



 , Melissa C. Schubbert 1, Maximilian M. Siewert 1, Michèle Ohnemüller 1, Frederic Kuwert 1, Ulla I. M. Gerling-Driessen 2, Katharina Pracht 1,*
, Melissa C. Schubbert 1, Maximilian M. Siewert 1, Michèle Ohnemüller 1, Frederic Kuwert 1, Ulla I. M. Gerling-Driessen 2, Katharina Pracht 1,*
1 Department of Translational Immunology, Nikolaus-Fiebiger Center of Molecular Medicine, Universitätsklinikum Erlangen, Friedrich-Alexander-University Erlangen-Nürnberg, 91054 Erlangen, Germany
2 Institute for Macromolecular Chemistry, Albert Ludwig University of Freiburg, 79104 Freiburg, Germany
Abstract
The enzyme N-glycanase 1 (NGLY1) regulates autophagic processes and endoplasmic reticulum (ER)-associated proteasomal degradation by de-N-glycosylation of misfolded glycoproteins. Mutations of NGLY1 that result in a loss of protein function cause congenital disorder of deglycosylation 1 (CDDG1), also known as NGLY1 deficiency. NGLY1 deficiency is associated with severe dysregulation of mitochondria and proteasomal degradation, which primarily manifests as impairments in the nervous system. However, recent studies also linked NGLY1 function to cellular processes associated with immunity and autoimmune diseases, such as rheumatoid arthritis. NGLY1 plays a distinct role in mitochondrial homeostasis, thereby potentially regulating interferon responses, cellular stress responses and innate immunity. It also controls the stability of the programmed cell death protein-1 (PD-1) receptor on T lymphocytes and cancer cells, influencing tumor immune evasion. Importantly, NGLY1 was shown to process foreign peptides destined for presentation on major histocompatibility complex (MHC) molecules, a process that enables cytotoxic T lymphocytes to identify pathogens or mutated cells. Finally, altered levels of NGLY1 may impair B lymphocytes, especially the formation and function of antibody-secreting cells. In this review, we aim to compile findings from the last three decades and explore the connections between NGLY1-controlled processes and immune cell function. Understanding NGLY1-mediated mechanisms may provide new insights into the modulation of immune responses and the development of therapeutic strategies for immune-related disorders.
Keywords
- NGLY1
- endoplasmic reticulum-associated degradation
- congenital disorders of deglycosylation (CDDG)
- immune system
- mitochondria
- T-lymphocytes
- B-lymphocytes
- inflammation
- rheumatoid arthritis (RA)
The cytosolic enzyme N-Glycanase 1 (NGLY1) de-N-glycosylates misfolded proteins, priming them for degradation via the endoplasmic reticulum (ER)-associated degradation (ERAD) pathway. So far, NGLY1’s function has been extensively investigated in the context of congenital disorder of deglycosylation 1 (CDDG1) also known as NGLY1 deficiency. The research mainly focused on NGLY1’s connection to mitochondrial function and proteasomal degradation, two processes which are described to be severely affected by NGLY1 deficiency. However, it has been demonstrated that NGLY1 affects several cellular pathways in various cell types, impacting not only protein degradation but also further processes. Little is known about whether NGLY1 influences immune cells and immunological pathways. However, previous studies have shown that NGLY1 is involved in processes that are either associated with or essential for immune cells. This highlights NGLY1 as a potential target for novel cancer or autoimmune therapies. This review will focus on the potential implications of NGLY1’s known functions for the immune system.
