




 , Larwsk Hayann 1, Juçara Gastaldi Cominal 1, Juliana do Rosario Silva de Sousa 1, Ana Paula Ramos 1, Saida Mebarek 2, Pietro Ciancaglini 1,*
, Larwsk Hayann 1, Juçara Gastaldi Cominal 1, Juliana do Rosario Silva de Sousa 1, Ana Paula Ramos 1, Saida Mebarek 2, Pietro Ciancaglini 1,*1 Departamento de Química, Faculdade de Filosofia, Ciências e Letras de Ribeirão Preto–FFCLRP, Universidade de São Paulo–USP, Ribeirão Preto, SP 140400-900, Brazil
2 Université Lyon 1, CNRS, Institut de Chimie et Biochimie Moléculaires et Supramoléculaires (ICBMS), UMR 5246, Lyon, France
Abstract
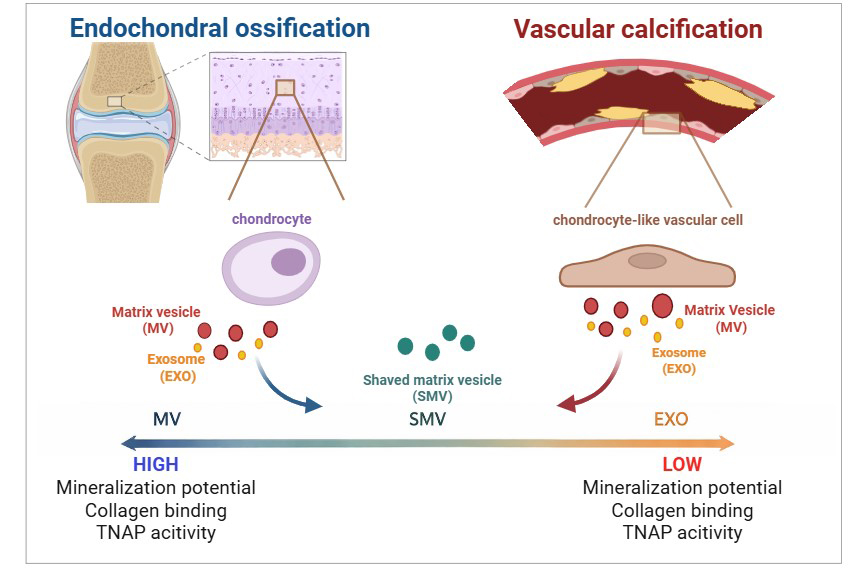
Pathological calcification of soft tissues is a hallmark of several diseases, including cardiovascular disorders and osteoarthritis. Macrocalcifications formed under pathological conditions share key features with physiological endochondral ossification. The initiation and progression of pathological calcification involve the transdifferentiation of resident soft-tissue cells into chondrocyte-like cells, which subsequently undergo hypertrophy. These hypertrophic cells release extracellular vesicles, including small-sized vesicles (exosomes, EXOs) and a specialized class of matrix-bound extracellular vesicles known as matrix vesicles (MVs). Previous studies have demonstrated that EXOs and MVs derived from the same mineralizing cells differ in lipid and protein composition, as well as in biological function.
In this study, we investigated the biochemical and physicochemical properties of EXOs and MVs, with particular emphasis on the role of the protein corona in modulating MVs mineralization capacity and collagen-binding ability. EXOs were directly purified from the extracellular medium, while MVs were isolated from a murine vascular smooth muscle cell line using enzymatic treatment. These vesicles were compared with those obtained from chondrocytes. To assess the contribution of the protein corona, MVs were treated with a high-ionic-strength buffer to remove surface-associated proteins, generating shaved matrix vesicles (SMVs).
EXOs, MVs, and SMVs displayed distinct electrophoretic protein profiles. Modulation of tissue-nonspecific alkaline phosphatase activity and turbidimetry assays indicated that SMVs retain mineralization capacity but exhibit delayed kinetics and reduced efficiency compared with native MVs.
These findings demonstrate that the protein corona plays a critical role in regulating MVs functionality, particularly by modulating mineralization efficiency and matrix interactions. This study establishes a versatile two-cell model platform for investigating pathological calcification and provides mechanistic insights into the regulation of hypertrophic chondrocyte-like cells, supporting the development of targeted therapeutic strategies.
Graphical Abstract
Keywords
- extracellular vesicles
- matrix vesicles
- vascular smooth muscle cells
- vascular calcification
- biomineralization
- protein corona
Bone mineralization, one of the most sophisticated biological processes, allows minerals to deposit within an organic matrix in a highly organized manner. This process underlies the outstanding physical and chemical properties of the bone, including its high mechanical strength and controlled ionic exchange. Extracellular matrix (ECM) mineralization is regulated by osteocompetent cells, such as mature osteoblasts, hypertrophic chondrocytes, cementoblasts, and odontoblasts. Mineralizing cells release different types of extracellular vesicles (EV), including small vesicles, enriched in exosomes (EXO) and matrix vesicles (MV), which exhibit distinct biochemical properties. Specifically, EXO have been considered the most important vesicles in cell-cell communication, while MV display high affinity for collagen fibers (hence the name matrix vesicles) that have biochemical machinery for accumulating phosphate and calcium [1, 2, 3, 4, 5].
MV usually consist of spherical, bilayered microstructures with diameters ranging from 100 to 300 nanometers. MV behave as nanoreactors and can initiate calcium phosphate nucleation, which facilitates mineral growth and propagation along collagen fibers. MV are the only EV that can bind to collagen [6, 7, 8, 9].
MV-mediated apatite nucleation relies on specific machinery. Such machinery consists of enzymes and transporters that are attached to the membrane and within the MV lumen. Calcium (Ca2+) and phosphate (Pi) ions are essential for this process and for MV biogenesis [10]. Proteomic analyses have identified key components in the MV membrane, which include the ectoenzyme GPI-anchored tissue nonspecific alkaline phosphatase (TNAP) and others [6, 11]. These proteins work synergistically to favor Ca2+ uptake and Pi acquisition. A complete review of MV machinery and the biophysical aspects of biomineralization can be found elsewhere [12]. Besides apatite nucleation and collagen mineralization, MV are implicated in other aspects of mineralized tissue physiology, such as osteogenic differentiation [1, 13].
EXO, the other EV subtype, is purified from culture media or biological fluids without the need for digesting collagenase. EXO are released into the ECM when intracellular multivesicular bodies with cell membrane diameters of 30–150 nm are fused [14, 15, 16, 17]. In summary, exosomes (EXO) originate from the inward budding of the endosomal membrane, resulting in the formation of intraluminal vesicles within multivesicular bodies. These multivesicular bodies function as central hubs in the cellular recycling system and can fuse with the plasma membrane to release exosomes into the extracellular space. Although exosomes share a common set of proteins, a subset is specific to the cell of origin, reflecting both cell type and pathological conditions. These unique proteins can be used to identify and to characterize EXO [18, 19, 20]. Vesicles can also acquire a protein corona, which modulates biological functions, including immune responses, coagulation, infections, diseases, and biomineralization [21, 22]. The protein corona, i.e., soluble proteins adsorbed at the vesicle surface, and other biological factors present in the vesicle microenvironment modify the physicochemical properties, thereby regulating how the vesicle interacts with cells and organelles, to target delivery and biodistribution [23]. Formation of a protein corona and its biological effects have been described for synthetic nanoparticles [24] and EV other than MV [23, 25, 26, 27, 28]. Cominal et al. (2025) [7] were the first to demonstrate that the protein corona is important for MV isolated from chick embryos to function properly during biomineralization. These authors revealed that the presence and composition of a protein corona enhance the TNAP enzymatic activity, thereby boosting mineralization and mineral propagation in the ECM [7].
Recently, studies have revealed that bone-related cells, like osteoclasts, osteoblasts, osteocytes, and bone marrow mesenchymal stem cells, also release EXO [29, 30, 31, 32]. Such bone-derived EXO play pivotal roles during bone remodeling: they transfer biologically active molecules to target cells, which favors osteoclast and osteoblast differentiation. Although the exact biological functions of EXO in the mineralization process have not been fully deciphered, EXO play a vital role in many cellular processes, such as cell-cell communication, coagulation, antigen presentation, waste management, and protein and nucleic acid transfer. As such, EXO contribute to understanding cellular physiology and pathology and can potentially be translated into various clinical applications, including disease diagnosis and prognosis [29, 33, 34, 35, 36, 37].
Certain pathologies can result in aberrant mineral deposition within soft tissues. Such aberrant deposition is named ectopic mineralization or pathological calcification. Ectopic mineralization is common in the cardiovascular system [38, 39], kidneys [40], and bone joints [41]. Notably, ectopic artery mineralization is often associated with reduced bone mineral density and disturbed bone turnover. Among the most common ectopic mineralization sites, vascular calcification (VC) has been the most explored. VC is characterized by blood vessels with altered nature due to accumulated calcium phosphate. VC reduces vascular elasticity and increases blood pressure [41, 42]. The molecular determinants that trigger VC have been investigated [43, 44], and inflammation is the common denominator [45, 46]. A review has underlined the role played by inflammation in the initial steps of ectopic mineralization [41]. The mechanisms that promote VC initiation and progression resemble the mechanisms of physiological bone formation and include osteogenic/chondrogenic trans-differentiation, reduced availability of calcification inhibitors, EV release, and extracellular matrix remodeling. Although the role EV in physiological mineralization has been described, how they participate in VC remains poorly explored. EV may promote or inhibit VC, communicate with cells and organs to regulate VC, or serve as therapeutic agents in VC [47]. In summary, EV can be easily detected in body fluids and may become a new therapeutic agent, a vehicle for drug administration, a diagnostic and prognostic biomarker, and a potential therapeutic target in the future.
Here, we report the characterization of two distinct types of extracellular vesicles (EVs) isolated from mouse-derived vascular smooth muscle cells (MOVAS) cultures under osteogenic conditions: exosomes (EXO), defined as free vesicles present in the culture medium that do not adhere to collagen fibers, and MV, which are released following the digestion of collagen fibers [48, 49, 50, 51]. To provide a physiological reference, chondrocyte-derived EXO and MV were included as a model of healthy biomineralization. Furthermore, we investigated the impact of protein corona removal from MV derived from both cell models on their physicochemical and biochemical properties, as well as on their ability to promote biomineralization. Advancing our understanding of these mechanisms is critical for the development of effective therapeutic strategies aimed at preventing and treating pathological calcification.
