



 , Caterina Gagliano 1,2, Cosimo Mazzotta 1,2, Fabiana D’Esposito 1,3, Alessandro Avitabile 4, Giuseppe Gagliano 4, Daniele Tognetto 5, Marco Zeppieri 5,6,*
, Caterina Gagliano 1,2, Cosimo Mazzotta 1,2, Fabiana D’Esposito 1,3, Alessandro Avitabile 4, Giuseppe Gagliano 4, Daniele Tognetto 5, Marco Zeppieri 5,6,*
1 Department of Medicine and Surgery, University of Enna “Kore”, Piazza dell’Università, 94100 Enna, Italy
2 Mediterranean Foundation “G.B. Morgagni”, 95125 Catania, Italy
3 Imperial College Ophthalmic Research Group (ICORG) Unit, Imperial College, NW1 5QH London, UK
4 Department of Ophthalmology, University of Catania, 95123 Catania, Italy
5 Department of Medicine, Surgery and Health Sciences, University of Trieste, 34129 Trieste, Italy
6 Department of Ophthalmology, University Hospital of Udine, 33100 Udine, Italy
Abstract
Diabetic retinopathy (DR) is increasingly recognized as a complex neurovascular degenerative disorder driven by intertwined immune and metabolic disturbances within the retinal microenvironment. Chronic hyperglycemia induces metabolic stress, mitochondrial dysfunction, and oxidative imbalance, which, in turn, activate innate and adaptive immune pathways. Key mechanisms—including complement dysregulation, microglial activation, leukostasis, cytokine and chemokine signaling, and advanced glycation end-product–mediated inflammation—contribute to endothelial injury, barrier breakdown, and progressive neuronal loss. Parallel alterations in lipid metabolism, amino acid utilization, and mitochondrial bioenergetics further amplify inflammatory cascades and shape the retinal immune landscape. This review synthesizes current evidence on how immune-metabolic crosstalk orchestrates early and late stages of DR, integrating findings from transcriptomic, proteomic, metabolomic, and epigenetic studies. We examine core signaling hubs that couple metabolic dysfunction to inflammatory amplification, including complement components, the advanced glycation end product (AGE)–receptor for AGE (RAGE) pathway, cytokine networks, and immune response regulation. Adopting a systems biology perspective, we highlight how convergent mechanisms can unify vascular, neuronal, and glial pathology under a shared framework of immune-metabolic imbalance. An extensive literature search was conducted (PubMed, accessed December 2025). By positioning DR as a model of inflammatory retinal degeneration, this review outlines a conceptual foundation for network-based diagnostics and therapeutics. Understanding the dynamic interactions among immune signaling, metabolic stress, and neurovascular instability may inform future strategies to restore retinal homeostasis and prevent vision-threatening disease progression.
Keywords
- diabetic retinopathy
- neurovascular unit
- microglia
- inflammation
- mitochondrial dysfunction
- advanced glycation end products
- complement system
- systems biology
Diabetic retinopathy (DR) has conventionally been regarded as a microvascular consequence of persistent hyperglycemia, characterized by capillary dropout, microaneurysm formation, and abnormal neovascularization. This vascular-centric model, although fundamental, is becoming inadequate to elucidate the early emergence of functional visual impairment, the disconnect between vascular lesions and neuroretinal dysfunction, and the diverse clinical trajectories seen in patients with comparable glycemic exposure. In the last ten years, accumulating experimental, translational, and clinical evidence has redefined DR as a progressive neurovascular degenerative disorder, wherein immune activation and metabolic stress interact dynamically within the retinal microenvironment, influencing disease onset and progression prior to the clinical manifestation of overt vascular pathology [1, 2, 3, 4, 5].
This reconceptualization acknowledges that chronic hyperglycemia functions not as a singular metabolic disturbance, but as a systemic catalyst for metabolic stress mechanisms that intersect with innate and adaptive immunological pathways. Chronic glucose surplus disrupts mitochondrial bioenergetics, elevates reactive oxygen species generation, and modifies redox-sensitive signaling pathways in retinal neurons, endothelial cells, Müller glia, and resident immune cells. These metabolic abnormalities trigger a condition of chronic, low-grade inflammation—commonly known as metaflammation—where nutrient-sensing pathways and immune signaling become functionally indistinguishable [6, 7, 8]. The retina, a tissue characterized by significant metabolic demands and limited regenerative capacity, exhibits immune–metabolic interactions that make the neurovascular unit especially susceptible to cumulative damage [9].
Microglial activation, complement cascade involvement, leukocyte adhesion, and pro-inflammatory cytokine production have been demonstrated in animal models and human diabetic retinas prior to clinically evident microangiopathy, suggesting early immune activation that is intimately linked to cellular metabolic abnormalities. In parallel, functional studies document neuronal dysfunction, impaired contrast sensitivity, and electrophysiological irregularities in diabetic patients without funduscopic evidence of retinopathy, consistent with a preclinical neurodegenerative phase driven by immune-metabolic stress [10, 11, 12].
The notion of DR as an inflammatory disorder is further substantiated by the discovery of immune effector pathways directly influenced by metabolic signals. The production of advanced glycation end products (AGEs) due to hyperglycemia, activation of the AGE–receptor for AGE (RAGE) axis, dysregulated lipid metabolism, and consumption of modified amino acids all influence cytokine and chemokine networks in the retina. These activities facilitate endothelial dysfunction, compromise the inner blood-retinal barrier, and exacerbate leukostasis, establishing self-perpetuating feedback loops between metabolic damage and immunological activation. Significantly, numerous pathways function independently of vascular endothelial growth factor (VEGF), elucidating the limited effectiveness of anti-VEGF therapy in a considerable fraction of patients and highlighting the necessity for more comprehensive pathophysiological models [13, 14, 15].
From a systems biology viewpoint, diabetic retinopathy arises not as a direct result of a singular predominant factor, but as an outcome of interrelated networks connecting metabolism, immunology, and neurovascular integrity. Transcriptomic, proteomic, metabolomic, and epigenetic investigations completed in the last decade have uncovered consistent modifications in inflammatory signaling, mitochondrial pathways, lipid metabolism, and immune cell regulation across many experimental platforms and patient populations. The data indicate the presence of common molecular hubs—such as complement components, inflammasome regulators, cytokine signaling nodes, and metabolic sensors—that orchestrate disease-related responses among many retinal cell types. Comprehending the dynamic interactions of these hubs across time is crucial for elucidating disease heterogeneity and pinpointing treatment opportunities [16, 17, 18].
This integrative framework has significant clinical implications. Diabetic retinopathy continues to be a predominant source of preventable visual impairment globally [19], although advancements in screening and intravitreal treatment have been made. The continued presence of visual impairment in patients undergoing guideline-directed care underscores the inadequacy of strategies that focus solely on specific downstream effectors rather than addressing upstream network dysfunction. By framing DR as a paradigm of immune-metabolic retinal degeneration, one can integrate vascular, neuronal, and glial pathologies into a cohesive conceptual framework that mirrors the disease’s intricacies encountered in clinical settings.
