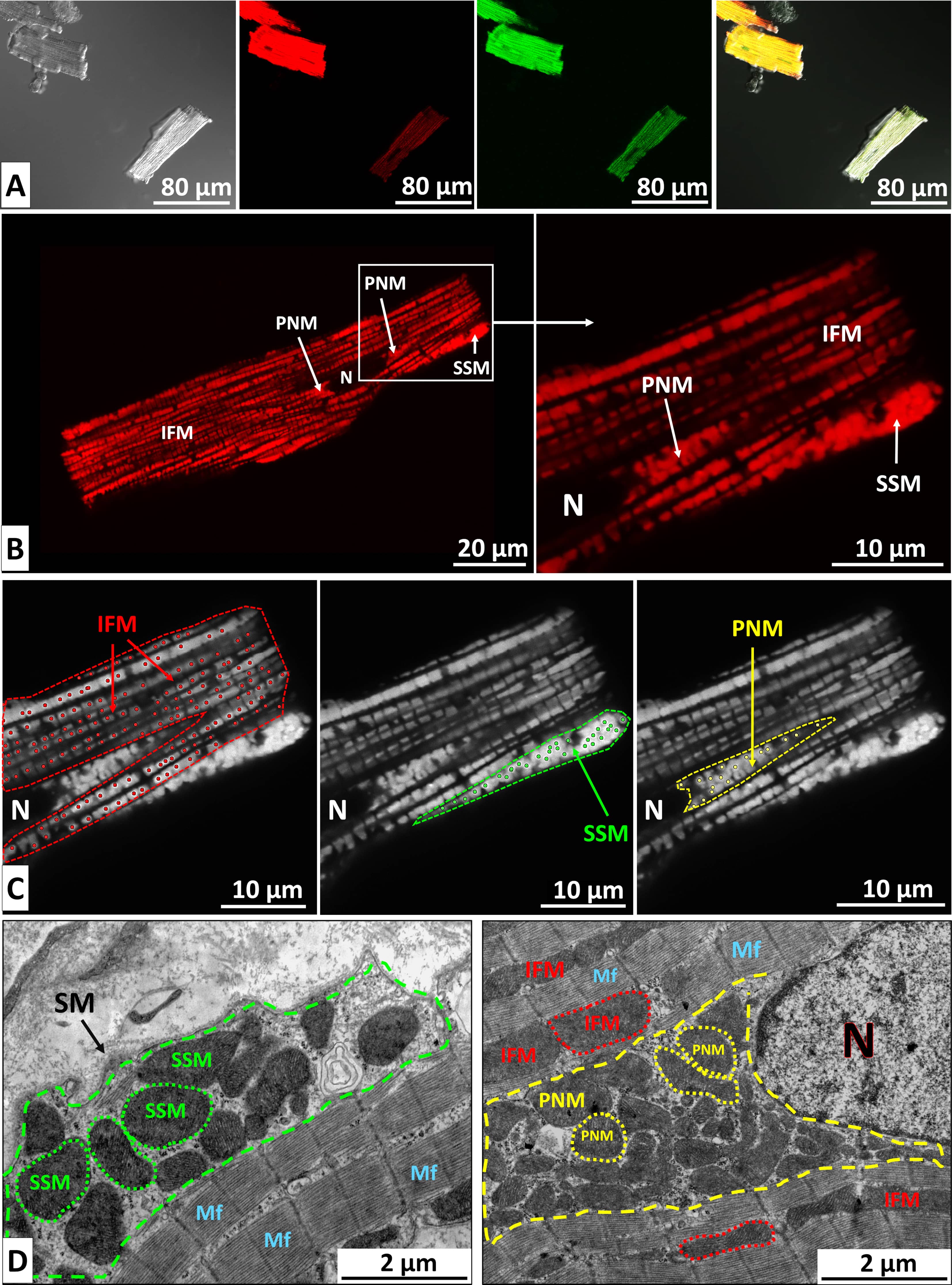
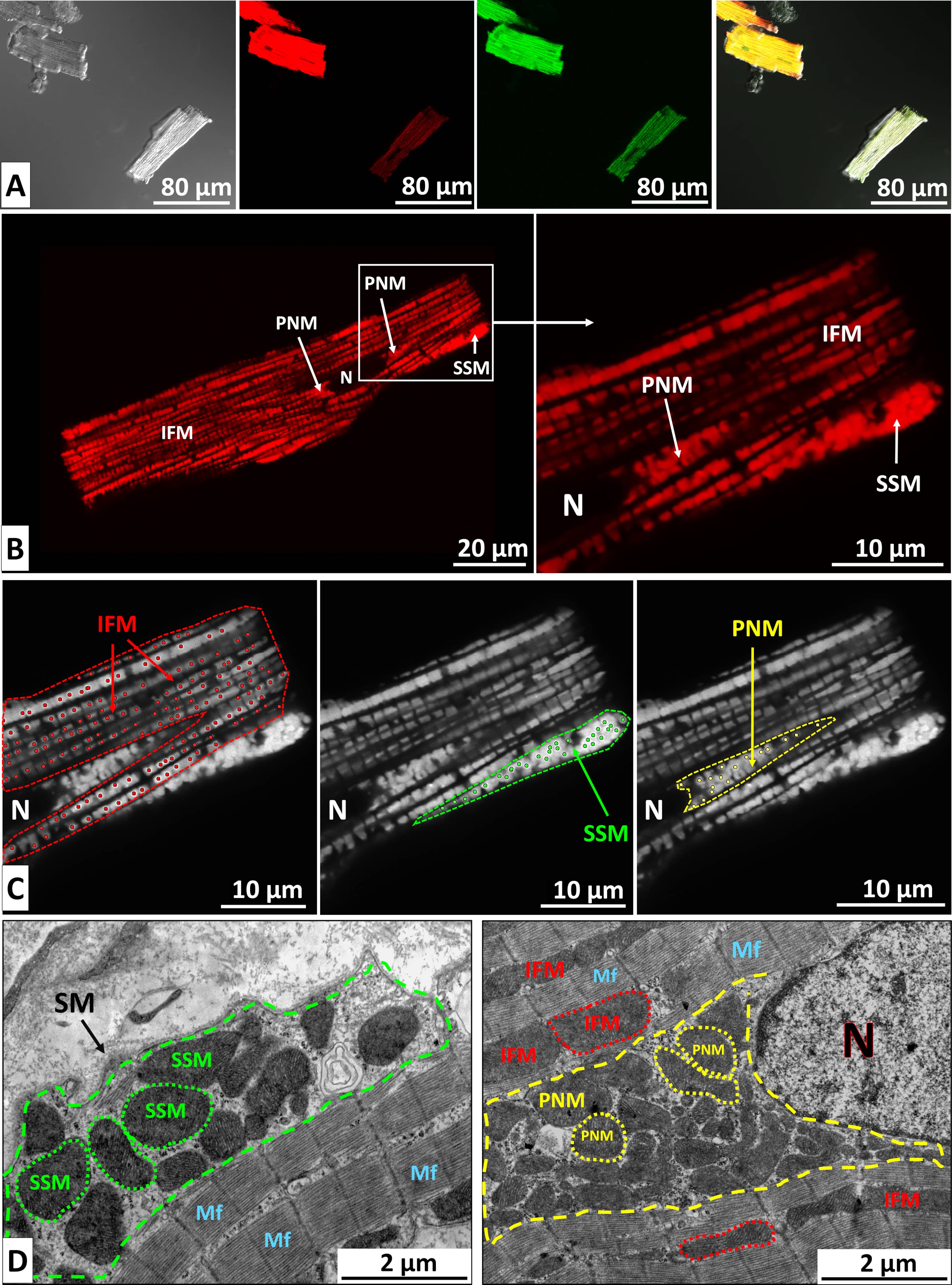 Fig. 1.