All the aqueous solutions were prepared by using ultrapure dust-free water from
a Milli-Q® system (resistivity 18.2 M
All the experiments were carried out according to the guidelines of the French Ministère de l’Agriculture (n° 87-848) and the E.U. Council Directive for the Care and Use of Laboratory Animals issued on November 24, 1986 (86/609/EEC). The experiments that involved animals were performed under authorization n°69-266-0501 (INSA-Lyon, DDPP-SV, Direction Départementale de la Protection des Populations–Services Vétérinaires du Rhône). MLC (n°692661241), AG (n°69266332), and COS (n°69266257) hold special licenses to experiment on living vertebrates; the licenses were issued by the French Ministry of Agriculture and Veterinary Service Department of France. All the procedures involving animal euthanasia were approved by the ethics committee of FFCLRP-Brazil (protocol 24.1.298.59.5.).
The experiments were carried out on animals euthanized by dislocation of the
cervical vertebra, which did not require surgery. Primary chondrocytes (CHONDRO)
were isolated from newborn (aged 5–6 days) SWISS mice by conducting successive
enzymatic digestion of the knee and femoral head cartilage, as described
previously [52, 53]. Mycoplasma testing was performed by PCR, and all samples
yielded negative results. Additionally, flow cytometry was conducted to confirm
the cellular phenotype, validating that the cells correspond to primary
chondrocytes based on their surface marker profile. The cells were then cultured
in DMEM (Dulbecco’s Modified Eagle Medium, Gibco, Thermo Fisher Scientific,
Logan, UT, USA) standard growth medium containing 10% (v/v) Fetal Bovine Serum
(FBS, Gibco, Thermo Fisher Scientific, Logan, UT, USA), 100 U mL-1
penicillin (Gibco, Thermo Fisher Scientific, Logan, UT, USA), and 100 µg
mL-1 streptomycin (Gibco, Thermo Fisher Scientific, Logan, UT, USA) at 37
°C, in humidified atmosphere of 95% air and 5% CO2. Cell
differentiation was achieved in osteogenic medium by supplementing the growth
medium with 50 µg mL-1 ascorbic acid and 10 mM
MOVAS (ATCC® CRL-2797™, ATCC, Manassas, VA, USA) were cultured in high
glucose (4.5 g
A differential ultracentrifugation method was used to isolate and to purify MV
from the ECM, as described previously [54, 55, 56]. Briefly, after the cells were
cultured for a certain time, they were washed with phosphate buffered saline
(PBS), pH 7.4. Then, MV were released from the ECM by enzymatic digestion with
2.5 mg
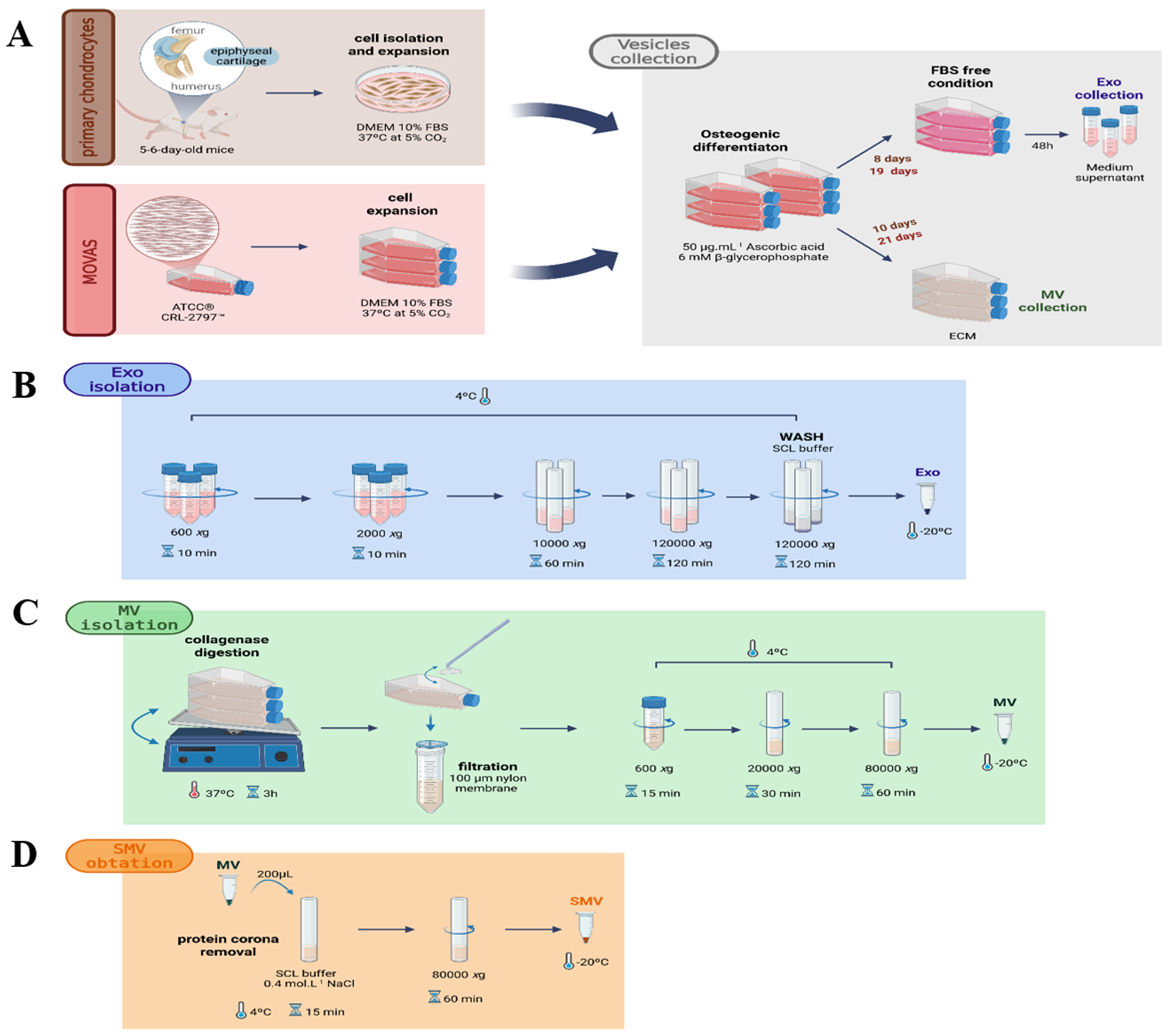
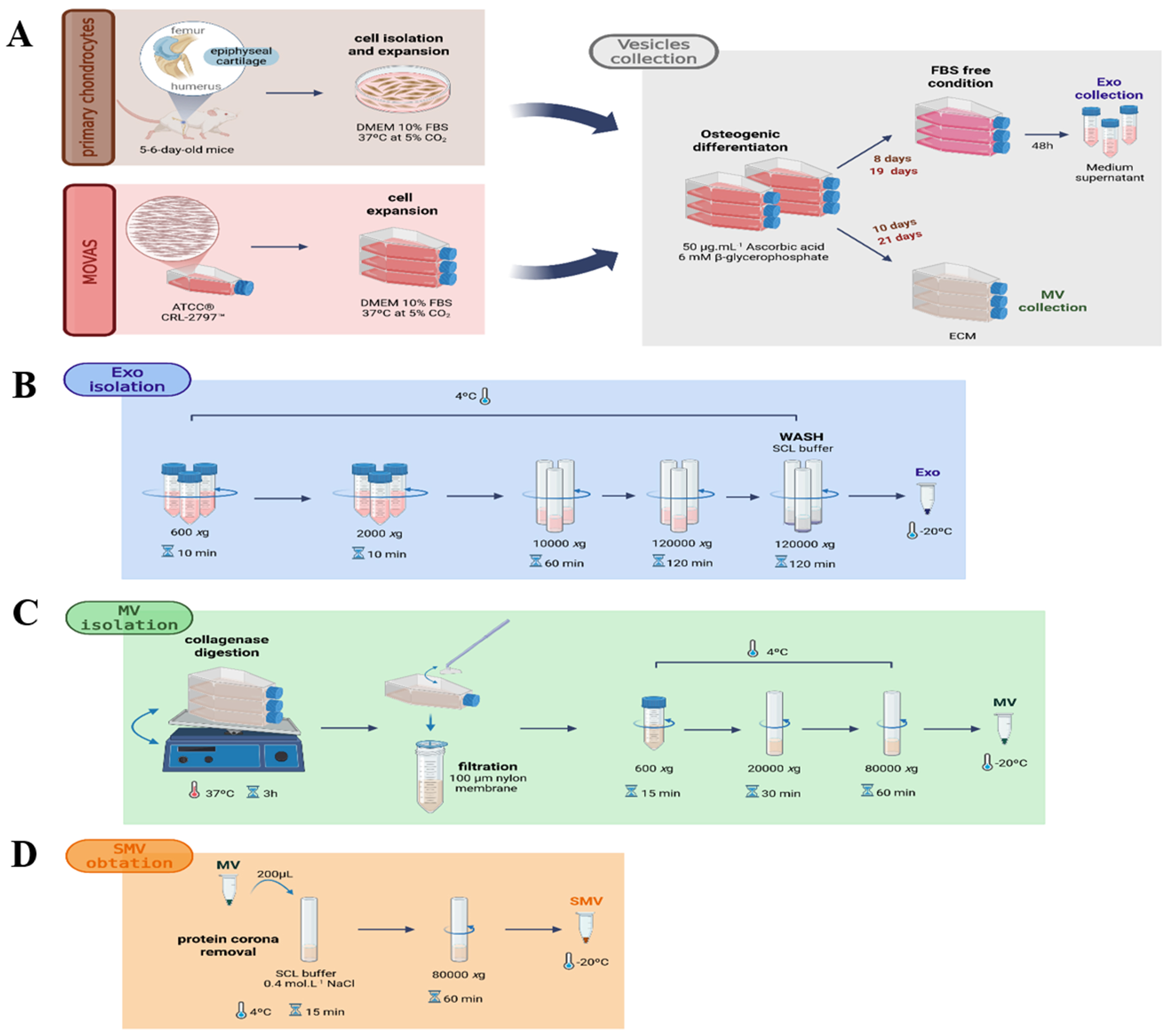 Fig. 1.
Fig. 1.
Experimental outline. (A) Schematic procedure for isolating primary chondrocytes from 5–6-day-old mice and MOVAS cells: Exosomes (EXO) isolated from culture medium and matrix vesicles (MV) were obtained following collagenase digestion. (B) EXO isolation from chondrocyte and MOVAS conditioned media. (C) Native MV isolation from chondrocytes and MOVAS cells. (D) Workflow for the generation of shaved matrix vesicles (SMV) following protein corona removal. Procedures are detailed in Materials and Methods. Created with BioRender.com.