This narrative review synthesizes current information that positions immune-metabolic crosstalk as a primary factor in diabetic retinopathy, focusing on studies published since 2020. Using human clinical data, translational models, and multi-omics studies, we investigate how metabolic stress initiates and perpetuates inflammatory signaling across disease phases. By employing a systems biology approach, we seek to consolidate these discoveries into a cohesive framework that can guide future diagnostic strategies and therapeutic approaches focused on restoring retinal homeostasis rather than merely mitigating late-stage vascular symptoms. Although the following sections discuss immune and metabolic mechanisms in thematic modules, these pathways should be interpreted as components of a sequential and interconnected pathogenic cascade. In this framework, chronic metabolic stress represents the initiating disturbance that alters retinal cellular metabolism and redox homeostasis. This metabolic imbalance promotes activation of innate immune pathways, including microglial activation, complement signaling, and inflammasome engagement. Subsequent amplification through cytokine networks, leukocyte recruitment, and adaptive immune responses progressively destabilizes the neurovascular unit, ultimately leading to neuronal dysfunction and microvascular degeneration. Viewing these processes as elements of a cascading immune–metabolic network helps reconcile the apparent overlap among mechanisms and emphasizes their coordinated contribution to disease progression.
Metabolic stress is the initial and most widespread catalyst of immune activation in diabetic retinopathy, occurring before observable microvascular damage and influencing the inflammatory characteristics of the retinal milieu. Chronic hyperglycemia exerts a persistent energetic strain on retinal cells [20], which depend significantly on oxidative metabolism for synaptic transmission, phototransduction, and barrier maintenance. Excess intracellular glucose flux modifies glycolytic efficiency and redirects substrates into secondary pathways, including the polyol pathway, the hexosamine biosynthetic route, protein kinase C activation, and advanced glycation end-product production. These metabolic deviations together disturb redox equilibrium, hinder mitochondrial activity, and produce reactive oxygen and nitrogen species, creating a pro-inflammatory biochemical environment that predisposes retinal tissue to immune activation [21, 22].
Mitochondrial dysfunction plays a pivotal role in this process [23]. Retinal
neurons, endothelial cells, and Müller glia exhibit early dysfunction in
mitochondrial dynamics, the electron transport chain, and mitophagy in diabetic
conditions. Impaired oxidative phosphorylation results in inadequate substrate
oxidation and excessive superoxide production, thereby activating redox-sensitive
transcription factors such as NF-
In addition to glucose metabolism, the disruption of lipid and amino acid processing exacerbates immunological activation. Diabetes is linked to qualitative alterations in retinal lipid composition, characterized by the buildup of harmful lipid intermediates and modified fatty acid oxidation, which can directly stimulate pattern-recognition receptors and inflammasome pathways [26, 27]. Similarly, disruptions in glutamine and branched-chain amino acid metabolism influence immune cell polarization and cytokine production, reinforcing a feed-forward relationship between metabolic dysregulation and retinal inflammation. Experimental investigations have shown that metabolic enzymes, such as BCAT1, engaged in branched-chain amino acid metabolism, can epigenetically modulate inflammatory signaling pathways and promote immune activation in diabetic retinopathy [28].
A characteristic aspect of inflammation generated by metabolic stress in the diabetic retina is its geographical and temporal variability. Single-cell transcriptome investigations have demonstrated that metabolic stress responses are heterogeneous across retinal cell populations [29]. Subsets of microglia, Müller glia, and endothelial cells exhibit unique transcriptional profiles characterized by activation of inflammatory mediators, complement components, and stress-response genes, despite the absence of apparent vascular disease [30, 31]. The findings substantiate the hypothesis that early diabetic retinopathy is propelled by localized immune–metabolic niches that gradually proliferate and interact as the disease progresses.
Metabolic stress prepares the retina for heightened immune responses to subsequent injuries. Hyperglycemia-induced epigenetic alterations, including histone acetylation and DNA methylation, can create a type of “metabolic memory” that perpetuates inflammatory gene expression even after glycemic control is achieved. This phenomenon elucidates the molecular basis of the enduring retinal inflammation observed in chronic diabetes and underscores why late therapeutic intervention frequently results in inadequate functional recovery [32, 33, 34]. From a systems biology perspective, metabolic stress is the initial disturbance that disrupts retinal homeostasis, lowers the threshold for immune activation, and lays the groundwork for progressive neurovascular degeneration.
Collectively, these data establish metabolic dysfunction as an active and organizing factor in the immunopathogenesis of diabetic retinopathy, rather than a passive background state. Metabolic stress links cellular energy imbalance to innate immune signaling, creating self-reinforcing networks that promote disease onset and advancement in retinal compartments. Comprehending these initial immune–metabolic interactions is crucial for identifying upstream intervention opportunities to modify the progression of diabetic retinopathy before permanent structural damage occurs.
The activation of the innate immune system serves as a primary mechanism by which metabolic stress is converted into persistent retinal inflammation in diabetic retinopathy. Retinal microglia, the complement system, and inflammasome signaling pathways are integral components of the innate immune system, interacting closely to respond to metabolic signals and contributing to both initial neurodegeneration and subsequent microvascular damage [35, 36, 37]. Instead of functioning as discrete routes, these systems constitute an interconnected inflammatory network that develops across disease stages and facilitates the chronicity and self-sustaining nature of retinal injury.
Microglia are the primary resident immune cells in the retina and function as
initial detectors of metabolic and oxidative stress [12]. Under normal
conditions, microglia preserve tissue homeostasis by conducting synaptic
surveillance, clearing debris, and providing trophic support. In diabetes,
prolonged hyperglycemia, mitochondrial dysfunction, and the accumulation of
damage-associated molecular patterns promote a transition to a pro-inflammatory
microglial phenotype [38]. Activated microglia exhibit morphological alterations,
increased migratory activity, and elevated inflammatory mediators, including
tumor necrosis factor-
Recent single-cell and spatial transcriptomic investigations have elucidated significant variation within the retinal microglial population. Various microglial subsets have divergent gene expression associated with oxidative stress responses, complement activation, antigen presentation, and phagocytosis. In diabetic settings, these subsets exhibit uneven expansion and preferential localization to areas of neuronal stress and capillary susceptibility, indicating that microglial activation is both metabolically influenced and spatially organized [41, 42, 43]. These findings contest the simplistic binary classification of microglia as either “pro-inflammatory” or “anti-inflammatory” and instead endorse a continuum of activation states influenced by local immune–metabolic contexts.