Fig. 1.
Visualization of image acquisition and analysis. (A) Viable CMCs, enzymatically isolated from the LV of an adult rat and simultaneously stained with the MMP–sensitive dye TMRM and the ROS indicator H2DCFDA. From left to right: phase contrast, TMRM fluorescence, H2DCFDA fluorescence, and merged image. Differences in fluorescence intensity between individual cells are clearly visible. Scale bar, 80 µm. (B) Confocal image of a live CMC isolated from a control rat and stained with TMRM. Distinct mitochondrial subpopulations are visible: perinuclear (PNM), subsarcolemmal (SSM), and intermyofibrillar mitochondria (IFM). N, nucleus. Scale bars, 20 µm (left) and 10 µm (right). (C) ImageJ analysis of mitochondrial fluorescence intensity. Cytoplasmic regions containing different mitochondrial subpopulations were manually delineated (dashed lines). Individual mitochondrial intensities were measured using the Find Maxima tool (dots), and mean fluorescence values were calculated. Scale bar, 10 µm. (D) Electron micrographs of CMCs from the control group showing subsarcolemmal (left), perinuclear, and intermyofibrillar mitochondria (right). Cytoplasmic regions containing PNM and SSM are outlined by dashed lines. Selected mitochondria are indicated with dotted contours. Scale bar, 2 µm. SM, sarcoplasmic membrane; Mf, myofibrils; CMCs, cardiomyocytes; MMP, Mitochondrial Membrane Potential; TMRM, tetramethylrhodamine methyl ester; ROS, reactive oxygen species; H2DCFDA, 2′,7′-dichlorodihydrofuorescein diacetate.
To evaluate mitochondrial energy status and ROS levels, freshly isolated live CMCs were stained for 30 min in the dark at room temperature with 10 nM tetramethylrhodamine methyl ester (TMRM; Thermo Fisher Scientific, Waltham, MA, USA), a potential-dependent dye, and 2 µM 2′,7′-dichlorodihydrofuorescein diacetate (H2DCFDA; Thermo Fisher Scientific, Waltham, MA, USA), a ROS indicator (Fig. 1A). Cells were imaged using an Axioscope microscope (Carl Zeiss, Oberkochen, Germany) equipped with a Leica DFC-360 monochrome camera (Wetzlar, Germany). Integrated cellular fluorescence intensity was quantified in ImageJ (version 1.54p, National Institutes of Health, Bethesda, MD, USA) [31]. Filter Set 15 LP 590 was used for TMRM imaging, and Filter Set 10 for H₂DCFDA imaging. Data from four control and three MI rats were analyzed, with 30–50 cells assessed per animal.
To assess mitochondrial energy status in distinct subpopulations, CMCs were imaged using an Olympus FV3000 confocal laser scanning microscope (Tokyo, Japan). TMRM fluorescence was excited at 561 nm, and H₂DCFDA at 488 nm. Z-stacks were acquired for each cell, and optical sections passing through the nucleus were selected for analysis (Fig. 1B). Regions of interest were manually outlined in ImageJ to quantify fluorescence from PNMs and SSMs. IFMs were analyzed by excluding PNMs, SSMs, and nuclear areas. Both average fluorescence within each region and the intensities of individual mitochondria were measured using ImageJ’s Find Maxima tool (Fig. 1C). CMCs from three control and three MI rats were analyzed (20 cells per rat).
For histological analysis, LVs were fixed in 4% neutral formaldehyde solution, embedded in paraffin, sectioned at 5–6 µm thickness, and stained with Meyer’s hematoxylin and eosin.
For TEM analysis, tissue samples were collected from distinct LV regions,
including the peri-infarct zone (1–2 mm from the scar border) and the intact
zone (5–6 mm from the scar border, primarily in the mid-lateral and posterior
walls, as the anterior wall was largely replaced by scar tissue). In control
rats, samples were obtained from corresponding locations in the mid-lateral and
posterior LV walls. LV myocardial tissue pieces were fixed in 2.5%
glutaraldehyde prepared in 0.01 M sodium cacodylate buffer (pH 7.2) supplemented
with 0.5 M sucrose. After primary fixation, samples were postfixed in 4% osmium
tetroxide for 1 h in the dark. Tissue pieces were then dehydrated through a
graded alcohol series and embedded in an Epon-Araldite mixture (Sigma, St. Louis,
MO, USA). Ultrathin longitudinal sections (~50 nm) were prepared
using a LKB III ultratome (Stockholm, Sweden) and collected on grids. Sections
were contrasted with 1% uranyl acetate, followed by 3% lead citrate. Electron
micrographs were acquired at 4000
The mitochondrial area and the proportion of the CMC cytoplasm occupied by different mitochondrial subpopulations (volume density) were quantified. Nuclei, perinuclear, and subsarcolemmal regions were manually delineated; the remaining CMC area was considered to contain IFMs (Fig. 1D). Hearts from three control and three infarct rats were examined. On average, 20 to 40 fields of view from LV tissue sampled across the different regions were analyzed per rat in each group.
Pairwise comparisons were performed using a two-tailed Student’s t-test
(GraphPad Prism 8.2.1, La Jolla, CA, USA) for normally distributed data (MMP and
ROS fluorescence values, and ECG parameters) while the Mann-Whitney test was
applied for data with a non-normal distribution (mitochondrial areas and volume
densities, and S-wave amplitude). A two-way analysis of variance (ANOVA) was used
to assess differences in the relative brightness of SSM and PNM subpopulations
between control and MI groups. Post hoc comparisons were conducted using the
Sidak test. Outliers were identified and excluded using the Tukey fence method (k
= 1.5). A p-value
To investigate subacute morphofunctional alterations in mitochondria within LV CMCs following ischemic injury, we performed permanent ligation of the left coronary artery in adult rats. Correct ligature placement was verified intraoperatively by electrocardiographic monitoring, with the onset of myocardial ischemia confirmed by ST-segment elevation (Fig. 2A).