Isolated native MV (200 µL) were incubated with 200 µL
of SCL buffer, pH 7.6, containing 0.4 mol
Exosomes (EXO) were isolated from cell culture supernatants using 10%
exosome-depleted fetal bovine serum (Gibco, Thermo Fisher Scientific, Logan, UT,
USA) and differential centrifugation, as recommended by the MISEV guidelines [58, 59] and as previously described by Yu et al. (2021)
[60]. After 24 h of culture in exosome-depleted medium, 10 mL of conditioned
medium was collected and sequentially centrifuged at 300
EV had their mean diameter and zeta potential determined by Dynamic Light Scattering (DLS) on ZetaSizer Nano ZS (Malvern Panalytical, Malvern, UK). The EV concentration was analyzed by Nanoparticle Tracking Analysis (NTA) on NS3000 (Malvern, UK). Atomic Force Microscopy (AFM) was employed to investigate EV morphology. For this purpose, an SPM-9600 Scanning Probe Microscope (Shimadzu Co., Kyoto, Japan) equipped with silicon probes (hardness: 34–50 N/m-1, resonance frequency: 324–369 Hz) and operating in the intermittent mode was used. Samples were prepared on freshly cleaved mica with 5.0 wt.% glutaraldehyde [7]. EV morphology and diameter were further examined by Transmission Electron Microscopy (TEM) on JEOL JEM-100 CXII. To record TEM images, an EV drop was dried on a conductive polymer-coated copper grid, treated with 1% phosphotungstic acid (PTA) for 15 min, and imaged. The total protein content in the EV pellets was measured with a Micro BCA Protein Assay Kit (Pierce, Thermo Scientific, Rockford, IL, USA) according to the manufacturer’s instructions; Bovine Serum Albumin (BSA) was employed as standard. To this end, pellets containing EV were resuspended in PBS containing 0.25% Triton X-100 (Fluka, Sigma-Aldrich). Samples and standards were mixed with working reagent and incubated at 37 °C for 30 min. Then, 10 µL of supernatant was completed to 200 µL of reactive medium. The reaction was stopped by thermal shock (plunging into ice). Absorbance was measured at 562 nm on a TECAN® Infinite M200 Pro microtiter plate reader. Calibration was obtained by using BSA as standard.
On the last day of differentiation, EV were harvested in 0.2% Nonidet P-40,
disrupted by sonication, and centrifuged at 2000
EV were lysed on ice with radio-immunoprecipitation assay buffer (RIPA; 25 mM Tris-HCl, 150 mM NaCl, 1 wt.% NP-40, 1 vol.% sodium deoxycholate, and 0.1 wt.% sodium dodecyl sulfate (SDS), pH = 7.4) for 30 min. The protein content was determined using the bicinchoninic acid (BCA, Thermo Fisher Scientific Inc., USA) assay, and samples were mixed with reducing Laemmli buffer (Bio-Rad, California, USA) and boiled in reducing Laemmli buffer (Bio-Rad, Hercules, CA, USA) for 5 min. Equal amounts of protein were loaded onto 12% SDS–polyacrylamide gels for electrophoretic separation. Proteins were then transferred to nitrocellulose membranes (GE Healthcare, Chicago, IL, USA), and the membranes were blocked in 5 wt.% BSA at room temperature for 2 h. To assess the efficiency and uniformity of protein transfer, membranes were stained with Ponceau S (Sigma Aldrich, USA) immediately after transfer by incubation in 0.1% Ponceau S solution (5% acetic acid) under gentle agitation at room temperature for 5 min. Membranes were then destained with Tris-buffered saline containing 0.05% Tween-20 (TBST; 50 mM Tris, 150 mM NaCl, pH 7.5) and rinsed with distilled water. Primary antibodies for TNAP (ALPL-Invitrogen, Thermo Fisher Scientific Inc., USA; 1:1000), CD9 (Abcam, UK; 1:1000), and Alix (Abcam, UK; 1:1000) were incubated at 4 °C overnight. After washing, blots were incubated with horseradish peroxidase (HRP)-conjugated goat anti-mouse or goat anti-rabbit secondary antibodies (Sigma-Aldrich, 1:5000) for 1 h at room temperature. Immunoreactive bands were detected using an enhanced chemiluminescence (ECL) detection system, and images were acquired using the ChemiDoc XRS+ imaging system (Bio-Rad).
The vesicles were incubated using 20 µg of total protein as
standard concentration in 96-well plates, for 22 h, at 37 °C, in SCL
buffer containing 2 mmol
Type I collagen was extracted from mouse tails and concentrated to approximately