Dysregulation of the complement system constitutes a concurrent and synergistic innate immune mechanism in diabetic retinopathy. The complement cascade, traditionally considered a part of host defense, is now acknowledged as a regulator of synaptic pruning, cellular clearance, and inflammation in the central nervous system and retina. Hyperglycemia and oxidative stress induce the overexpression of complement components such as C1q, C3, and C5, while concurrently diminishing the expression of complement regulatory proteins [36, 44]. This imbalance promotes persistent complement activation, resulting in endothelial damage, heightened vascular permeability, and exacerbation of microglial inflammatory responses. Research on human vitreous samples and retinal tissue has revealed increased levels of complement activation products in individuals with diabetic retinopathy [45, 46].
The inflammasome pathway, especially the NLRP3 inflammasome, establishes a vital
molecular link between metabolic stress and enhanced innate immunity. NLRP3
activation is initiated by many metabolic danger-signals pertinent to diabetes,
including mitochondrial failure, reactive oxygen species, potassium efflux, and
the buildup of AGEs. In retinal cells, the activation of the NLRP3 inflammasome
results in caspase-1-mediated maturation of interleukin-1
Significantly, microglial activation, complement signaling, and inflammasome engagement function in an interdependent manner. Complement fragments can directly stimulate microglia, whereas cytokines produced by microglia enhance the expression of complement components and inflammasome-priming signals [47]. This mutual reinforcement establishes a feed-forward inflammatory circuit that is increasingly autonomous as the disease advances. From a systems biology viewpoint, these innate immune components form an interrelated module that assimilates metabolic stress signals and disseminates inflammatory responses among retinal cell types.
Innate immune signaling in the diabetic retina manifests as a metabolically calibrated, spatially structured, and self-perpetuating network that exacerbates both neurodegeneration and microvascular disease. Microglia, complement components, and inflammasomes serve as intermediaries between metabolic stress and tissue damage, playing pivotal roles in the immune–metabolic framework of diabetic retinopathy. Clarifying the interactions among various pathways over time is crucial for formulating therapies that can halt disease progression in its initial stages.
Innate immune systems trigger inflammatory signaling in diabetic retinopathy, whereas prolonged metabolic stress increasingly activates adaptive immune responses and fosters detrimental interactions between circulating immune cells and the retinal vasculature. This transformation signifies a shift from localized, tissue-specific inflammation to a more extensive immunovascular disease, in which leukocytes play an active role in endothelial damage, capillary non-perfusion, and disruption of the blood-retinal barrier. Adaptive immune activation does not merely signify a delayed or secondary response; rather, it appears to develop alongside disease progression, exacerbating the chronicity and irreversibility of retinal injury [48].
Leukostasis serves as a crucial pathway connecting immune activation to microvascular impairment in diabetes. Hyperglycemia, oxidative stress, and inflammatory cytokines promote the upregulation of endothelial adhesion molecules, such as intercellular adhesion molecule-1 and vascular cell adhesion molecule-1, on retinal capillaries. These alterations promote robust adherence of neutrophils, monocytes, and lymphocytes, resulting in capillary obstruction, localized ischemia, and endothelial cell apoptosis [49, 50].
In addition to mechanical blockage, adhering leukocytes directly damage the retinal endothelium by releasing reactive oxygen species, proteases, and pro-inflammatory cytokines. These mediators undermine tight-junction integrity, induce endothelial cell apoptosis, and increase vascular permeability. Studies demonstrate that the pathogenic significance of leukostasis is underscored by genetic or pharmacological inhibition of adhesion molecules, which diminishes vascular leakage and capillary degeneration in diabetic models, thereby highlighting immune–vascular coupling as a primary factor in microangiopathy rather than a mere bystander [51, 52].
T lymphocytes, a kind of adaptive immune cell, are increasingly acknowledged as
contributors to this inflammatory environment. Diabetic circumstances promote a
pro-inflammatory T-cell phenotype marked by increased Th1 and Th17 responses and
diminished regulatory T-cell function. Increased concentrations of
interferon-
Endothelial cells actively engage in adaptive immune responses. In diabetic and inflammatory states, retinal endothelial cells and activated microglia can express major histocompatibility complex class II molecules and co-stimulatory signals, facilitating local antigen presentation and prolonged T-cell activation. This phenomenon obscures traditional boundaries between immune and vascular compartments, hence reinforcing the notion of the neurovascular unit as a cohesive immunological entity. These interactions sustain chronic inflammation and compromise the integrity of the inner blood-retinal barrier, a critical factor in the development of diabetic macular edema.
Metabolic reprogramming enhances immune-vascular coupling. Activated leukocytes predominantly rely on glycolytic metabolism, thereby improving their inflammatory potential under glucose-abundant conditions, whereas endothelial cells under chronic hyperglycemia exhibit mitochondrial impairment and diminished nitric oxide availability [56, 57]. These concurrent metabolic alterations foster a milieu conducive to prolonged immune activation and vascular damage, strengthening feedback loops among inflammation, ischemia, and barrier compromise. From a systems biology standpoint, leukostasis and adaptive immunity function as amplifiers that disseminate localized metabolic stress into extensive microvascular disease. In addition to retinal-resident immune components, bone marrow–derived immune cells contribute to the inflammatory environment of the diabetic retina. Circulating monocytes, macrophages, and neutrophils can be recruited to retinal tissue through adhesion molecules and chemokine gradients induced by hyperglycemia and endothelial activation. Once present in the retinal microenvironment, these infiltrating immune cells interact with resident microglia and glial cells, amplifying cytokine production, oxidative stress, and vascular injury [10, 58].
The activation of adaptive immunity and the contacts between leukocytes and endothelial cells signify a crucial expansion of immune–metabolic imbalance in diabetic retinopathy. These processes contribute to disease development and treatment resistance by linking systemic immune-metabolic changes to localized vascular damage. In a network-based perspective, leukostasis and adaptive immunity serve as critical nodes that amalgamate metabolic stress, innate inflammation, and microvascular degradation into a cohesive pathogenic process. In addition to T-cell–mediated mechanisms, B lymphocytes and antibody-mediated immune responses may also contribute to the inflammatory milieu of diabetic retinopathy, although these pathways remain less extensively characterized. Experimental and clinical studies have reported alterations in circulating B-cell subsets in diabetes and increased levels of immunoglobulins and immune complexes in ocular fluids of patients with advanced disease. B cells may influence retinal inflammation both through antibody production and through antigen presentation and cytokine secretion, thereby modulating T-cell responses and innate immune activation. Although the precise role of humoral immunity in diabetic retinopathy remains incompletely defined, emerging evidence suggests that B-cell–dependent pathways may participate in the broader immune–metabolic network underlying retinal degeneration [59, 60].
The buildup of AGEs is a critical biochemical outcome of persistent hyperglycemia and a significant catalyst of immune–metabolic enhancement in diabetic retinopathy. AGEs result from non-enzymatic interactions between reducing sugars and proteins, lipids, or nucleic acids, resulting in structurally and functionally modified macromolecules that accumulate in retinal tissue and the bloodstream. In the diabetic retina, AGEs serve as indicators of metabolic stress and as active signaling ligands that activate inflammatory pathways via interaction with their primary receptor, RAGE. The AGE–RAGE axis serves as a pivotal biochemical connection between metabolic imbalance and prolonged immunological activation, leading to progressive neurovascular damage [61].