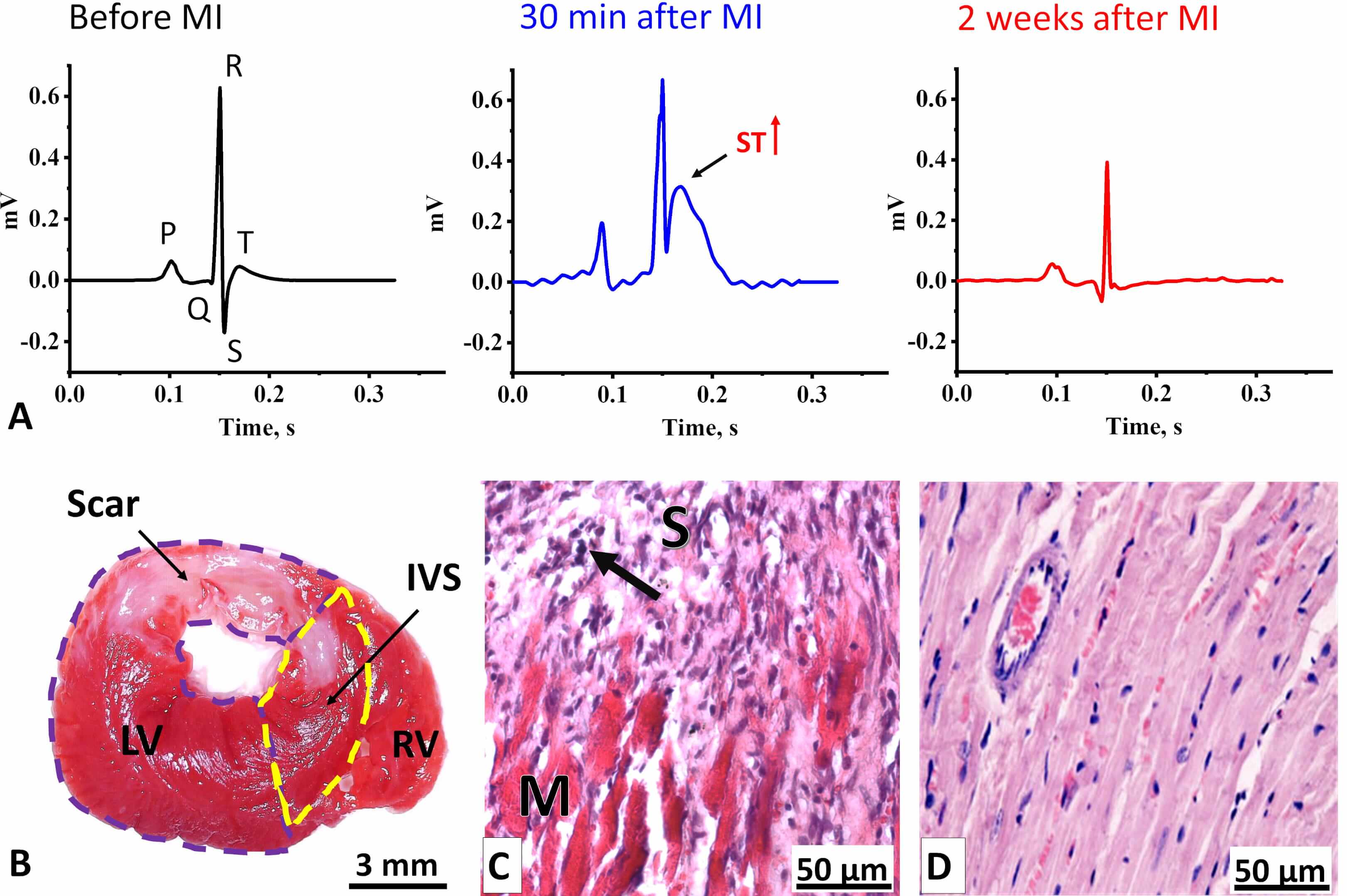
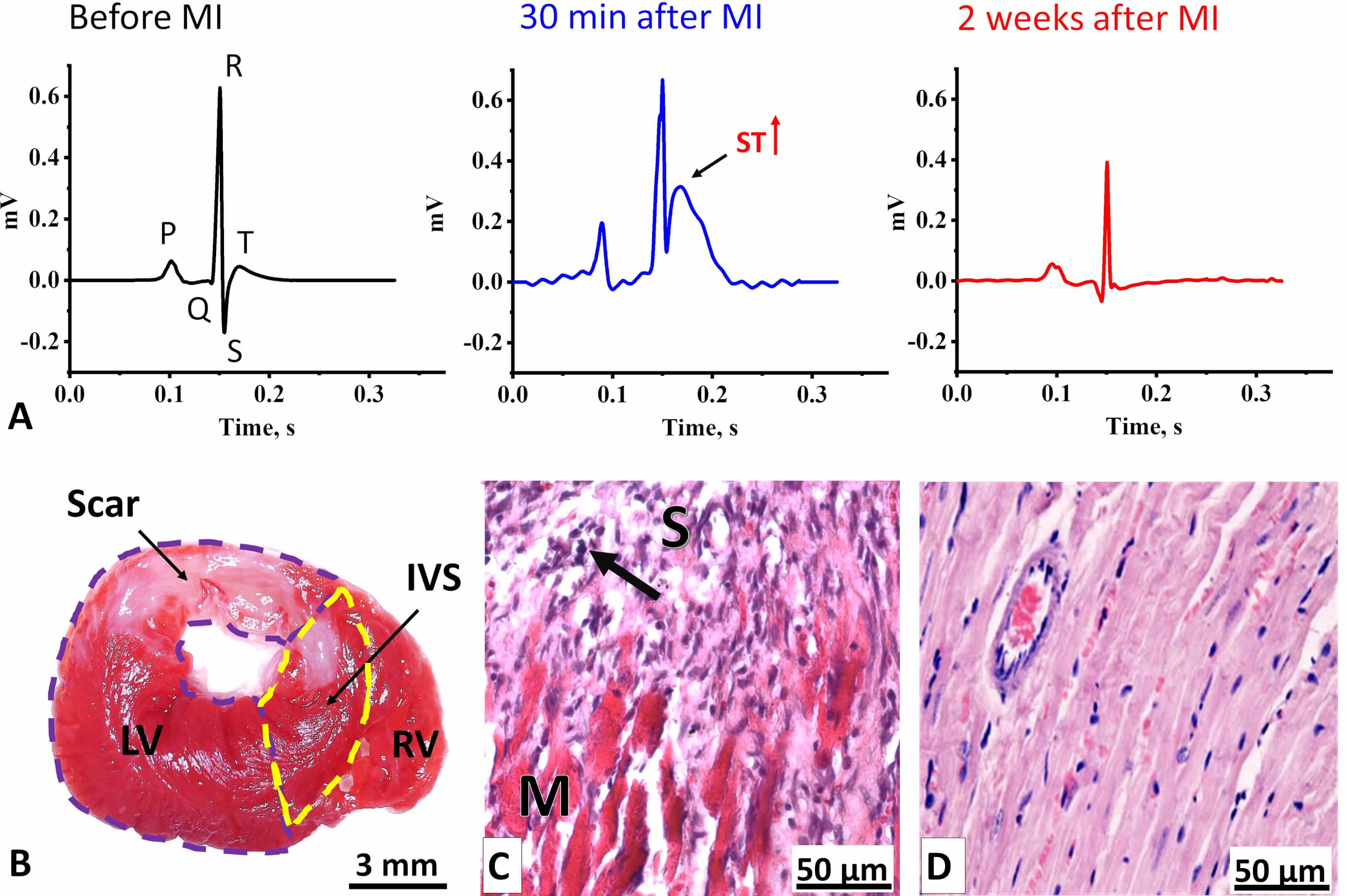 Fig. 2.
Fig. 2.