NGLY1 is an almost ubiquitously expressed, evolutionary conserved de-N-glycosylating enzyme, present in most eukaryotes [1, 2, 3] and also known as peptide: N-glycanase (PNGase) in drosophila and yeast [4, 5]. The protein consists of three main domains: the transglutaminase domain, which has catalytic function; the “peptide:N-glycanase and UBA or UBX-containing proteins” (PUB) domain, which is located at the N-terminus and binds ubiquitin; and the C-terminal “present in PNGases and other worm proteins” (PAW) domain, which binds to high-mannose structures [6, 7] (Fig. 1A). The PUB domain can bind to the ATPase p97, which is a key player in the protein quality control machinery and functions as an adaptor protein facilitating the interaction with a variety of ERAD substrates [6, 8, 9, 10]. Meanwhile, the PAW domain is necessary to bind retro-translocated, misfolded glycoproteins via their high-mannose structures [6]. In contrast to the PUB and PAW domain, which are primarily found in higher eukaryotes [6, 11], the transglutaminase domain is conserved throughout different species and catalyzes the cleavage of N-glycans from glycoproteins. Therefore, NGLY1 plays an essential role in cytosolic degradation of glycosylated unfolded or misfolded proteins and ensures proper ERAD processing [8, 12] (Fig. 1B). ERAD is essential for maintaining cellular protein homeostasis, as it disposes of proteins that are no longer functional and have been targeted for degradation by the ubiquitin-proteasome system (UPS). Thus, the UPS-ERAD axis plays a key role in cellular protein quality control and, for glycoproteins at least, depends on NGLY1 function.
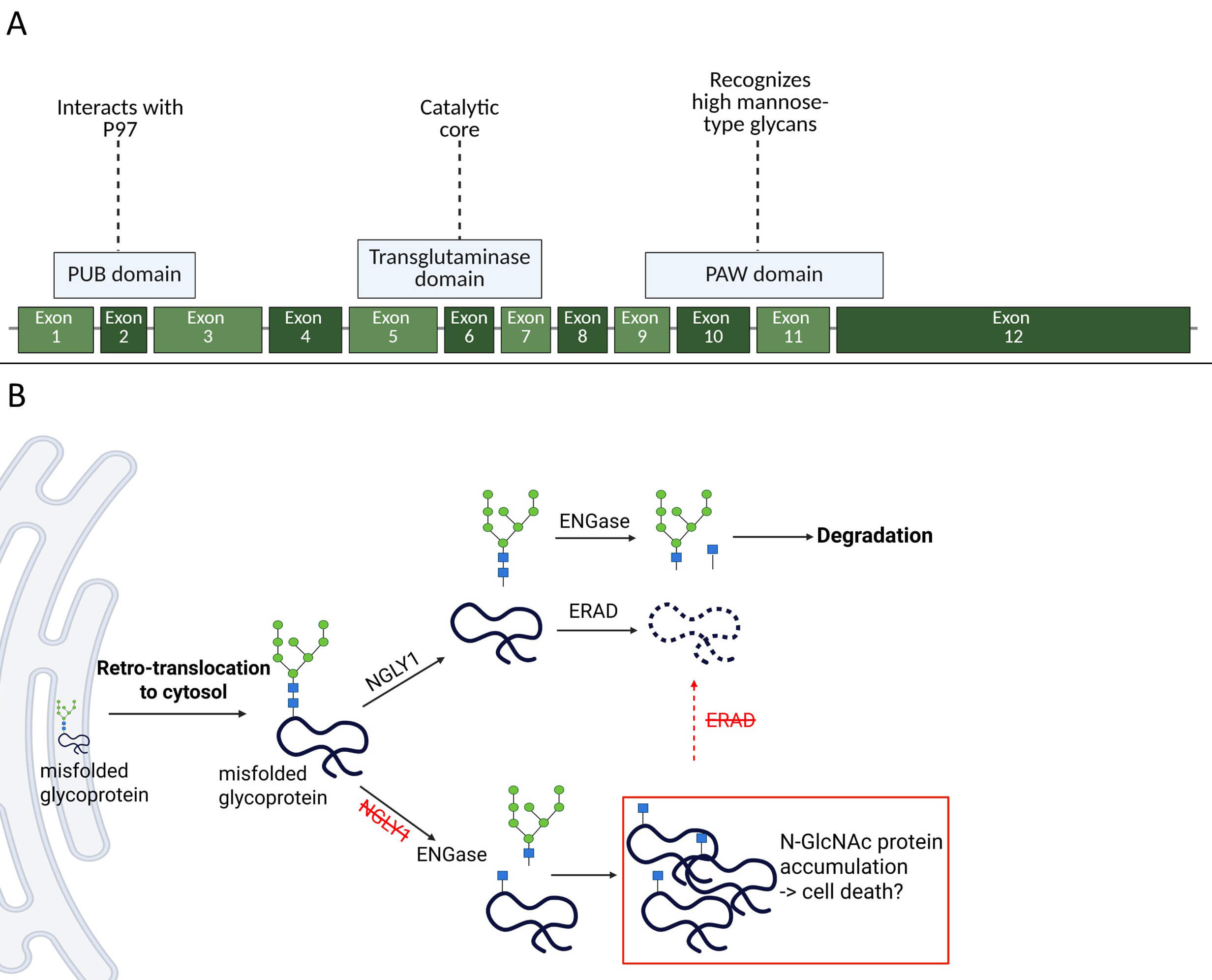 Fig. 1.