RAGE is a multiligand pattern-recognition receptor found in retinal endothelial
cells, Müller glia, pericytes, neurons, and microglia. In diabetic
circumstances, there is a significant increase in both AGE buildup and RAGE
expression, fostering an environment conducive to persistent receptor engagement.
The binding of AGEs to the receptor RAGE stimulates downstream signaling
cascades, particularly NF-
In retinal endothelial cells, AGE–RAGE signaling facilitates vascular
dysfunction through multiple mechanisms. The activation of NF-
The AGE–RAGE axis significantly impacts retinal immune cells beyond its direct
vascular effects. RAGE activation in microglia and Müller glia elicits a
pro-inflammatory phenotype marked by heightened production of tumor necrosis
factor-
The cytokine and chemokine networks activated downstream of AGE–RAGE signaling
form a complex, interconnected inflammatory network in the diabetic retina.
Principal mediators, such as tumor necrosis factor-
From a systems biology standpoint, the AGE–RAGE axis serves as a potent feed-forward amplifier within the immune–metabolic network of diabetic retinopathy. The accumulation of AGEs elevates RAGE expression, whereas RAGE activation stimulates cytokine production, which in turn exacerbates oxidative stress and promotes further AGE synthesis [59]. This self-perpetuating cycle sustains retinal inflammation despite improvements in glycemic control, offering a molecular explanation for the metabolic memory phenomena noted in chronic diabetes. Epigenetic alterations triggered by AGE-mediated signaling enhance the stability of inflammatory gene expression, integrating immune activation into the retinal transcriptional framework [67].
Clinically, addressing the AGE–RAGE–cytokine axis is an appealing, although inadequately developed, treatment approach. Although therapies targeting the reduction of AGE production or the inhibition of RAGE signaling have demonstrated effectiveness in preclinical research, their translation into clinical practice has been constrained by pharmacological obstacles and the redundancy of downstream inflammatory pathways [68]. Nonetheless, acknowledging this axis as a pivotal immune-metabolic center has revitalized interest in combination strategies that concurrently target metabolic stress and inflammatory signaling, especially in patients with early or progressive disease insufficiently managed by anti-angiogenic therapies alone.
The AGE–RAGE axis and its related cytokine networks constitute a fundamental immune-metabolic amplifier in diabetic retinopathy. This pathway transforms chronic metabolic stress into persistent inflammatory signaling, so unifying vascular dysfunction, immunological activation, and neurodegeneration into a singular pathogenic mechanism. In a network-based perspective, AGE–RAGE signaling serves as a pivotal link between upstream metabolic harm and downstream immunological and vascular dysfunction, rendering it a crucial target for future disease-modifying therapies.
In addition to elevated glucose, diabetic retinopathy is characterized by significant changes in lipid and amino acid metabolism, as well as mitochondrial energy production, that fundamentally influence retinal immune responses. These metabolic pathways interface with inflammatory signals at various levels, affecting immune cell activation, endothelial integrity, and neuronal survival. Growing data suggest that dysregulated lipid and amino acid metabolism are increasingly recognized as mechanistic contributors that interact bidirectionally with inflammation [26, 68, 69, 70]. Lipid metabolism is significantly modified in the diabetic retina, indicating both systemic dyslipidemia and tissue-specific metabolic reprogramming. The accumulation of lipotoxic intermediates, such as ceramides and oxidized low-density lipoproteins, has been observed in diabetic retina [26, 71]. These lipid species serve as powerful inflammatory triggers by activating pattern-recognition receptors, generating endoplasmic reticulum stress, and facilitating oxidative damage. Lipotoxic stress in retinal endothelial cells compromises membrane integrity and tight-junction architecture, whereas in immune cells it promotes pro-inflammatory polarization and cytokine secretion.
Fatty acid metabolism additionally influences immune modulation in the retina.
Compromised
Mitochondrial dysfunction is pivotal to the integration of lipid and amino acid metabolism with immunological activation. Mitochondria function as metabolic centers and innate immunological signaling platforms, integrating energy generation with inflammatory responses [70, 73]. In the diabetic retina, persistent metabolic overload disrupts mitochondrial dynamics, diminishes respiratory chain efficacy, and fosters excessive production of reactive oxygen species. Compromised mitochondria release mitochondrial DNA and other danger-associated molecular patterns that activate inflammasome pathways and enhance cytokine production [74].
Mitochondrial dysfunction also plays a role in the occurrence of metabolic memory in diabetic retinopathy. Chronic mitochondrial DNA damage, compromised mitophagy, and oxidative stress-induced epigenetic alterations can perpetuate inflammatory signaling despite the amelioration of metabolic parameters. Human studies have shown enduring changes in mitochondrial gene expression and function in diabetic retinal tissue, offering a mechanistic rationale for ongoing disease progression despite postponed glycemic optimization [75].
From a systems biology standpoint, lipid, amino acid, and mitochondrial metabolism intersect to establish an immune-metabolic core that regulates retinal inflammatory dynamics. Disruptions in these pathways traverse interwoven signaling networks, affecting immune cell function, vascular integrity, and neural robustness. Significantly, these metabolic pathways interact bidirectionally with inflammatory mediators, establishing feedback loops that perpetuate chronic conditions and hinder spontaneous remission.
Clinically, identifying these metabolic factors broadens the therapy options beyond glucose-focused approaches. Interventions targeting dyslipidemia, mitochondrial dysfunction, or specific metabolic pathways may influence retinal inflammation and enhance current vascular-targeted therapies. Translating these discoveries into effective treatments requires integrated approaches that restore metabolic equilibrium across multiple retinal cell types, rather than merely targeting single metabolic deficiencies.
Modified lipid management, amino acid metabolism, and mitochondrial bioenergetics are pivotal in influencing immune responses in the diabetic retina. These pathways associate systemic metabolic failure with local inflammatory activation, hence facilitating the persistence and advancement of diabetic retinopathy. In a network-based architecture, they signify essential nodes that enable the transformation of metabolic stress into chronic immune-mediated retinal degeneration.
The neurovascular unit (NVU) embodies the functional and anatomical integration of retinal neurons, endothelial cells, pericytes, glial cells, and resident immune cells, all of which are collectively tasked with preserving metabolic balance and visual performance. In diabetic retinopathy, this meticulously regulated mechanism is disrupted by persistent metabolic stress and prolonged immunological activation, resulting in progressive neurovascular deterioration. NVU failure arises from impaired communication among its components, rather than from isolated cellular damage, influenced by immune–metabolic signaling loops [76]. This network-based disruption elucidates why the first functional deficiencies may arise without evident vascular disease. It also offers a framework for understanding the heterogeneous and often unpredictable progression of diabetic retinopathy among people with similar metabolic profiles. From this viewpoint, NVU degradation is more accurately understood as an emergent characteristic of interacting pathogenic mechanisms rather than a sequential disease progression [77].