10 mg
Data are presented as the mean
We characterized MV by multiple techniques to examine their biological and physicochemical properties (Summarized in Table 1) and to investigate how they interact with collagen fibrils. To investigate how the protein corona contributes to MV-driven mineralization, we removed the protein corona from the MV surface by using a buffer with high ionic strength, to obtain shaved matrix vesicles (SMV).
| Parameters | CHONDRO-MV | CHONDRO-SMV | MOVAS-MV | MOVAS-SMV |
| NTA (particle/mL) | 5.2 |
2.7 |
1.6 |
1.7 |
| Protein (µg/mL) | 3. 31 |
1.82 |
1.44 |
1.00 |
| µg protein/particle ×10–9 | 6.30 |
6.70 |
9.20 |
6.30 |
| TNAP specific activity (U/mg) | 29.00 |
14.00 |
1.34 |
0.70 |
| DLS diameter (nm) | 173.73 |
253.10 |
213.85 |
253.60 |
| Zeta potential (mV) | –28.40 |
–23.50 |
–30.40 |
–21.10 |
| AFM diameter (nm) | 201.00 |
224.00 |
188.00 |
316.00 |
| AFM medium height (nm) | 2.12 |
1.14 |
1.25 |
1.06 |
| AFM ratio roughness (nm) | 0.76 |
1.02 |
0.81 |
1.24 |
AFM, Atomic force microscopy; DLS, dynamic light scattering; NTA, nanoparticle
tracking analysis; TNAP, tissue nonspecific alkaline phosphatase. Data reported
as the mean
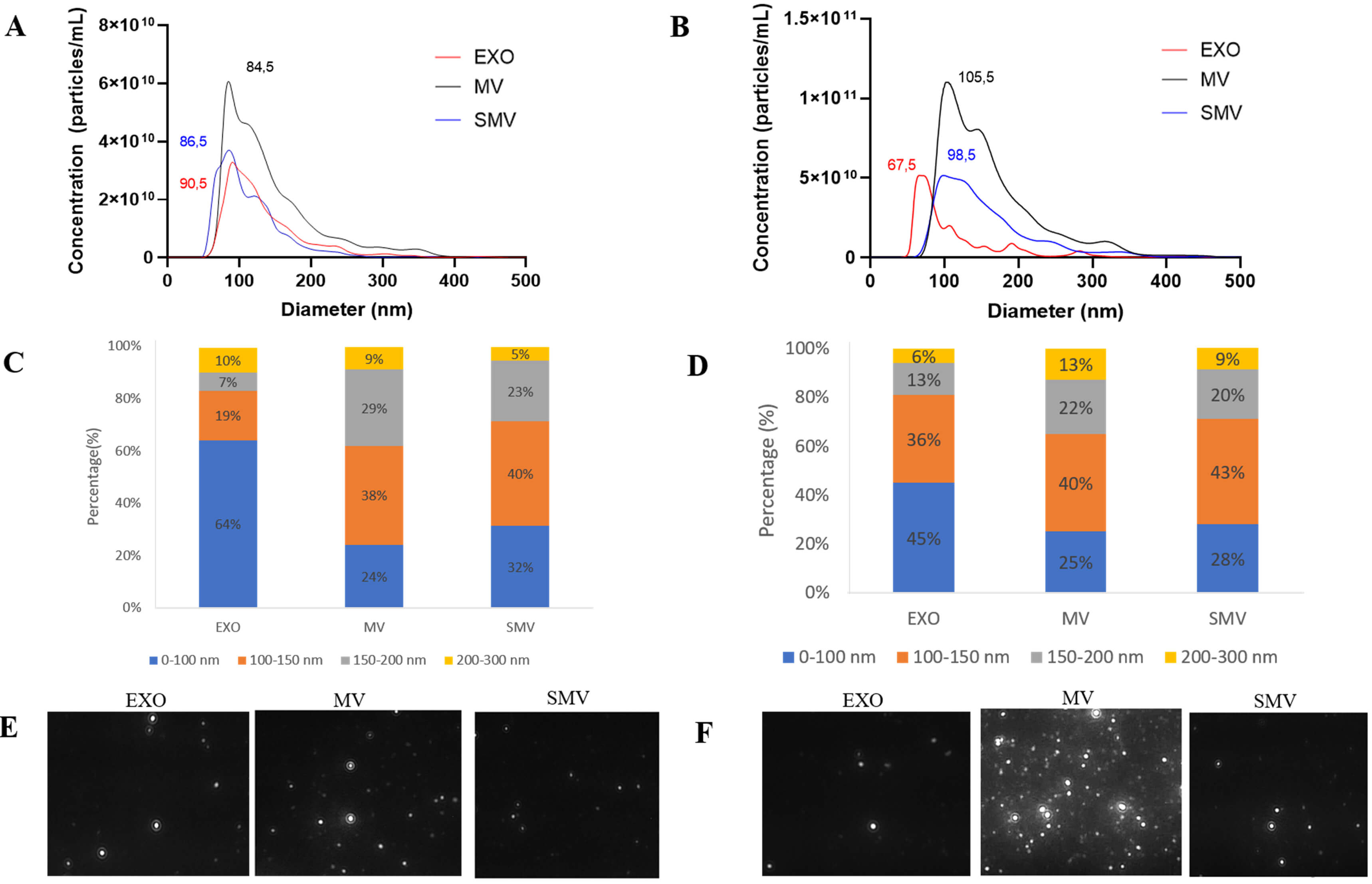
NTA analysis revealed that CHONDRO-MV and MOVAS-MV presented similar size
distribution profile, and that the predominant particle size (
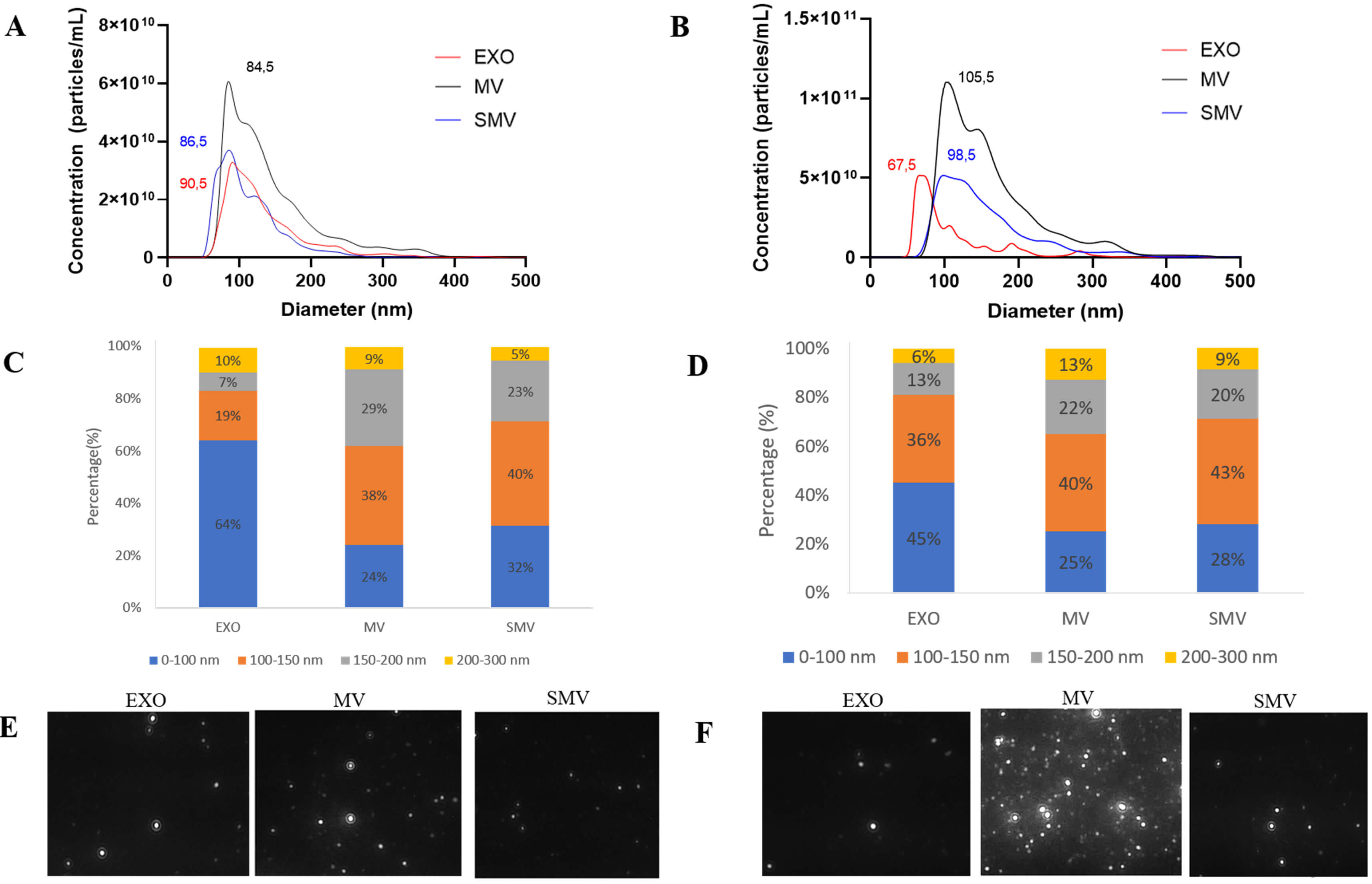 Fig. 2.
Fig. 2.
Biophysical characterizations of extracellular vesicles obtained from chondrocyte and MOVAS cells, respectively. (A–D) Size distribution as measured by nanoparticle tracking analysis (NTA NS3000, Malvern). (E,F) Screenshot of the corresponding video for the exosomes (EXO), matrix vesicles (MV), and shaved matrix vesicles (SMV).
In the case of CHONDRO and MOVAS, MV exhibited the highest protein content per particle, while SMV displayed the lowest protein content per particle (Table 1). Using a solvent with high ionic strength during centrifugation removed 41% and 31% of peripheral proteins from CHONDRO-MV and MOVAS-MV, respectively, which was also consistent with previous findings [7].
Zeta potential analysis helped to assess surface charge. Removing the protein corona increased the zeta potential from –28.4 mV in CHONDRO-MV to –23.5 mV in CHONDRO-SMV, and from –21 mV in MOVAS-MV to –30 mV in MOVAS-SMV. These shifts indicated that MV and SMV had different surface protein composition.
We used TEM to investigate EV morphology (Fig. 3). EV were spherical and displayed disc-shaped and cup-shaped structures. The diameters ranged from 170 to 250 nm, which agreed with NTA.
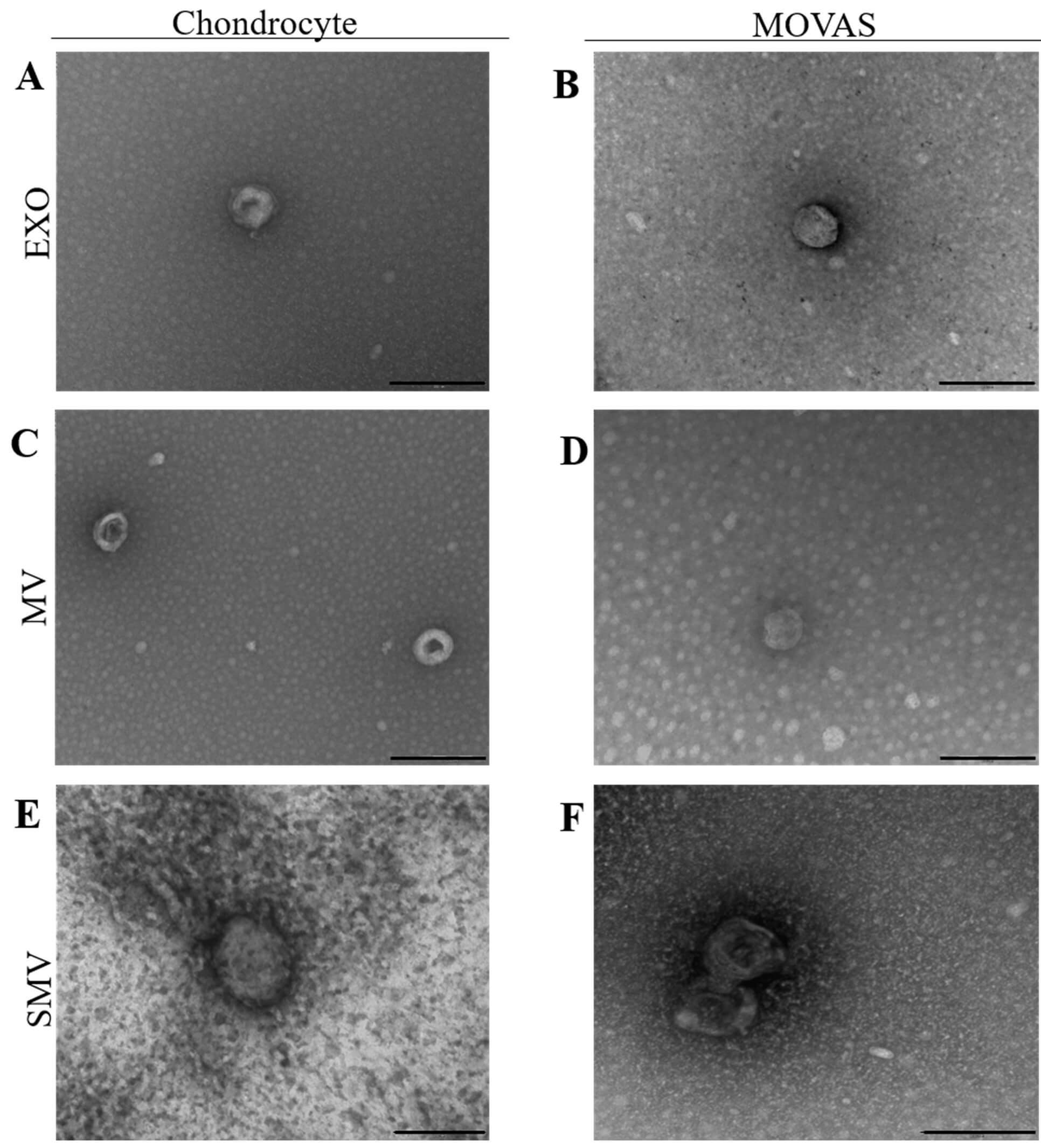 Fig. 3.
Fig. 3.
Transmission electron microscopy analysis of extracellular vesicle samples obtained from chondrocyte and MOVAS cells, respectively. TEM images of the vesicles (A,B) exosomes (EXO) (C,D) native MV (E,F) shaved MV (SMV). The black scale bar represents 200 nm.
The morphology of the vesicles was also analyzed using AFM (Table 1 and Fig. 4). In general, EV placed onto mica surfaces displayed flat, round morphologies. Topographic analysis revealed that MV were smaller than SMV, which corroborated with the DLS and TEM findings. As illustrated in Fig. 4, the overall architecture of MV showed a circular structure with particle populations around 200 nm. CHONDRO-SMV and MOVAS-SMV measured 220 and 315 nm, respectively. SMV had 40% larger diameter than MV for both CHONDRO and MOVAS, which could be attributed to SMV partially aggregating in solution because they do not have the protein corona. The height, amplitude of cantilever oscillation, and phase images determined by AFM highlighted that MV and SMV exhibited different surface topography and biophysical contrast. SMV displayed less rough surface than MV. Notably, CHONDRO-SMV and MOVAS-SMV had roughness of 1.02 and 1.24 nm, respectively. When we evaluated surface topography, a critical yet often underexplored EV biophysical feature, by AFM phase imaging, we detected differences between the MV and SMV surfaces, as indicated by the presence of protrusions (darker areas on the EV surface), which were associated with distinct viscoelastic properties (Table 1, Fig. 4).