Fig. 1.
Structure and function of the cytosolic enzyme N-Glycanase 1 (NGLY1). (A) The NGLY1 sequence contains 12 exons and is located on chromosome 14 in mice and on chromosome 3 in humans. NGLY1 comprises 3 structural domains. The “peptide:N-glycanase and UBA or UBX-containing proteins” (PUB) domain interacts with ATPase p97, functioning as an adaptor towards endoplasmic reticulum (ER)-associated degradation (ERAD) substrates. The transglutaminase domain performs NGLY1’s catalytic function, while the “present in PNGases and other worm proteins” (PAW) domain facilitates binding to misfolded substrates by recognizing their high-mannose glycans. (B) NGLY1’s canonical function. Once misfolded proteins are retro-translocated into the cytosol, NGLY1 breaks the linkage between the innermost N-acetylglucosamines (GlcNAc) and the asparagin (Asn) residue. The free glycan is further degraded by the endo-beta-N-acetylglucosaminidase (ENGase), while the deglycosylated protein can be degraded in the proteasome. In NGLY1-deficient cells, ENGase is proposed to cleave within the glycan structure. This leaves the innermost GlcNAc still attached to the protein and causes the accumulation of misfolded, improperly deglycosylated proteins.
Huang and colleagues [13] hypothesized that, in the absence of NGLY1, endo-beta-N-acetylglucosaminidase (ENGase) would process N-glycans that were still attached to proteins. Typically involved in non-lysosomal glycan degradation, ENGase cleaves between the two truncal N-acetylglucosamines (N-GlcNAc) of the chitobiose core. This process results in partially glycosylated proteins that carry only one N-GlcNAc. These proteins are more difficult for the proteasome to degrade properly and can therefore potentially aggregate in the cytosol. However, even in the absence of the ENGase, glycoproteins are often partially deglycosylated in NGLY1-deficient cells. Incompletely deglycosylated proteins are aberrantly recognized and ubiquitinated by the ubiquitin ligase SKP1–CUL1–F-box protein (SCF) with the glycoprotein recognition component F-box protein 2 (FBS2) (SCFFBS2). These ubiquitinated products may readily accumulate and ultimately lead to proteasome dysfunction [14].
Silencing NGLY1 in mammalian cells using small interfering RNAs demonstrated that some proteins can undergo degradation independently of their glycosylation state [15]. Although it is not yet fully clear how these aggregates can be disposed of, the data suggest that the degradation of accumulated proteins may involve autophagic processes [16]. However, it is speculated that their, at least temporary, accumulation following aberrant ubiquitination by SCFFBS2 induces cytotoxicity through proteasome dysfunction and interferes with O-GlcNAc-mediated signaling [12, 16, 17].
NGLY1 is essential for processing the transcription factor nuclear factor
(erythroid-derived 2)-like 1 (NFE2L1), also known as NF-E2-related factor 1
(NRF1) [18, 19, 20]. The NRF1 referred to here should not be confused with the
nuclear respiratory factor 1, which also uses NRF1 as its abbreviation [19].