Neuronal dysfunction represents an early and clinically significant indication of NVU disruption in diabetes. In diabetic conditions, retinal ganglion cells, amacrine cells, and photoreceptors exhibit compromised mitochondrial activity, altered synaptic signaling, and heightened susceptibility to apoptosis [78, 79]. The neuronal alterations are induced by intrinsic metabolic stress and by inflammatory mediators generated by activated microglia and Müller glia. Functional studies in diabetic patients have revealed deficits in contrast sensitivity, dark adaptation, and electrophysiological responses prior to the emergence of microvascular lesions, thereby substantiating the notion of initial neurodegeneration [80, 81]. Neuronal damage contributes to the inflammatory network by generating danger-associated molecular patterns that further stimulate immune pathways. This reciprocal interaction enhances tissue susceptibility and hastens NVU disintegration.
Glial cells serve a pivotal coordinating function in converting immune-metabolic stress into neurovascular unit dysfunction. Müller glia, which provide metabolic and structural support across the retinal layers, exhibit reactive gliosis in response to persistent hyperglycemia and inflammation. This condition is characterized by altered energy metabolism, heightened synthesis of pro-inflammatory cytokines, and diminished release of neurotrophic factors vital for neuronal survival. Müller cells consequently forfeit their ability to modulate extracellular glutamate concentrations and ionic equilibrium, ultimately exacerbating excitotoxic stress on neurons. Astrocytes, despite being less prevalent in the retina, similarly participate in inflammatory signaling and compromised vascular supply. Collectively, glial dysfunction serves as a crucial intermediate by which immune–metabolic imbalance undermines neural integrity and vascular stability [82].
The vascular elements of the neurovascular unit are especially susceptible to immune-metabolic disturbances. Retinal endothelial cells subjected to prolonged hyperglycemia and inflammatory cytokines demonstrate mitochondrial dysfunction, heightened oxidative stress, and decreased expression of tight junction proteins. These alterations compromise endothelial barrier integrity and facilitate vascular permeability, a characteristic of diabetic macular edema. Pericytes, crucial for capillary integrity and blood flow regulation, experience selective loss in diabetic retinopathy, exacerbating microvascular instability. The loss of pericytes increases endothelial susceptibility and impairs endothelial–glial communication, exacerbating neurovascular unit dysfunction. These vascular changes not only impair tissue perfusion but also intensify metabolic stress in the retina, resulting in self-reinforcing cycles of damage [83, 84, 85].
The disruption of neurovascular coupling signifies a significant functional outcome of neurovascular unit degeneration. In a healthy retina, neural activity dynamically modulates local blood flow to correspond with metabolic requirements, thereby maintaining effective delivery of oxygen and nutrients. In diabetic retinopathy, inflammatory signals and endothelial dysfunction hinder adaptive responses, leading to imbalances between energy supply and neural demand. This uncoupling leads to localized hypoxia, the buildup of metabolic waste, and increased immunological activity, thereby exacerbating tissue damage [86]. Advanced imaging investigations indicate that these problems manifest early and exhibit spatial heterogeneity, contributing to the irregular distribution of retinal lesions found clinically [87, 88]—impaired neurovascular coupling functions as both an indicator and a catalyst of advancing neurovascular unit dysfunction.
From a systems biology perspective, NVU degeneration signifies the stabilization of maladaptive network states influenced by immune–metabolic interactions. Inflammatory mediators diminish metabolic flexibility, metabolic stress amplifies immunological activation, and vascular dysfunction restricts nutritional supply, thereby undermining the robustness of the retinal network. The nonlinear feedback loops elucidate why enhancing glycemic control alone frequently proves inadequate to reverse established disease. Upon the establishment of pathological network topologies, the NVU gets entrenched in a degenerative trajectory that defies spontaneous recovery. Identifying NVU degeneration as a network-level phenomenon underscores the need for therapeutic approaches that restore coordinated activity among retinal cell types, rather than focusing on isolated pathways. Fig. 1 depicts the systems-level immune–metabolic network through which chronic metabolic stress initiates and perpetuates inflammatory signaling, ultimately leading to degeneration of the neurovascular unit in diabetic retinopathy.
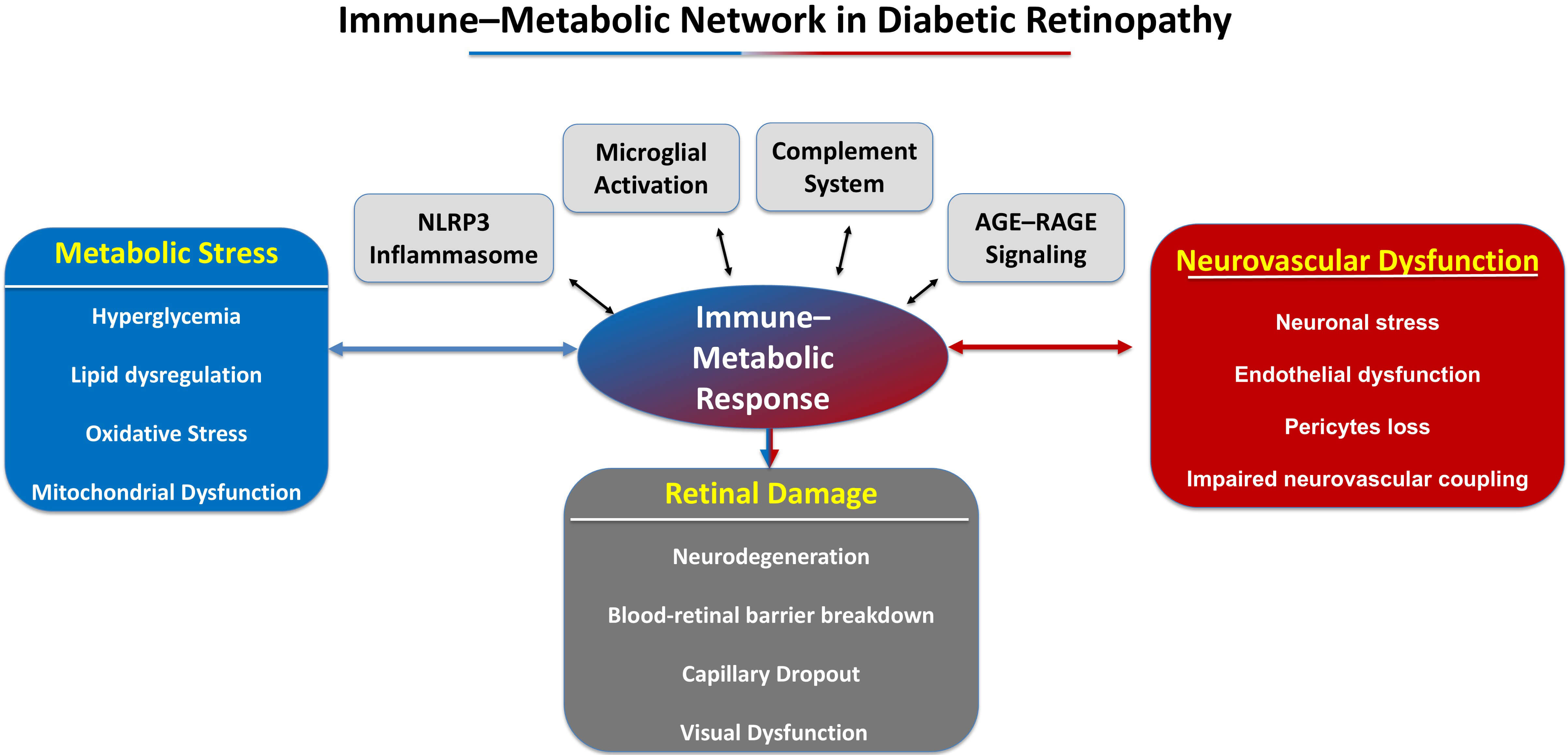 Fig. 1.