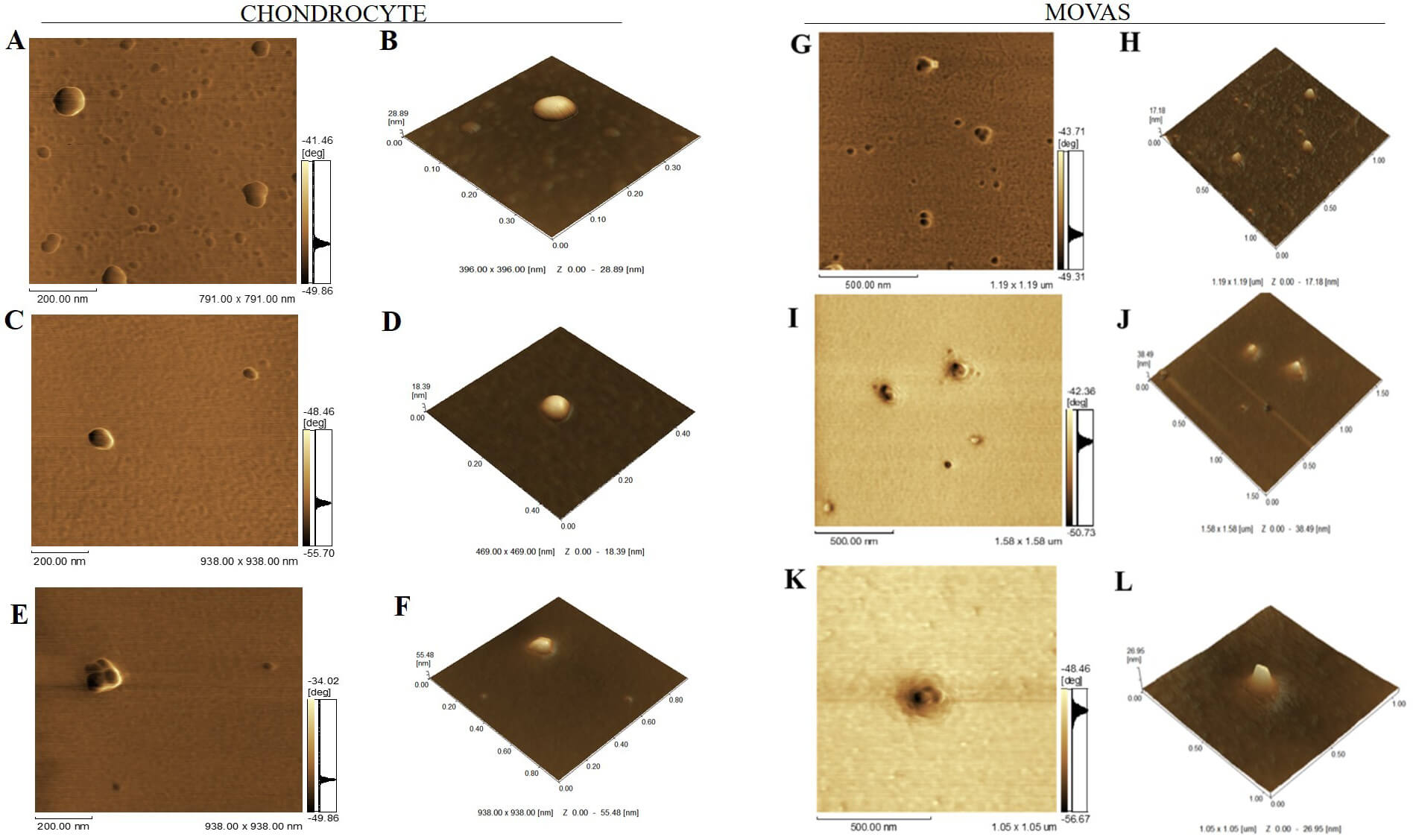 Fig. 4.
Fig. 4.
Atomic force microscopy (AFM) micrographs of vesicles groups. (A,B) EXO, (C,D) MV, and (E,F) SMV for chondrocytes and (G,H) EXO, (I,J) MV, and (K,L) SMV for MOVAS. (A,C,E,G,I,K) Phase image showing short range of phase shift in vesicle topology, revealing high protein complexity . Scale bars: 200 nm (A,C,E) and 500 nm (G,I,K). (B,D,F,H,J,L) 3D topographic image showing vesicle topology. Images were obtained with a Shimadzu SPM-9600 Scanning Probe Microscope (Shimadzu, Japan). For each sample, 100 vesicles were analyzed.
We observed different biochemical properties for the cell models (CHONDRO and MOVAS) and their respective MV subtypes (MV and SMV). The presence of TNAP was observed in the cell lysate, MV and SMV isolated from both MOVAS and CHONDRO cells. Notably, CHONDRO-SMV exhibited 50% lower TNAP activity than CHONDRO-MV, and MOVAS-SMV exhibited ~48% lower TNAP activity than MOVAS-MV (Table 1, Fig. 5). This attested that the presence of soluble proteins adsorbed at the EV surface was necessary for the mineralization biochemical machinery to function. The differences between MV and SMV in terms of TNAP activity might result from changes in the steric hindrance of the TNAP active site or loss of specific interactions with the protein corona components that modulate the TNAP function (Fig. 5E).
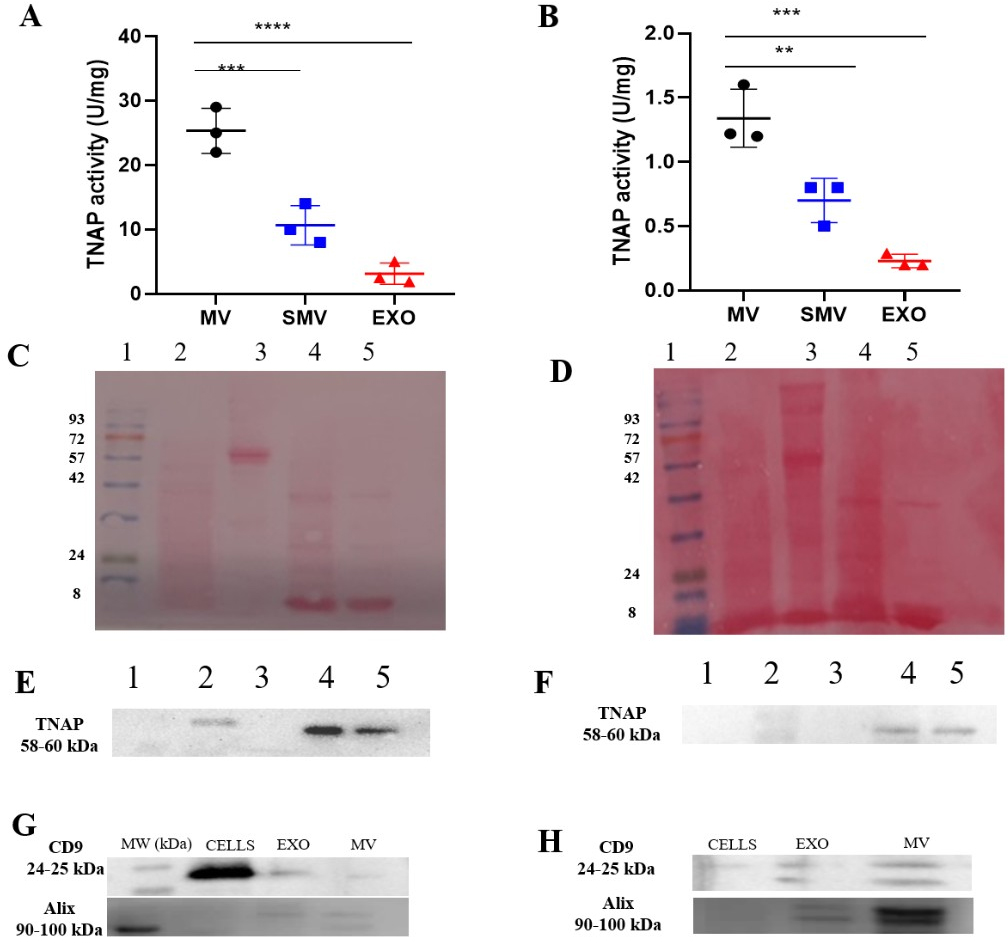 Fig. 5.
Fig. 5.
TNAP activity and protein profile analysis of
extracellular vesicles and cells. (A,B) Specific tissue non-specific alkaline
phosphatase (TNAP) activity. (C,D) Protein profiles by Ponceau Staining on 12%
polyacrylamide gel. (E,F) TNAP specific activity on 7% polyacrylamide renatured
gel. Lane numbers at the top of the gels correspond to: (1) Molecular standard;
(2) Cells; (3) EXO; (4) MV, and (5) SMV. (G,H) Western blotting analysis of EV
markers CD9 and ALIX in cells, exosomes (EXO), and matrix vesicles (MV). Multiple
statistical comparisons were performed by one-way ANOVA, **p
Conducting digestion by collagenase to isolate MV from the ECM typically yields a large amount of MV with specific TNAP activity ranging between 4 and 30 U⋅mg-1 [8, 56, 63]. Cominal et al. [7] previously described specific TNAP activity of 4.2 and 4.5 U⋅mg-1 for MV isolated from chick embryos and the corresponding SMV, respectively. The relatively small difference in specific TNAP activity is likely attributable to the methodology used by Cominal et al. (2025) [7], in which MV were extracted directly from tissue.
Western blot analysis revealed a clear enrichment of CD9 in EXO and MV fractions isolated from both MOVAS cells and primary chondrocytes, supporting the presence of small extracellular vesicles consistent with exosomes (Fig. 5G,H). CD9 was also detectable in whole-cell lysates, in agreement with the well-established intracellular localization of tetraspanins within endosomal compartments and multivesicular bodies prior to vesicle secretion.
Notably, ALIX was consistently detected in cell lysates as well as in EXO and MV fractions, reflecting its role in ESCRT-mediated vesicle biogenesis. The combined presence of EV-enriched markers, such as CD9 together with ALIX, is consistent with current MISEV 2023 guidelines for small extracellular vesicle characterization. These findings support the successful isolation of vesicle populations enriched in exosome-like EVs derived from both conditioned medium and mineralized extracellular matrix.
However, in line with MISEV recommendations, it is important to emphasize that these markers are not exclusively specific to a single EV subtype. Their detection across different fractions indicates the presence of heterogeneous and partially overlapping EV populations, suggesting that the isolated samples likely contain a mixed vesicle population rather than strictly defined subtypes. This observation directly relates to a key methodological limitation of the present study, as EV classification remains operational and dependent on the isolation strategy employed. Therefore, while our approach enables the enrichment of distinct EV fractions, it does not fully exclude co-isolation of vesicles with shared molecular signatures, which should be considered when interpreting the functional differences observed between EXO and MV.