Electrophysiological and morphological changes in the LV myocardium two weeks after coronary occlusion. (A) ECG Lead II averaged cardiocomplexes of rats (n = 10) before MI (left) and two weeks after MI (right). In the center, a representative averaged cardiocomplex from a single rat recorded 30 min after MI is shown. The arrow indicates an elevated ST-segment. (B) Transverse section of a rat heart stained with TTC. Viable myocardium appears red, while the formed scar is unstained. LV, left ventricle (outlined by a purple dashed line); IVS, interventricular septum (outlined by a yellow dashed line); RV, right ventricle. Scale bar, 3 mm. (C) Histological section of the LV at the border between post-infarct scar tissue (S) and viable myocardium (M). The arrow indicates leukocyte infiltration. Scale bar, 50 µm. (D) Histological section of the LV myocardium from a control rat. Scale bar, 50 µm. Sections in (C) and (D) are stained with Mayer’s Hematoxylin-Eosin. ECG, Electrocardiograms; MI, myocardial infarction; TTC, 2,3,5-triphenyl-tetrazolium chloride.
Two weeks after surgery, the signs of acute MI became less pronounced (Fig. 2A). The predominant alterations involved changes in the amplitudes of the QRS-complex waves (Supplementary Table 1). Specifically, we observed an almost eightfold increase in Q-wave amplitude, a reduction in R- and S-wave amplitudes, and a decrease or inversion of the T-wave, while the durations of the RR, QT, and QTc intervals remained unchanged. Such changes are indicative of a transmural myocardial infarction (Q-wave enlargement, T-wave decrease/inversion), localized mainly in the basal region of the anterior LV wall and the interventricular septum [32, 33, 34].
Histological analysis confirmed the ECG findings, revealing a transmural scar predominantly in the LV and partially in the interventricular septum two weeks after MI (Fig. 2B). The scar occupied up to 36% of the LV area, with a mean value of 25%. The scar consisted mainly of loose connective tissue (Fig. 2C) and contained numerous leukocyte infiltrates, particularly at the border between the scar and adjacent myocardium (Fig. 2C), indicating incomplete myocardial remodeling and potential for future scar expansion. Furthermore, pronounced eosinophilic staining of the myocardium in infarcted rats compared to controls (Fig. 2D), reflected hypercontraction associated with ATP depletion, suggesting insufficient mitochondrial activity [35].
To assess functional changes in mitochondria within LV CMCs after acute MI, we enzymatically isolated live CMCs from control and MI rat hearts and stained them with the voltage-sensitive dye TMRM (Fig. 3A). Two weeks post-MI, CMCs showed a significant 24% decrease in integrated fluorescence intensity, indicating reduced mitochondrial membrane potential (Fig. 3B). Oxidative stress was evaluated by measuring ROS levels using H₂DCFDA staining. Cytofluorimetric analysis revealed that the ROS levels in CMCs isolated from post-MI hearts were nearly double those of controls (Fig. 3B).
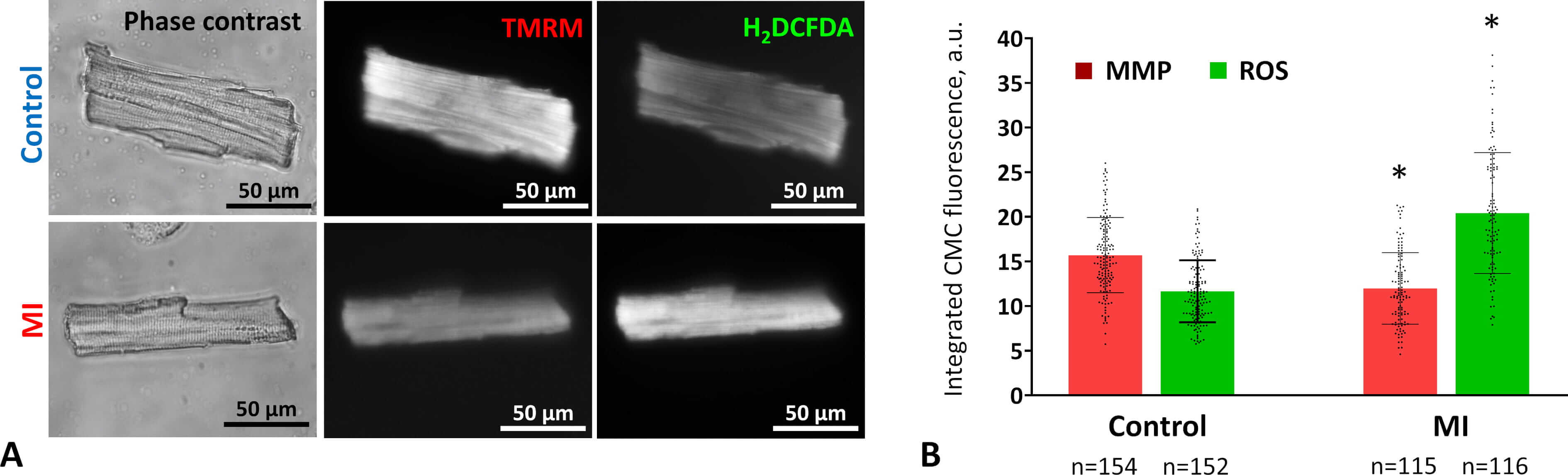
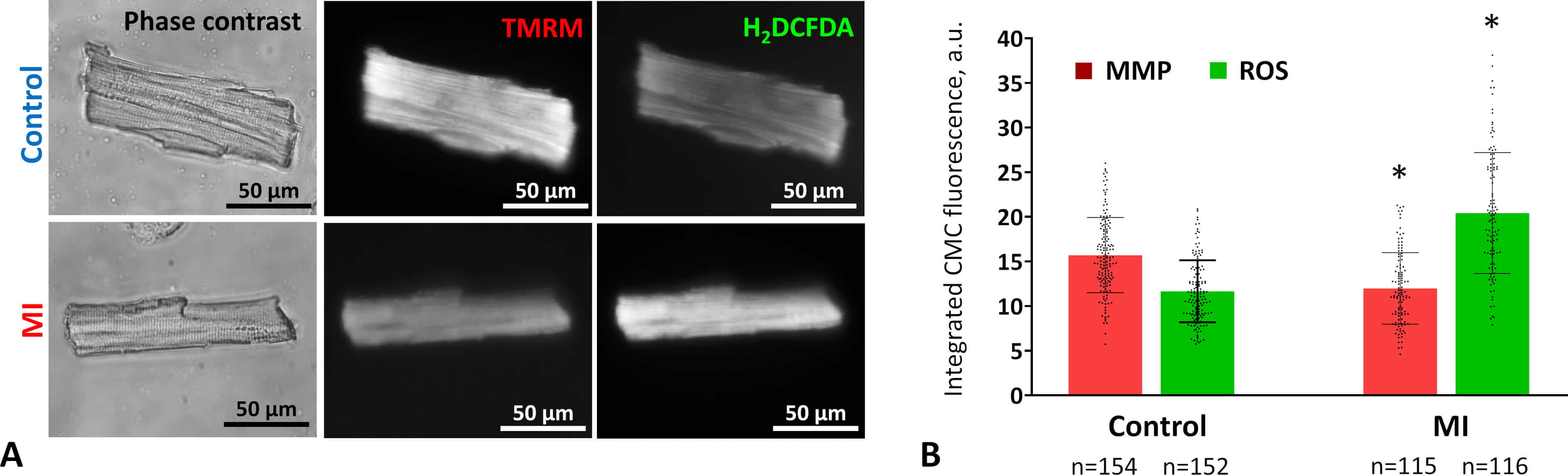 Fig. 3.