Fig. 1.
Immune-metabolic network in diabetic retinopathy. This schematic diagram depicts the interrelated mechanisms connecting metabolic stress to retinal degeneration in diabetic retinopathy. The upper portion of the picture illustrates the metabolic determinants of disease, encompassing chronic hyperglycemia, mitochondrial dysfunction, oxidative stress, and lipotoxicity. These metabolic anomalies stimulate immunological signaling pathways in the retinal microenvironment. The middle section of the diagram depicts essential immune–metabolic hubs, encompassing microglial activation, complement cascade dysregulation, AGE–RAGE signaling, inflammasome activation, and cytokine network amplification. These processes engage in bidirectional interactions with elements of the neurovascular unit. Chronic inflammatory signaling impairs neurovascular coupling and undermines the integrity of the blood-retinal barrier. The image also emphasizes feed-forward inflammatory loops and metabolic memory mechanisms that perpetuate the development of illness despite improvements in metabolic conditions. The synergistic effects of these pathways lead to progressive retinal dysfunction, characterized by neuronal death, vascular leakage, capillary dropout, and visual impairment. AGE–RAGE, advanced glycation end product–receptor for AGE.
Omics technologies have significantly enhanced the understanding of diabetic retinopathy by uncovering coordinated immune-metabolic changes that are not detected by traditional histopathological or single-pathway methods. Transcriptomic, proteomic, metabolomic, and epigenetic analyses, combined, reveal that diabetic retinopathy is marked by extensive reprogramming of gene expression, protein signaling pathways, and metabolic processes across several retinal cell types [89]. Significantly, omics data substantiate the notion that inflammation, metabolic stress, and neurovascular dysfunction arise from interrelated regulatory networks rather than from discrete molecular events. Integrating this information enables conceptualizing diabetic retinopathy as a systems-level disease driven by a coordinated immune–metabolic imbalance. Transcriptomic investigations of human retinal tissue and experimental models have repeatedly revealed increases in inflammatory, complement-related, and stress-response genes in diabetes [16, 90, 91]. Bulk RNA sequencing investigations have demonstrated elevated production of cytokines, chemokines, adhesion molecules, and elements of innate immune pathways, even in the first stages of the disease. Recently, single-cell RNA sequencing has enhanced these findings by uncovering transcriptional programs specific to cell types associated with diabetic stress. Various subsets of microglia, Müller glia, endothelial cells, and pericytes display transcriptional profiles that signify immunological activity, oxidative stress, and metabolic reprogramming [92, 93].
Proteomic investigations have enhanced transcriptome data by elucidating post-transcriptional regulation and protein-level signaling dynamics in diabetic retinopathy [94, 95]. Examinations of vitreous fluid and retinal tissue from individuals with diabetic retinopathy have demonstrated increased concentrations of inflammatory cytokines, complement components, extracellular matrix proteins, and angiogenic mediators [96, 97].
Metabolomic investigations have yielded direct evidence of altered metabolic states associated with diabetic retinopathy and have identified compounds that may actively modulate immune responses. Research on plasma, vitreous, and retinal tissue has revealed alterations in glucose-derived metabolites, lipids, amino acids, and oxidative stress markers that are associated with disease severity. Disruptions in glutamine, arginine, and branched-chain amino acid metabolism are associated with endothelial dysfunction and inflammatory activation. In contrast, modified lipid profiles indicate both systemic dyslipidemia and retinal-specific metabolic stress. Metabolomic patterns frequently differentiate patients with diabetic retinopathy from those with diabetes without retinal pathology, suggesting potential utility for risk assessment [98, 99, 100].
Epigenetic research has clarified the processes by which immune–metabolic imbalance becomes chronic in diabetic retinopathy. Hyperglycemia and oxidative stress alter DNA methylation, histone modifications, and chromatin accessibility, thereby modifying the transcriptional profile of retinal cells. These epigenetic modifications can perpetuate inflammatory gene expression even after metabolic conditions are improved, providing a biological basis for metabolic memory. Recent research has emphasized the significance of non-coding RNAs, such as microRNAs and long non-coding RNAs, in modulating inflammatory and metabolic pathways pertinent to diabetic retinopathy. The dysregulation of these epigenetic regulators affects cytokine production, mitochondrial function, and endothelial stability, hence exacerbating immune–metabolic imbalance within retinal networks [33, 34, 101, 102].
Collectively, omics datasets converge on a restricted number of interrelated pathways that function as hubs within the immune–metabolic network of diabetic retinopathy. Complement activation, cytokine signaling, mitochondrial dysfunction, and metabolic reprogramming frequently appear as pivotal elements across transcriptomic, proteomic, and metabolomic analyses. The integration of this information at the systems level has uncovered feedback loops connecting metabolic stress to immunological amplification and neurovascular injury, elucidating the persistence and heterogeneity of the disease. Nonetheless, obstacles persist, including discrepancies in study design, tissue sources, and analytical methodologies, which hinder direct comparisons among datasets. Notwithstanding these constraints, omics-derived insights have radically transformed the comprehension of diabetic retinopathy and established a basis for network-oriented diagnostics and therapeutic approaches.