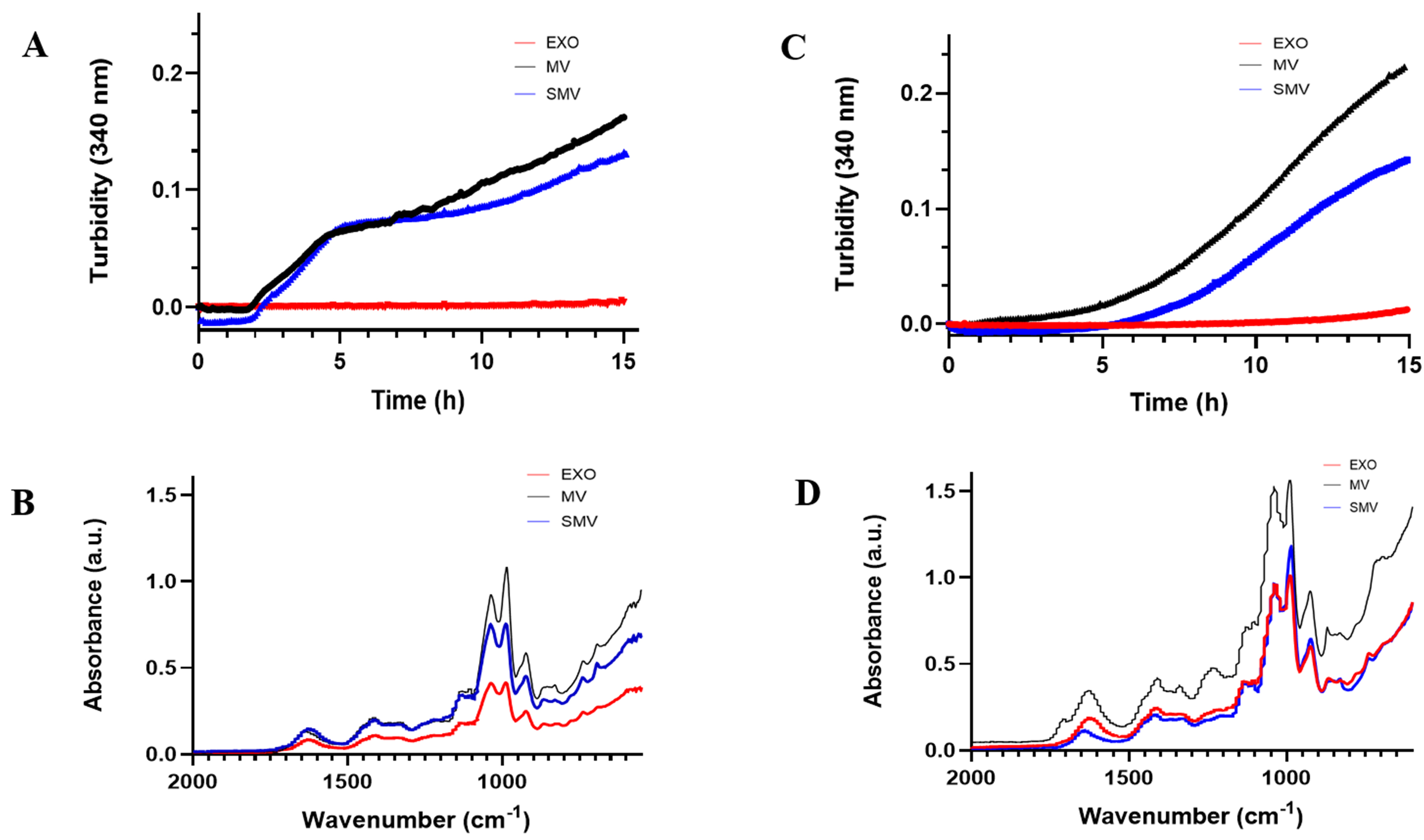
We evaluated the mineralization potential of extracellular vesicles (EVs) by performing turbidimetry assays (Fig. 6A,C), which monitor the formation and precipitation of calcium phosphate. We observed a sigmoidal curve for both MOVAS-MV and MOVAS-SMV, which indicated a single-step mineralization process (Fig. 6C). Under conditions that favored nucleation (presence of Ca2+ and ATP), mineral formation was delayed, and mineral propagation was 25% slower for MOVAS-SMV compared to MOVAS-MV. Nucleation started at 5.08 h for MOVAS-MV and 6.75 h for MOVAS-SMV. In contrast, CHONDRO-MV and -SMV underwent two-step mineralization (Fig. 6A), so the cell models (CHONDRO and MOVAS) displayed different nucleation behavior.
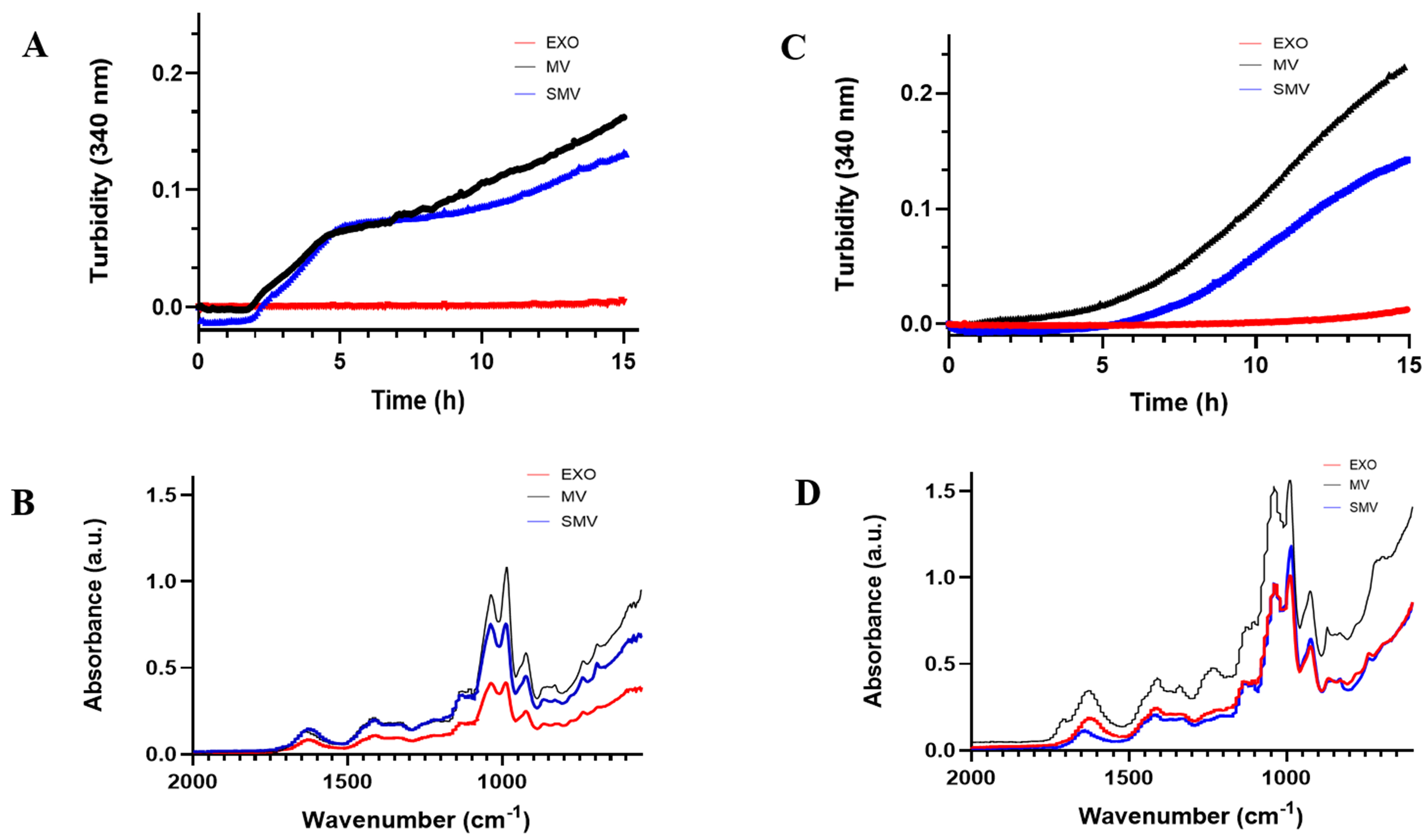 Fig. 6.
Fig. 6.
In vitro mineralization and mineral characterization of extracellular vesicle groups. (Red) exosomes (EXO); (Blue) shaved MV; (Black) native MV. (A,C) Mineralization curves in the presence of ATP. (B,D) ATR-FTIR espectra of minerals produced by vesicles in the presence of ATP (adenosine triphosphate). Data reported as the mean of triplicate measurements.
Furthermore, we used ATR-FTIR spectroscopy to characterize the mineral that was formed after we incubated the EV with SCL and ATP (Fig. 6B,D). Biological apatite, a calcium phosphate mineral with composition close to the composition of Hap.
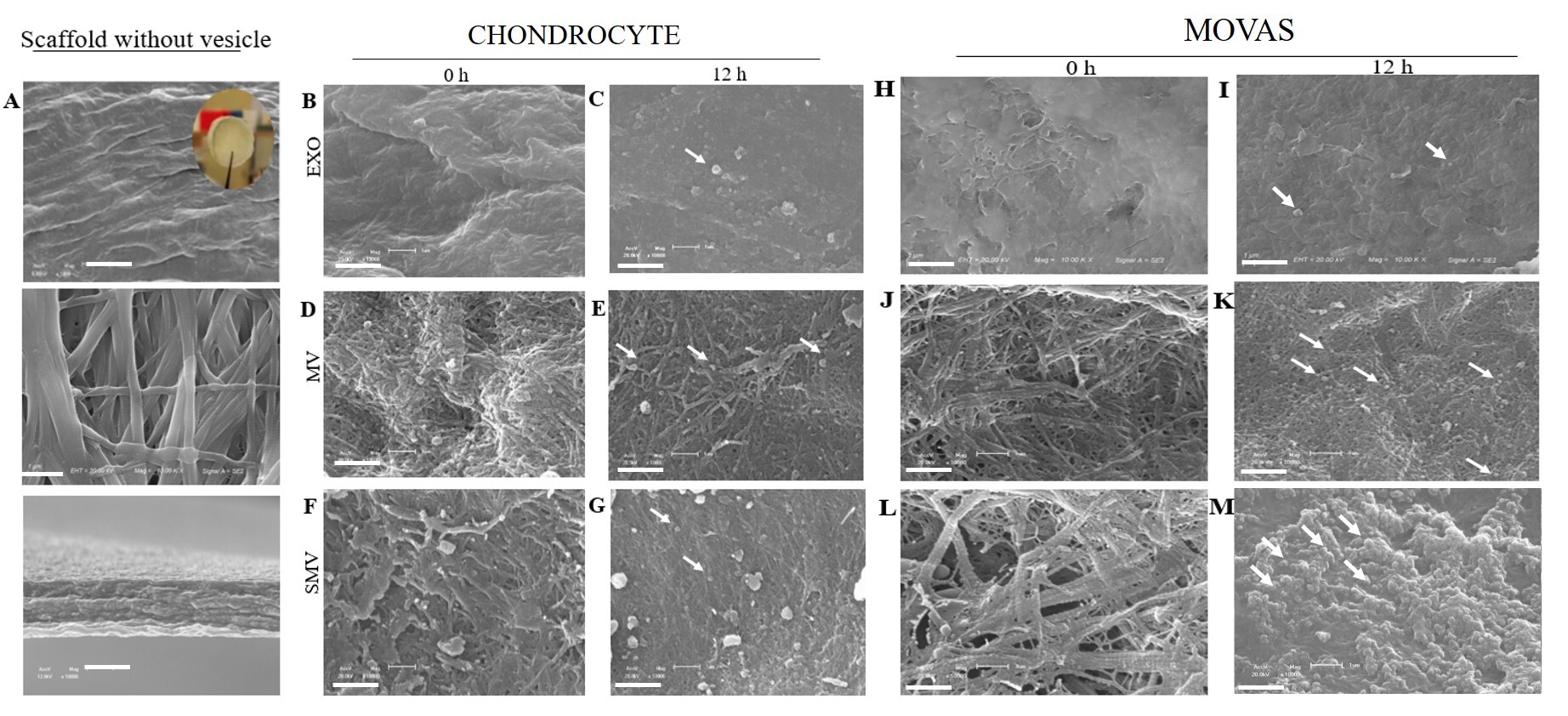
To assess whether MV and SMV were able to anchor to a collagen-rich ECM, we evaluated their specific interaction with type I collagen-based biomimetic scaffolds. In the absence of vesicles, electron microscopy of the scaffold surface revealed uniformly sized, compact collagen fibrils organized into a dense and interconnected network (Fig. 7A, upper panels). Analysis of the lateral faces of the scaffolds, obtained by transverse fracture, showed fibrils arranged in parallel arrays, closely resembling the organization observed in demineralized human bone (Fig. 7A, lower panels). After incubation of the scaffolds with ATP in the presence of MV or SMV for 12 h, clear morphological changes were observed (Fig. 7E,K). These changes were indicative of mineral propagation, which was particularly pronounced in scaffolds incubated with CHONDRO- and MOVAS-derived MVs. In scaffolds treated with CHONDRO-MVs, vesicles were found embedded within the collagen matrix (Fig. 7E), suggesting an accelerated mineralization process promoted by MV anchoring to collagen.