Fig. 3.
Mitochondrial activity and oxidative stress in CMCs two weeks
after MI. (A) Representative images of viable CMCs from control (upper row) and
MI (lower row) rat myocardium. Cells were stained with the MMP–sensitive dye
TMRM (center) and the ROS detector H2DCFDA (right). Phase-contrast images
are shown on the left. Fluorescence images were acquired using an Axioscope
microscope with Filter Set 15 (TMRM) and Filter Set 10 (H₂DCFDA). Scale bar, 50
µm. (B) Changes in MMP and ROS levels in CMCs two weeks post-coronary
occlusion. Data are presented as mean
These results indicate that two weeks after MI, mitochondrial activity remains impaired and oxidative stress persists, suggesting that myocardial energy status is not fully restored and cellular damage is not yet resolved.
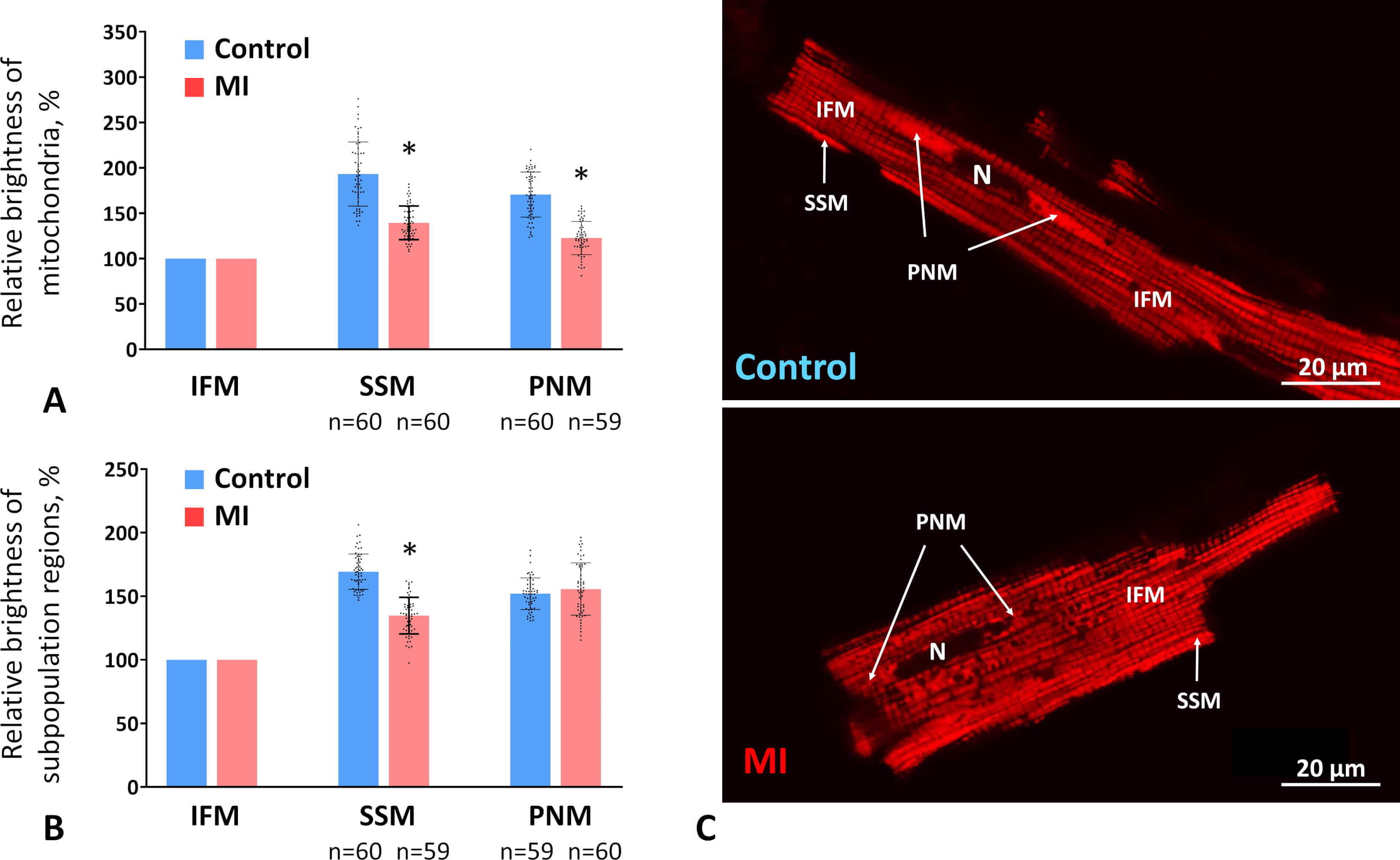
To investigate the activity of different mitochondrial subpopulations two weeks after MI, we measured the maximum fluorescence intensity of individual mitochondria stained with membrane potential–sensitive dye TMRM. Because staining efficiency and confocal microscope settings varied between control and experimental CMCs, mitochondrial fluorescence in each subpopulation was averaged per cell and expressed as a percentage relative to IFMs.
In control CMCs, PNMs and SSMs exhibited brightness levels 71% and 93% higher than IFMs, respectively. After MI, the relative brightness of PNMs and SSMs decreased, approaching that of IFMs, exceeding it by only 23% and 39%, respectively (Fig. 4A,C). Overall, the average relative brightness of subsarcolemmal regions compared with intermyofibrillar regions also decreased by 20% in MI versus control CMCs (Fig. 4B). In contrast, the average relative brightness of perinuclear regions remained unchanged (Fig. 4B), which may reflect the preservation of PNM volume density in the MI group at control levels (see Section 3.4).