The evidence examined herein advocates for a conceptual transition in diabetic retinopathy from primarily a microvascular complication of diabetes to a multifaceted, immune-metabolic neurodegenerative disease. In clinical, translational, and experimental research, chronic metabolic stress is identified as the primary disturbance that disrupts retinal homeostasis and triggers innate and adaptive immune responses [3, 4, 8, 35, 48]. These immune responses are not ephemeral or secondary; rather, they become permanently integrated within retinal tissue through reciprocal interactions involving neurons, glial cells, vascular cells, and immune mediators. The resultant inflammatory environment fosters ongoing deterioration of the neurovascular unit, ultimately presenting as the structural and functional impairments that define vision-threatening conditions. Analyzing diabetic retinopathy from this perspective provides a clear rationale for initial neuronal impairment, heterogeneous disease progression, and the limited effectiveness of treatments targeting specific downstream pathways. A systems biology approach is especially beneficial in harmonizing the seemingly diverse pathogenic pathways associated with diabetic retinopathy. Instead of depicting isolated contributors, pathways that involve microglial activation, complement dysregulation, AGE–RAGE signaling, cytokine networks, metabolic reprogramming, and mitochondrial dysfunction converge on common regulatory hubs and feedback loops [12, 43, 44, 62]. These hubs amalgamate metabolic inputs with immunological outputs, orchestrating responses among various retinal cell types. Omics-based research substantiates this perspective by consistently revealing convergent immune-metabolic markers across transcriptomic, proteomic, metabolomic, and epigenetic platforms [16, 90, 91, 92, 93, 94, 95, 96, 97, 98, 99, 100, 101, 102]. This convergence indicates that disease development is influenced less by singular dominant causes and more by the entrenchment of maladaptive network states that resist spontaneous resolution once formed. This integrative paradigm elucidates numerous unclear clinical observations. Functional vision impairment often occurs prior to the development of evident microvascular lesions, aligning with the initial neuronal and glial engagement induced by immune–metabolic stress. The ongoing retinal inflammation and neurodegeneration, despite enhanced glycemic control, indicate the existence of metabolic memory and epigenetically reinforced inflammatory pathways. The limited efficacy of anti-VEGF therapy in a significant portion of patients underscores the inadequacy of approaches that focus solely on angiogenic outcomes while neglecting upstream immune–metabolic factors [14, 15]. Conversely, perceiving diabetic retinopathy as a network condition underscores the necessity for earlier intervention and combinatorial strategies that concurrently address immune activation, metabolic dysregulation, and neurovascular coupling. From a translational perspective, immune–metabolic interactions present a fertile ground for biomarker identification and therapeutic advancement. Circulating and vitreous biomarkers indicative of inflammatory activation, complement involvement, lipid dysregulation, and mitochondrial stress have demonstrated potential in stratifying disease risk and progression [45, 46, 55, 63, 96, 99, 100]. Furthermore, identifying core network nodes offers the opportunity to target common upstream regulators rather than specific downstream mediators. Such tactics may be especially pertinent in the early phases of disease, when network adaptability is sufficient to restore retinal homeostasis. Translating these discoveries into clinical practice necessitates meticulous consideration of safety, timing, and patient selection, due to the critical physiological functions of numerous immunological and metabolic pathways. Taken together, the evidence discussed throughout this review suggests that diabetic retinopathy may be conceptualized as a progressive cascade of interconnected immune-metabolic events. Chronic metabolic stress initiates innate immune activation, which subsequently amplifies inflammatory signaling and ultimately disrupts the neurovascular unit. A simplified schematic representation of this proposed pathogenic sequence is presented in Fig. 2.
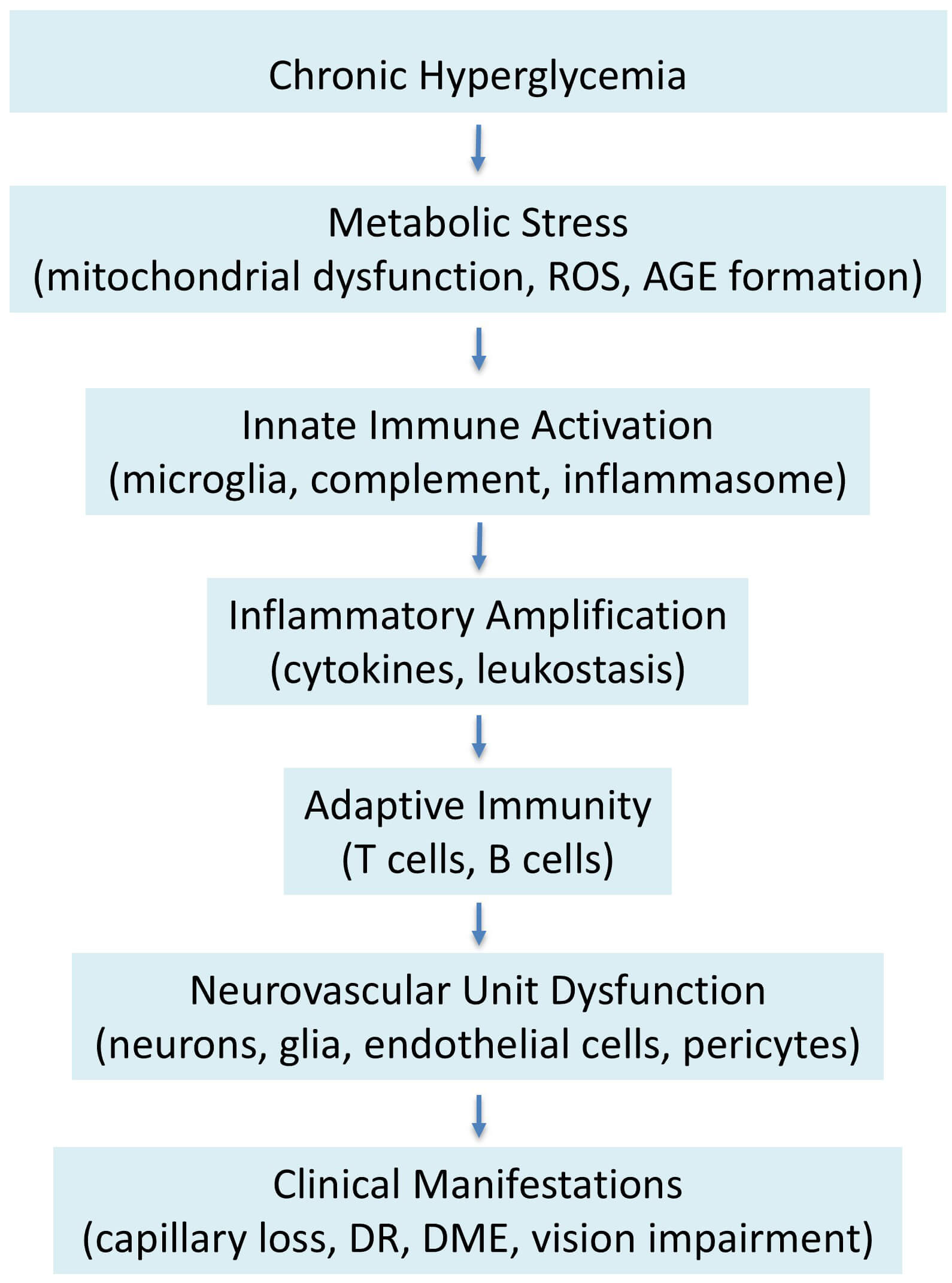 Fig. 2.
Fig. 2.