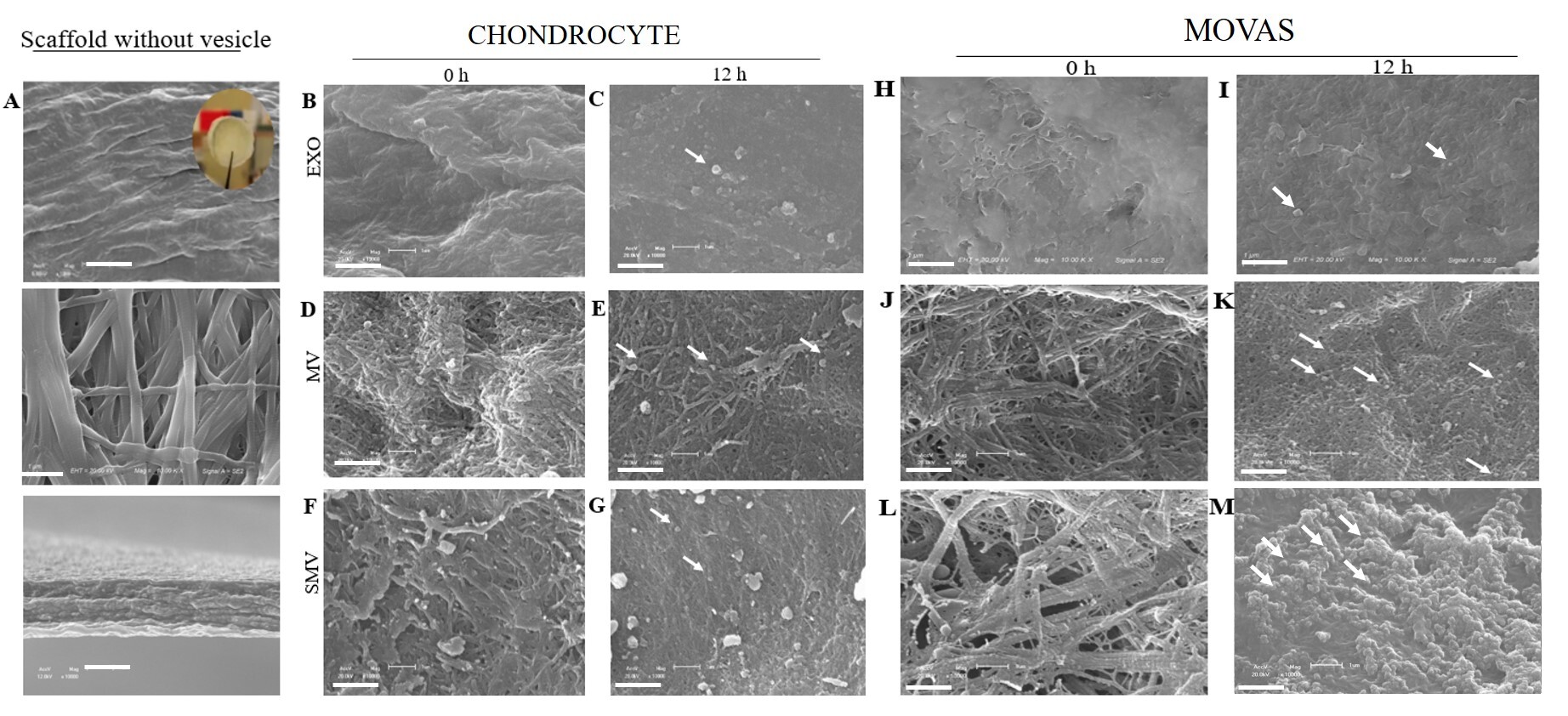 Fig. 7.
Fig. 7.
Electron microscopy images of the collagen-based scaffolds
incubated with extracellular vesicle groups isolated from chondrocytes and
MOVAS. The assay was performed on the collagen scaffold control in the absence
of vesicles (A) and immediately after incubation (0 h) with ATP for vesicles
obtained from chondrocytes (B,D,F) and MOVAS cells (H,J,L) and after incubation
(12 h) with ATP for vesicles obtained from chondrocytes (C,E,G) and MOVAS
(I,K,M). Micrographs obtained at 10,000
Interestingly, removing the protein corona did not impair the SMV anchoring capacity (Fig. 7G,M), so the ability of vesicle to bind to collagen was preserved. Anchoring of MV to collagen likely supports a significant stage of mineral deposition, notably HAp precursors, as attested by FTIR spectroscopy (Fig. 6B,D).
In agreement with the turbidimetry assays, CHONDRO-MV and CHONDRO-SMV exhibited earlier onset of mineralization and higher TNAP activity than their MOVAS counterparts (Fig. 5, Table 1). The collagen-based scaffolds embedded with MV or SMV contained agglomerates that resembled mineral structures (Fig. 7D–G), which might reflect the presence of supersaturation sites for minerals to deposit. We observed that MOVAS-derived vesicles displayed a similar behavior; however, MOVAS-MV and MOVAS-SMV exhibited the presence of numerous attached vesicles and fewer mineral clusters, which corroborates the delayed onset of mineralization compared to CHONDRO-MV, as demonstrated in the turbidimetry assay (Fig. 7K,M).
We examined the EXO mineralization potential to investigate whether EXO contribute to pathological VC. For this purpose, we characterized and compared CHONDRO-EXO and MOVAS-EXO (see Table 2).
| Parameters | CHONDRO-EXO | MOVAS-EXO |
| NTA (particle/mL) | 2.8 |
3.0 |
| Protein (µg/mL) | 2.82 |
3.22 |
| µg protein/particle ×10–9 | 1.01 |
9.30 |
| TNAP specific activity (U/mg) | 2.22 |
0.21 |
| DLS diameter (nm) | 133.40 |
132.00 |
| Zeta potential (mV) | –25.80 |
–22.10 |
| AFM diameter (nm) | 74.00 |
71.00 |
| AFM medium height (nm) | 0.80 |
0.73 |
| AFM ratio roughness (nm) | 0.52 |
0.46 |
AFM, Atomic force microscopy; DLS, dynamic light scattering; NTA, nanoparticle
tracking analysis; TNAP, tissue nonspecific alkaline phosphatase. Data reported
as the mean
CHONDRO-EXO and MOVAS-EXO had mean diameter of 133
The TEM images revealed disc-shaped particles with diameters ranging from 95 to 100 nm and clear backgrounds (Fig. 3A,B). The AFM images confirmed these observations and showed uniform round particles without significantly different height or surface roughness (Fig. 4A,B,G,H). Notably, MV and SMV but not EXO surfaces presented protrusions, which suggested different protein or lipid composition.
As expected, EXO exhibited markedly lower TNAP activity than MV. CHONDRO-EXO exhibited average TNAP activity of 2.2 U mg-1, which corresponded to 92% reduction compared to CHONDRO-MV. MOVAS-EXO displayed even lower activity (0.21 U mg-1), representing an 81% reduction relative to MOVAS-MV (Table 2). Furthermore, TNAP was not detected in EXO by Western blot analysis, suggesting that only minimal amounts of this enzyme were present in the EXO membrane, as inferred from the low but measurable enzymatic activity (Fig. 5A,B).
In agreement with these findings, EXO did not exhibit any ability to mineralize. The turbidimetry assay did not detect any calcium phosphate nucleation or formation of mineral precursors (Fig. 6). Additionally, EXO failed to anchor to collagen-based scaffolds, which is critical for minerals to deposit and to propagate. In the absence of anchorage, no mineral phase emerged (Fig. 7).
Comparison between CHONDRO-MV and MOVAS-MV agreed with findings of previous studies, which reported MV diameters ranging from 100 to 300 nm for MV isolated from mature osteoblasts and hypertrophic chondrocytes [6, 12, 22, 71, 72, 73, 74]. Cominal et al. (2025) [7] also reported average diameter of 225 nm for SMV isolated from chicken embryos. For both cell models (CHONDRO and MOVAS), the polydispersity index (PDI) was approximately 0.3, which suggested a relatively homogeneous EV population (Table 1). Additionally, the TEM images presented blurred background and more impurities due to the isolation method, which was consistent with previous observations [75, 76].