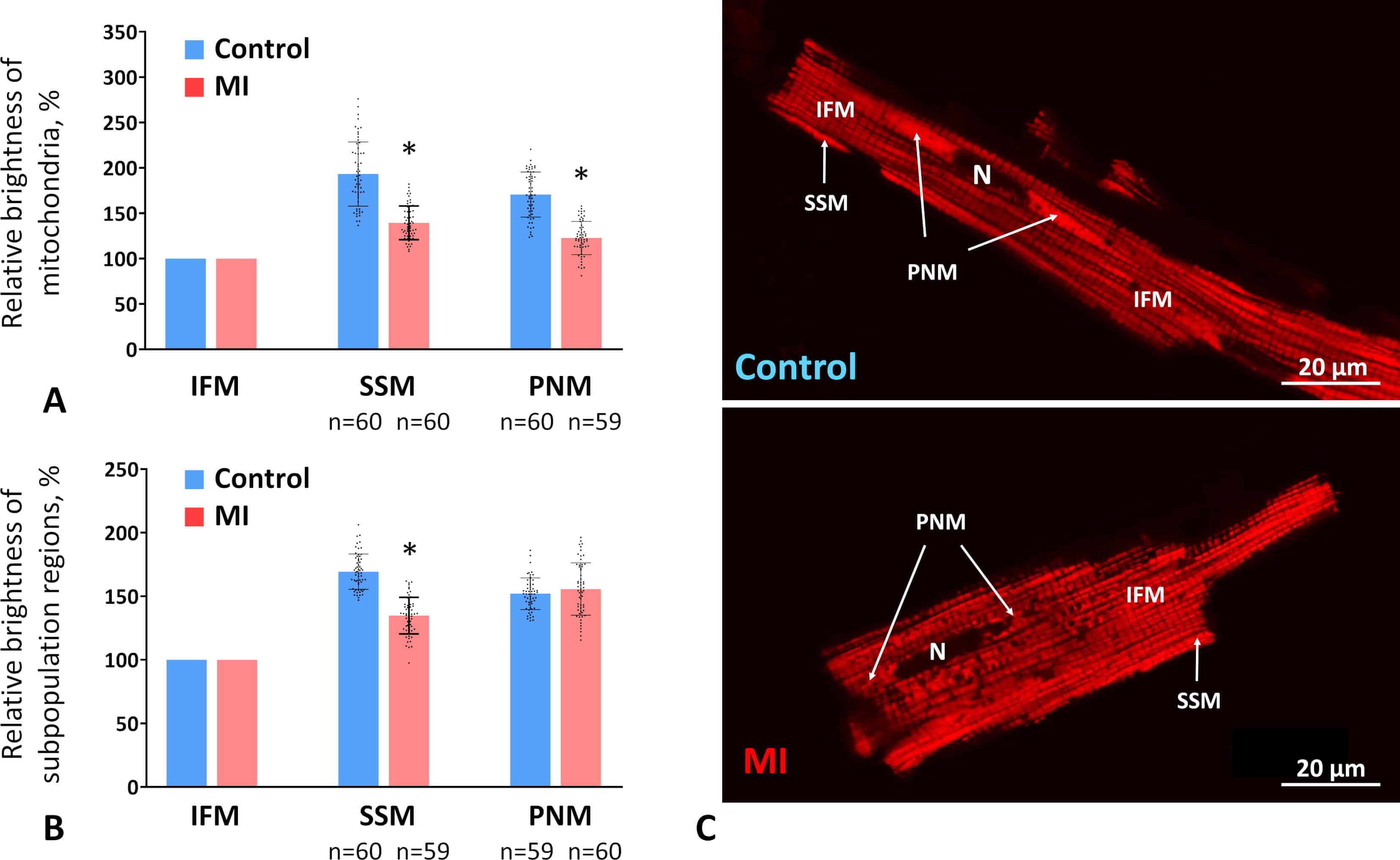 Fig. 4.
Fig. 4.
The MMP in different subpopulations two weeks post-MI. (A)
Brightness levels of individual mitochondria in the perinuclear (PNM) and
subsarcolemmal (SSM) subpopulations relative to the brightness level of
intermyofibrillar mitochondria (IFM) in control and MI groups expressed as a
percentage. (B) Average brightness levels of the PNM and SSM regions of the CMCs
relative to regions containing IFMs. Data are presented as mean
These results indicate that SSMs and PNMs experience a greater decline in activity than IFMs at the subacute stage post-infarction.
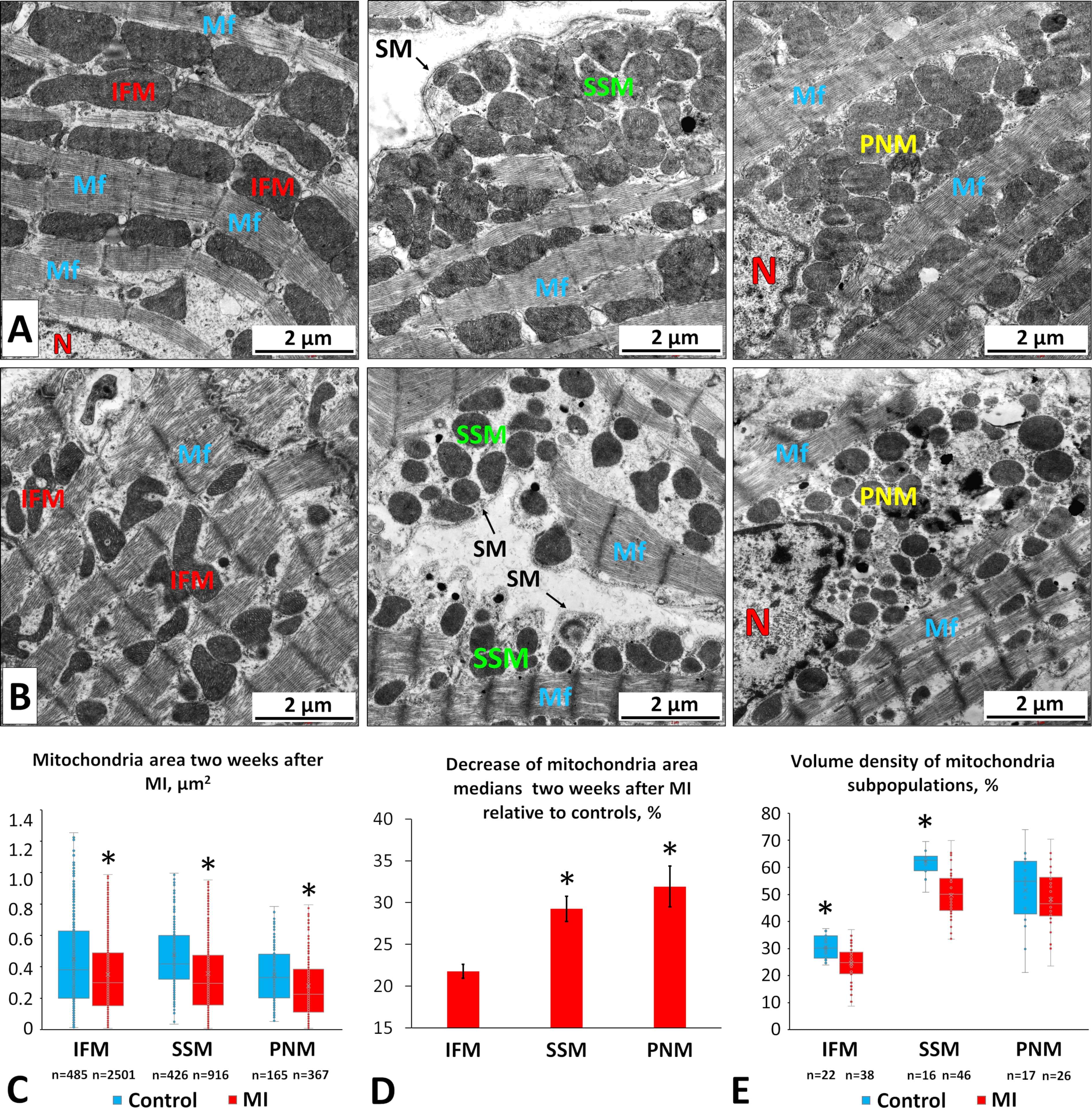
To evaluate morphological changes in mitochondrial subpopulations two weeks post-MI, we conducted a TEM study. Morphometric analysis of electronograms revealed a reduction in mitochondrial area across all subpopulations (Fig. 5A–C), with perinuclear mitochondria (PNMs) and subsarcolemmal mitochondria (SSMs) showing the greatest declines (Fig. 5D). Specifically, PNM and SSM areas decreased by 32% and 29%, respectively, whereas the area of IFMs decreased by 22% (Fig. 5C,D) compared with controls.