Proposed pathogenic sequence of immune–metabolic dysregulation in diabetic retinopathy. Chronic hyperglycemia induces metabolic stress characterized by mitochondrial dysfunction, oxidative imbalance, and accumulation of advanced glycation end products. These metabolic disturbances promote activation of innate immune pathways, including microglial activation, complement signaling, and inflammasome engagement. Subsequent inflammatory amplification through cytokine production and leukocyte recruitment contributes to progressive dysfunction of the retinal neurovascular unit. The cumulative effects of these processes ultimately lead to structural retinal damage and clinical manifestations such as diabetic macular edema, capillary dropout, and visual impairment. This schematic illustrates the conceptual progression linking metabolic perturbation to immune activation and neurovascular degeneration. ROS, Reactive Oxygen Species.
Notwithstanding these advancements, many significant shortcomings hinder the current comprehension of immune–metabolic interactions in diabetic retinopathy. A substantial portion of the mechanistic information comes from animal models that inadequately replicate the chronicity, heterogeneity, and comorbidities of human diabetes. Human retinal tissue is infrequently accessible at early disease stages, constraining direct validation of preclinical results. Omics studies, although potent, are typically cross-sectional, exhibit variability in tissue sources and analytical methodologies, and often lack longitudinal clinical linkage. Furthermore, differentiating primary immune-metabolic drivers from secondary adaptive responses is particularly problematic in advanced disease, where many pathogenic processes coexist. These constraints highlight the necessity for comprehensive, longitudinal human studies that integrate molecular profiling with functional and clinical outcomes. Despite an expanding corpus of literature endorsing the involvement of immune–metabolic dysregulation in diabetic retinopathy, numerous facets remain inadequately elucidated. Experimental models consistently exhibit microglial activation and inflammatory signaling in the early stages of disease; yet, clinical research has shown inconsistent relationships between inflammatory biomarkers and disease severity. These inconsistencies may indicate variations in study populations, illness phases, or analytical techniques. Furthermore, numerous mechanistic investigations rely on animal models that simulate hyperglycemia but fail to reflect the chronic systemic complexities of human diabetes fully. Thus, distinguishing causative factors from subsequent inflammatory reactions remains difficult. Subsequent research should focus on amalgamating longitudinal clinical data with mechanistic studies to identify the immune–metabolic pathways that are primary predictors of illness development. Another unresolved question concerns the relative importance of local retinal immune responses versus systemic immune-metabolic dysregulation. While increasing evidence suggests that circulating immune cells and systemic metabolic inflammation influence retinal pathology, the mechanisms governing their recruitment and interaction with retinal-resident immune cells remain incompletely understood. Addressing these gaps will require longitudinal human studies integrating single-cell and spatial transcriptomics, metabolomic profiling, and advanced retinal imaging. Such approaches could clarify causal relationships among metabolic stress, immune activation, and neurovascular degeneration and help identify testable hypotheses regarding key regulatory nodes within the immune–metabolic network.
This paper has several limitations considered it was prepared as a narrative review. The manuscript does not adhere to a formal systematic search approach or established inclusion criteria. Although significant databases and essential contributions were evaluated to guarantee comprehensiveness and relevancy, certain relevant studies may have been inadvertently excluded. The selection of studies was influenced by clinical significance and conceptual coherence, potentially introducing a level of selection bias characteristic of narrative synthesis.
Future research should emphasize network-based methodologies that amalgamate multi-omics data with sophisticated imaging, functional assessments, and clinical phenotyping. Longitudinal studies utilizing single-cell and spatial transcriptomics, in conjunction with metabolomic and epigenetic profiling, may elucidate the evolution of immune–metabolic networks throughout disease stages and pinpoint therapeutic opportunities. Future research should include longitudinal clinical cohort studies to validate immune–metabolic biomarkers and network patterns across the natural history of diabetic retinopathy. Combining genetic profiling with comprehensive clinical phenotyping, retinal imaging, and functional visual evaluations would enable researchers to determine whether specific immune–metabolic signatures predict disease onset, progression, or therapeutic response. Such prospective research is crucial for converting systems-level findings into clinically applicable strategies. Longitudinal validation across varied patient groups would elucidate whether immune–metabolic network states vary among phenotypic subtypes of diabetic retinopathy and could facilitate the formulation of tailored therapy regimens. From a translational perspective, identifying priority therapeutic nodes within the immune–metabolic network remains an important challenge. Among the pathways discussed, several mechanisms appear particularly promising as potential intervention targets. Complement signaling represents an attractive candidate due to its upstream role in inflammatory amplification and its emerging therapeutic tractability in ocular diseases. Likewise, mitochondrial dysfunction and oxidative stress represent central metabolic disturbances that influence both neuronal survival and immune activation. The AGE–RAGE axis also constitutes a key regulatory hub linking metabolic stress with inflammatory signaling and vascular injury. Rather than targeting isolated downstream mediators, future therapeutic strategies may benefit from focusing on these upstream immune-metabolic regulators, potentially in combination with existing vascular-targeted treatments such as anti-VEGF therapy.
Diabetic retinopathy is increasingly recognized as a multifaceted neurovascular degenerative disorder shaped by the interplay between metabolic stress and immunological activation within the retinal milieu. Evidence from clinical investigations, experimental models, and multi-omics analyses indicates that persistent hyperglycemia initiates a cascade of immune-metabolic disturbances that extend beyond vascular pathology to involve neurons, glial cells, and the entire neurovascular unit. These interconnected processes are coordinated through shared signaling hubs and feedback loops that reinforce maladaptive inflammatory and metabolic network states, thereby contributing to disease persistence and progression. Importantly, immune–metabolic dysregulation emerges early during disease development and is associated with functional impairment that precedes overt microvascular abnormalities. Adopting a systems biology framework provides a cohesive model that integrates vascular, neural, and immunological mechanisms into a unified pathogenic context. This perspective helps explain clinical heterogeneity, partial therapeutic responses, and the phenomenon of metabolic memory observed in diabetic retinopathy. Moving the focus from isolated molecular targets toward network-level dysfunction may facilitate the identification of upstream regulatory nodes with greater potential for disease modification. Ultimately, improving the management of diabetic retinopathy will likely require therapeutic strategies that restore immune–metabolic balance and support neurovascular integrity, with the goal of preserving retinal function and preventing irreversible vision loss.
FC, CG, and MZ designed the research study. FC, CM, FD, CG, AA, GG, DT and MZ performed the research and prepared the draft. FC, CM, FD, CG, AA, GG, DT and MZ contributed to the literature search and interpretation. FC, CG and MZ wrote the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research received no external funding.
The authors declare no conflicts of interest. The Mediterranean Foundation “G.B. Morgagni” is the affiliated institution of Francesco Cappellani, Caterina Gagliano and Cosimo Mazzotta, and this relationship did not influence the judgments in data interpretation or manuscript writing.
During the preparation of this work, the authors used ChatGPT by OpenAI (GPT-5.5 model) to check spelling and grammar. After using this tool, the authors reviewed and edited the content as needed and took full responsibility for the content of the publication.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.