The effect of high-ionic-strength treatment on extracellular vesicles to remove protein corona, indicates that the conditions employed in this study do not compromise their physicochemical integrity (Table 1). Compared to the higher NaCl concentrations reported by Försönits et al. [77], the milder ionic strength used here was sufficient to modulate the protein corona without inducing structural damage. This is supported by transmission electron microscopy (TEM in Fig. 3) and atomic force microscopy (AFM in Fig. 4), which confirmed the preservation of the characteristic spherical morphology of the vesicles. In addition, particle size measurements obtained by dynamic light scattering (DLS) and nanoparticle tracking analysis (NTA) remained consistent, with no evidence of disruption. Together, these results demonstrate that the applied conditions effectively modify the protein corona while maintaining vesicle stability, ensuring the reliability of downstream functional analyses. In addition, Kong et al. (2020) [78] showed that crosstalk between proteins and enzymes is needed to maintain basic biological functions. In this context, the observed reduction in TNAP activity (Table 1) following protein corona removal is likely multifactorial. Increased ionic strength may directly affect TNAP conformation and catalytic efficiency, while also disrupting the membrane microenvironment and associated lipid–protein interactions. Additionally, the removal or displacement of surface-associated proteins may lead to the loss of stabilizing interactions or cofactors that contribute to optimal enzymatic activity. Considering that TNAP is a membrane-associated enzyme whose function is highly dependent on its lipid environment, even subtle alterations in membrane organization induced by ionic conditions can significantly impact its activity. Therefore, the decrease in TNAP activity observed in shaved matrix vesicles (SMVs) likely reflects a combined effect of protein corona removal and ionic strength–mediated modulation of enzyme structure and membrane context, rather than a single isolated mechanism [66].
A recent study by Li et al. [79] showed that MV serve as nucleating foci for microcalcification to initiate. The MV origin and composition determine the MV calcification potential [4, 71, 72, 80, 81]. ATR-FTIR spectroscopy to characterize the mineral formed by EV incubated with SCL and ATP (Fig. 6B,D) revealed a calcium phosphate mineral with composition close to the composition of HAp, commonly occurs in mineralized tissues [82, 83]. In mammalian tissues, substituting phosphate for carbonate and replacing calcium with another cation generates biological apatite [83, 84].
Based on our results, despite the removal of the protein corona, SMV retained
their mineralization capacity, albeit slower and with a lower mineral yield, as
observed in the turbidimetry and FTIR graphs (Fig. 6). In agreement with the
observations of Sauer and Wuthier [85], the intensity and position of P–O
absorption bands in the 1130–1030 cm-1 region provide important insights
into the maturation state of minerals formed by MV. Notably, MV exhibited a
stronger band at 1040 cm-1, corresponding to the
The lower wavenumber region of the FTIR spectrum also provided a characteristic fingerprint of hydroxyapatite (HAp), confirming the mineral nature of the deposits. Specifically, the band at 1040–1030 cm-1 corresponds to PO43- asymmetric stretching, indicative of apatite, whereas the band at 985 cm-1 is associated with HPO42-. Additionally, a weak band at 925 cm-1 was detected, attributed to P–O–P groups [85, 86]. While less crystalline mineral phases such as amorphous calcium phosphate (ACP) typically display a single broad band in this region, our spectra showed well-resolved bands, suggesting the formation of a more crystalline mineral phase. This phase could potentially mature into octacalcium phosphate (OCP) or hydroxyapatite, particularly in the presence of an intact protein corona in MV.
But important point to comment is that the collagen-based scaffolds embedded with MV or SMV contained agglomerates that resembled mineral structures (Fig. 7), which might reflect the presence of supersaturation sites for minerals to deposit. A critical function of MV is to initiate apatite formation and to facilitate apatite deposition onto ECM [13, 88, 89, 90]. Studies have demonstrated that MV have strong binding affinity for collagen fibrils, which further supports our findings that EV-ECM interactions play a pivotal role in regulating the mineralization dynamics [54, 74].
Finally, CHONDRO and MOVAS consistently and homogenously produced EXO (Table 2), which demonstrated that the isolation protocol was effective. Given that EXO biogenesis, release, and characteristics heavily depend on the cell conditions [91], the similar physical properties point out that a conserved mechanism underlies EXO formation and function. Although EXO morphologically resembled MV, EXO did not display mineralization activity. Therefore, EXO do not play a direct role in physiological or pathological mineralization. Instead, they seem to have a different function from the function of MV, which are actively involved in mineral deposition and are typically anchored to the ECM. This contrast reinforces the hypothesis that MV and EXO released by mineralizing cells serve distinct roles in mineralized tissues, with MV being structurally and functionally adapted for initiating and propagating mineralization.
This study presents some limitations that should be considered when interpreting the results. First, extracellular vesicle (EV) isolation approaches differed between populations: exosomes (EXO) were collected from conditioned medium, whereas matrix vesicles (MV) were obtained through enzymatic digestion of the extracellular matrix. Although this strategy reflects their distinct biological origins, it may introduce methodological bias, potentially affecting vesicle purity, yield, and composition. We have expanded the Discussion to better reflect the limitations of our isolation approach, in line with MISEV 2023 recommendations [59]. In particular, the distinction between EXO and MV in this study is primarily based on isolation strategy (conditioned medium vs. collagenase-released ECM vesicles), which may not fully exclude overlap between vesicle subtypes. MV fractions may therefore contain heterogeneous vesicle populations, including membrane fragments or larger vesicular structures. In addition, commonly used exosomal markers such as CD9 and CD63 may also be detected in MV preparations, further highlighting the challenge of obtaining highly pure and well-defined EV subpopulations [92]. These observations reinforce that EV classification remains operational and dependent on methodological context, as emphasized by MISEV guidelines. Furthermore, the limited yield of vesicles represents an additional technical constraint, making quantitative and high-sensitivity analyses—such as fluorescence-based approaches—more challenging and potentially affecting the robustness of comparative measurements.
Second, the study relies predominantly on in vitro assays, including turbidimetry and enzymatic activity measurements, to assess mineralization capacity. While these methods provide controlled and reproducible conditions, they may not fully recapitulate the complexity of the in vivo microenvironment, particularly with respect to extracellular matrix composition, mechanical forces, and cellular interactions.
Additionally, the use of a murine vascular smooth muscle cell line and chondrocytes may limit the direct translation of findings to human physiology and disease contexts. Species-specific differences in EV composition and mineralization processes should therefore be considered.
Finally, although differences in protein profiles and functional outcomes were identified, a comprehensive proteomic and lipidomic characterization of the vesicle populations and their coronas was not performed. Such analyses would provide deeper mechanistic insight into the molecular determinants underlying the observed functional differences.
Together, these limitations may impact the generalizability and mechanistic interpretation of the findings, and highlight the need for further studies incorporating advanced EV characterization, in vivo models, and human-derived systems.
In summary, while we acknowledge the importance of functional studies, we respectfully emphasize that the novel characterization of shaved matrix vesicles and their protein corona represents a meaningful and necessary contribution to the field, particularly given the limited data currently available for EVs derived from mineralizing cells. We thank the reviewer for this valuable suggestion, which has helped us better clarify the scope, significance, and future directions of our work.
In conclusion, this study provides a comprehensive comparative analysis of EXO and MV derived from murine vascular smooth muscle cells and primary chondrocytes, highlighting their distinct biochemical, structural, and functional properties in the context of mineralization. By establishing a reproducible method to remove the protein corona from MV, we generated shaved matrix vesicles (SMV) and directly assessed the contribution of the protein corona to MV-mediated mineralization and extracellular matrix interactions.
Our results demonstrate that MV exhibit robust mineralization capacity and an intrinsic ability to anchor to collagen fibrils, key features shared with physiological endochondral ossification and pathological soft tissue calcification. Removal of the protein corona reduced TNAP activity and delayed mineralization kinetics, yet SMVs retained functional characteristics such as collagen-binding capacity, indicating that the protein corona modulates rather than is strictly required for MV function. In contrast, EXO displayed distinct protein profiles and biophysical properties and lacked mineralization potential and matrix anchoring, highlighting their divergent biological roles despite originating from the same mineralizing cells.
These findings reinforce the concept that pathological calcification recapitulates key aspects of hypertrophic chondrocyte behavior and vesicle-mediated mineralization observed during skeletal development. The two-cell model presented here provides a versatile platform for mechanistic studies of ectopic mineralization and offers a foundation for future investigations aimed at targeting the protein corona or vesicle properties as potential therapeutic strategies.
Finally, we acknowledge the limitations of this study, including the use of a VSMC cell line instead of primary vascular cells, the absence of an in vivo ectopic calcification model, and the lack of functional assays evaluating cellular responses to EVs. These limitations will be addressed in future studies to further elucidate the biological roles and therapeutic potential of MV and SMV.
EV, extracellular vesicles; EXO, exosomes; MV, matrix vesicles; SMV, shaved matrix vesicle; SCL, synthetic cartilage lymph; TNAP, Tissue-nonspecific alkaline phosphatase.
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
MTM (Formal analysis, Investigation, Methodology, Writing—review & editing); JGC (Formal analysis, Data curation, Writing-review); LH (formal analysis, review); JRSS (Formal analysis); SM, APR, and PC (Conceptualization, Resources, Supervision, Writing—review & editing). All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
All the experiments were carried out according to the guidelines of the French Ministère de l’Agriculture (n° 87-848) and the E.U. Council Directive for the Care and Use of Laboratory Animals issued on November 24, 1986 (86/609/EEC). The experiments that involved animals were performed under authorization n°69-266-0501 (INSA-Lyon, DDPP-SV, Direction Départementale de la Protection des Populations–Services Vétérinaires du Rhône). MLC (n°692661241), AG (n°69266332), and COS (n°69266257) hold special licenses to experiment on living vertebrates; the licenses were issued by the French Ministry of Agriculture and Veterinary Service Department of France. All the procedures involving animal euthanasia were approved by the ethics committee of FFCLRP-Brazil (protocol 24.1.298.59.5.). All animal experiments were conducted in accordance with national and international guidelines for the care and use of laboratory animals and were approved by the relevant institutional and national ethics committees in France and Brazil.
We thank you to Ivana Borin from the AFM Core Facility, of the FFCLRP-USP multiuser platform and to José Augusto Maulim, Maria Dolares Seabra Ferreira from the Central multi-user electron microscopy equipment unit (LMME), of the FMRP-USP.
The authors acknowledge funding from Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP), grants 2019/08568-2 (to PC); 2024/12254-1 (to MTM); 2019/25054-2 (to APR); 2022/04885-6 (to JGC); 2023/17960-9 and 2024/17743-0 (to LH); Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) grants Finance Code 001 (to APR and PC). Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq) grants 305426/2021-4 and 200506/2022-6 (to PC); and Brazil-France USP-COFECUB Uc Sv 184/20 grant.
The authors declare no conflicts of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.