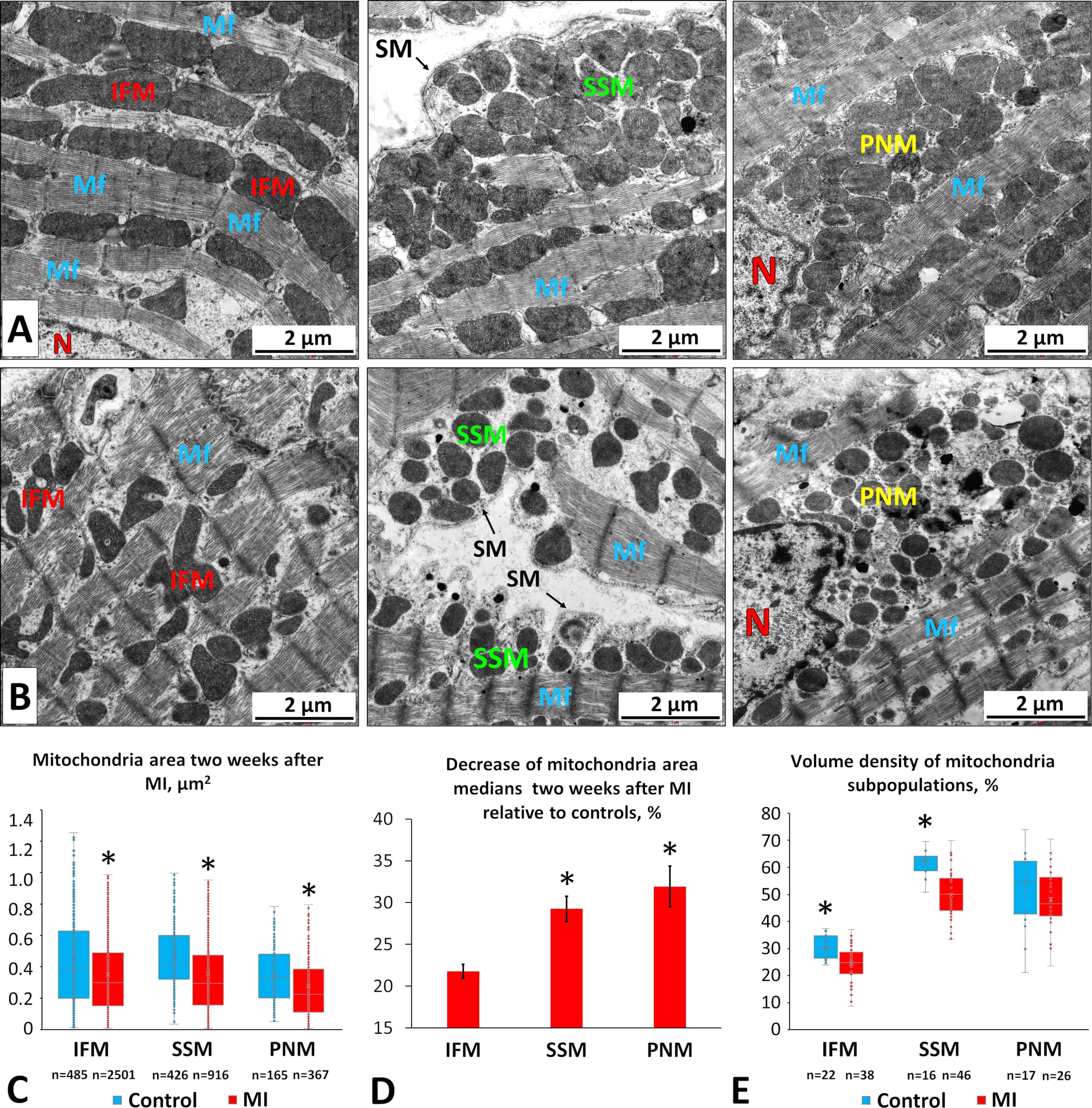 Fig. 5.
Fig. 5.
Morphological changes in different mitochondrial subpopulations
in LV CMCs two weeks after coronary occlusion. (A) Electron micrographs of CMCs
from control rats showing intermyofibrillar (left), subsarcolemmal (middle), and
perinuclear (right) mitochondria. Scale bar, 2 µm. (B) Electron micrographs
of CMCs from MI rats showing intermyofibrillar (left), subsarcolemmal (middle),
and perinuclear (right) mitochondria. SM, sarcoplasmic membrane; N, nucleus; Mf,
myofibrils. Scale bar, 2 µm. (C) Mitochondrial areas of different
subpopulations under normal and post-MI conditions. (D) Percent decrease (
Although the mitochondrial area decreased, energy output could theoretically be maintained if the mitochondrial number increased. Therefore, mitochondrial volume density, the proportion of cytoplasmic area occupied by mitochondria, serves as a more informative indicator of cellular energy status. We measured volume density for each subpopulation and found significant decreases in IFM and SSM regions by 18% and 20%, respectively (Fig. 5E). In contrast, the perinuclear region showed no significant change relative to controls.
These findings suggest that mitochondrial fragmentation and degradation occur in IFM and SSM subpopulations, without sufficient compensation through mitochondrial synthesis. In the perinuclear regions, fission appears to predominate during post-infarction remodeling, but degradation is less pronounced, or new mitochondria are synthesized more actively. This observation aligns with our data showing that the relative brightness of perinuclear regions in MI CMCs remains at control levels (Fig. 4B).
Following MI, the heart undergoes a dynamic remodeling process that includes necrosis, inflammation, granulation tissue formation, and ultimately mature scar development. Each stage is accompanied by characteristic structural and functional changes at the tissue, cellular, and molecular levels within the surviving myocardium, and these changes largely determine whether the heart recovers or progresses toward heart failure. Mitochondria play a central role in this process by supplying the energy required for CMC contraction and by regulating cell death pathways and redox homeostasis [36]. The dynamics of mitochondrial morphology have been shown to play a critical role in post-infarction remodeling and represent potential therapeutic targets [37]. Mitochondrial dynamic proteins, including Drp1, Fis1, Mfn1/2, and OPA1, regulate fission and fusion processes, thereby maintaining mitochondrial structure and function and enabling CMCs to adapt to stress conditions [6].
In the early phase following MI, within hours to a few days, CMC mitochondria experience significant fragmentation, swelling, loss of cristae integrity, bioenergetic failure, excessive ROS production, and the initiation of cell death pathways [6, 10, 18, 38, 39]. In contrast, studies investigating the later stages of post-infarction remodeling, occurring weeks to months after MI, report more complex changes, including increased mitochondrial functional heterogeneity, the accumulation of damaged mitochondria, and the activation of compensatory mechanisms such as mitochondrial biogenesis induction, selective mitophagy [6, 40, 41], and compensatory mitochondrial hypertrophy [42]. In our previous studies, we showed that six months after permanent coronary occlusion in rats, heart failure was associated with an increased mitochondrial volume density in the peri-infarction zone and enlarged mitochondria in the intact myocardium, accompanied by CMC hypertrophy [42, 43]. Simultaneously, the overall mitochondrial membrane potential of CMCs remained reduced compared to controls [30], likely reflecting alterations in the pathological architecture of CMC cristae, which were less tightly packed and exhibited more irregular morphology.
Despite this knowledge, the subacute phase of remodeling, typically spanning two to four weeks post-infarction, remains less well characterized [6, 44]. This period is particularly critical as it represents a transitional window during which the balance between mitochondrial injury and adaptive responses may significantly influence the progression toward heart failure with reduced ejection fraction or mitigate the extent of ischemic myocardium [24, 25, 26]. In our previous works, we demonstrated that during the scar maturation process two weeks after experimental MI, a complex hypotrophic response of CMCs is observed throughout the entire volume of the LV, including the remote intact regions and border zone adjacent to the infarct [42, 43]. This hypotrophy, manifested by reduced dry mass and cell size, was accompanied by a decrease in average mitochondrial area across all subpopulations, suggesting a potential functional relationship between the condition of the contractile apparatus and the level of energy metabolism in CMCs. In the present study, we aimed to determine which mitochondrial subpopulations are most severely affected at this stage of myocardial remodeling, because different mitochondrial subpopulations have different vulnerabilities to ischemic injury and can undergo different changes during scar formation [9].
We found that two weeks after post-infarction LV remodeling, CMCs exhibit a significant reduction in overall mitochondrial activity accompanied by a marked increase in intracellular ROS levels (Fig. 3). Similar alterations have been widely reported in post-infarction myocardium across various stages of ischemia-reperfusion [18, 19, 45, 46], as well as during subacute and chronic myocardial damage [20, 21]. Because IFMs constitute the dominant mitochondrial subpopulation, representing approximately 80% of all CMC mitochondria [47], the overall decrease in integral fluorescence intensity is likely driven primarily by a reduction in their membrane potential. However, the magnitude of membrane potential loss may differ among mitochondrial subpopulations. By comparing the average fluorescence intensities of SSMs and PNMs relative to IFMs under normal conditions and after MI, we observed that the membrane potentials of the SSMs and PNMs decreased by approximately 30% more than that of IFMs (Fig. 4A,C). Morphological analyses further revealed that the sizes of SSMs and PNMs were reduced to a greater extent than the sizes of IFMs (Fig. 5C,D). Thus, the size and activity of individual mitochondria in the perinuclear and subsarcolemmal subpopulations declined more profoundly than those of the intermyofibrillar subpopulation.
The existing literature on this topic remains highly contradictory. Lesnefsky et al. [12], using an isolated buffer-perfused rabbit heart model, reported that after 45 min of ischemia, oxidative phosphorylation was impaired in SSMs, but not in IFMs. Similarly, Leistner et al. [48], employing an isolated rat heart model of ischemia-reperfusion, observed more pronounced damage in SSMs. By contrast, in a rat model of heart failure induced by transverse aortic constriction, IFMs displayed a loss of mitochondrial content and respiratory capacity, whereas SSMs were only mildly affected [23]. A subsequent study by the same group, using an acute pre-clinical porcine model of ischemia-reperfusion, found no significant functional differences between IFMs and PNMs, despite clear evidence of global mitochondrial dysfunction, including impaired ATP production, altered state 3 and state 4 respiration, and reduced calcium retention capacity [15]. Studies examining PNMs specifically are relatively limited. However, Voglhuber et al. [16], using a mouse model of pressure overload, demonstrated that PNMs were more susceptible than IFMs to mitochondrial membrane depolarization, elevated ROS generation, and impaired calcium uptake. Overall, despite inconsistencies across experimental models and species, the majority of studies indicate that all mitochondrial subpopulations are affected by ischemic injury, with SSMs and PNMs often exhibiting more severe dysfunction than IFMs. These observations are in line with our findings.
However, a reduction in the size and functional activity of individual mitochondria can potentially be compensated by an increase in their number, thereby maintaining the required level of cellular energy. For this reason, one of the most informative morphometric indicators is mitochondrial volume density, which reflects the proportion of the CMC volume occupied by mitochondria. In our study, we detected a decrease in the volume density of SSMs and IFMs (Fig. 5E), whereas no significant changes were found in the volume density of PNMs. The proximity of PNMs to the nucleus may facilitate a more rapid restoration of fragmented and degraded mitochondria compared with other regions of the CMC cytoplasm, partially compensating for the reduction in activity and size of individual mitochondria within this subpopulation. Consequently, the average brightness of perinuclear regions of the CMC cytoplasm decreased in the MI group proportionally to the decrease observed in regions containing the contractile apparatus and IFMs. In contrast, the average brightness of submembrane regions containing SSMs declined more markedly than that of intermyofibrillar regions (Fig. 4B). Thus, based on our data, SSMs appear to be the most vulnerable mitochondrial subpopulation at studied stage of myocardial remodeling, as they exhibit a simultaneous reduction in membrane potential, mitochondrial area, and volume density, changes that collectively contribute to diminished overall energy production in these cytoplasmic regions.
During the post-infarction period, SSMs and PNMs in CMCs may be particularly
susceptible due to their localization and regulatory dependencies. In both
populations, mitochondrial dynamics proteins such as DRP1 and OPA1 play a
critical role. Stress-induced translocation of DRP1 and its imbalance with OPA1
promote excessive fragmentation, especially in mitochondria closely linked to
nuclear and sarcolemmal stress pathways [6, 16, 49]. The increased vulnerability of
SSMs in post-infarction CMCs may result from persistent oxidative stress, chronic
Ca2⁺ overload, and structural remodeling. Due to their proximity to
Further investigation of the underlying regulatory mechanisms, including the signaling pathways controlling mitochondrial dynamics, mitophagy and mitochondrial biogenesis, calcium homeostasis and interactions with sarcolemmal and nuclear domains on subacute stage of postinfarction remodeling, will enhance understanding of different mitochondrial subpopulations remodeling processes and guide the identification of potential therapeutic targets.
Morphofunctional analysis of intermyofibrillar, subsarcolemmal, and perinuclear mitochondrial subpopulations in LV CMCs under conditions of CTO of the left coronary artery revealed a decrease in both the activity and size of these organelles during the subacute stage of the disease. Among them, SSMs emerged as the most sensitive mitochondrial subpopulation to ischemic injury. Thus, the comparative assessment of mitochondrial damage performed in this study expands current knowledge of mitochondrial remodeling in CMCs during the subacute phase of MI, and serves as preliminary research supporting the development of therapeutic approaches to limit scar expansion.
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
SAF: validation, formal analysis, visualization, writing — original draft, writing — review and editing. AVS: methodology, investigation, writing — review and editing. GAS: methodology, investigation, writing — review and editing. MLV: methodology, investigation, visualization, writing — review and editing. EVB: conceptualization, methodology, data curation, investigation, validation, supervision, funding acquisition, formal analysis, visualization, project administration, resources, writing — original draft, writing — review and editing. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
All animal experiments were conducted in accordance with the Treaty of Lisbon amending the Treaty on European Union and the Treaty establishing the European Community, which entered into force on 1 December 2009. The study was approved by the Ethical Committee for Animal Research of the Institute of Cytology of the Russian Academy of Science (Protocol No. 3; date of approval 28 June 2016) and the Animal Care and Use Committee at the L.A. Orbeli Institute of Physiology NAS RA (Protocol No. 07.02.2025/1; date of approval 19 April 2025).
We would like to express our gratitude to Mr. Yury Filippov for his assistance with the analysis of rat ECGs.
These studies have been supported by the Higher Education and Science Committee of the Republic of Armenia, grant # 23/2PostDoc-1F001, and the ERA Chair Project (NAR-SAR-IPH) funded by the European Union.
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/FBL47046.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.