NGLY1-mediated deglycosylation of NFE2L1 enables the DDI1 homolog 2 (DDI2)
protein to cleave the N-terminal domain of NFE2L1, removing its ER anchor and
allowing it to translocate into the nucleus [19]. NFE2L1 is involved in the
transcription of a variety of genes, including those that control autophagy and
mitophagy [21]. This contributes to the regulation of cellular metabolic
homeostasis. NFE2L1 further regulates the expression of proteasomal subunit genes
[19, 22, 23, 24]. Interestingly, cells that are deficient in the DDI2 enzyme that
cleaves NFE2L1, or that have an impaired DDI2-NFE2L1 interaction, exhibit global
hyperubiquitination and increased cell death [25]. Furthermore, NFE2L1-mediated
activation of proteasome subunit gene expression protects from ferroptosis [25].
Ferroptosis is described as an iron-dependent form of regulated cell death, being
distinguished from other types of cell death like apoptosis by the accumulation
of lipid peroxides [26, 27]. By promoting the pathological death of individual
cells in autoimmune and autoinflammatory diseases, the dysregulation of
ferroptosis can contribute to prolonged inflammation and tissue injury. This
process also affects immune cells. Their ferroptosis-induced death or the
secretion of damage-associated molecular patterns (DAMPs) from dying cells can
either curb or trigger immune reactions, thus impacting disease progression [28].
Furthermore, it has been shown that the expression of the pro-inflammatory
cytokine tumor necrosis factor alpha (TNF
NGLY1 deficiency (NGLY1-CDDG) is caused by biallelic variants of NGLY1
and was the first congenital disorder of deglycosylation (CDDG1). It was
clinically described by Enns and colleagues [29] and first discovered in 2012
through exome sequencing of a three-year-old boy with various symptoms [30]. The
thorough examination of 12 NGLY1-CDDG patients that took place in 2017 aimed for
a comprehensive characterization of the disease [31]. This investigation led to
the identification of 13 distinct mutations that have the potential to induce
NGLY1 deficiency. The most common mutation observed in patients with NGLY1-CDDG
was c.1201A
NFE2L1, the transcription factor regulated by NGLY1 function, has a crucial role in maintaining the health and function of neurons and its deletion results in severe neurodegeneration [33, 34, 35, 36]. Postmortem analyses of patients with Alzheimer’s (AD) disease or Parkinson’s disease show reduced NFE2L1 expression compared to healthy subjects [37, 38]. Diminished NFE2L1 function reduces proteasome activity and the degradation of non-functional proteins [39, 40]. This can lead to the formation of aggregates and ER stress, both of which may contribute to the pathogenesis of AD and Parkinson’s disease [35]. A recent case report describes a 59-year-old patient with Parkinson’s disease and an autoimmune disorder who was found to carry a heterozygous pathogenic NGLY1 variant. The authors hypothesize that this heterozygous variant could contribute to Parkinson’s disease susceptibility in the context of immune dysregulation [41]. Another study examined the function of ER stress-related genes in brain tissue, finding 17 ER stress-related differentially expressed genes in AD patients’ samples compared to healthy controls. Interestingly, NGLY1 was one of the genes found to be significantly downregulated in AD patients, directly linking NGLY1 function to AD pathogenesis [42].
A recent study found that genes associated with ER stress, autophagy and proteasome processes were upregulated in induced pluripotent stem cell-derived neurons from NGLY1-CDDG patients, compared to cells from the same samples in which NGLY1 abundance was restored by reintroducing a functional NGLY1 sequence [17]. Furthermore, the expression of various genes associated with the oxidative stress response and a variety of mitochondrial processes was modified in the NGLY1-deficient cells, too. This resulted in defective clearance of protein aggregates, as determined by staining the cells with ProteoStat, a dye that detects protein aggregates. Additionally, the presence of severely fragmented mitochondria and impaired mitochondrial function was detected using Mitochondrial Encode Cytochrome C Oxidase II immunostaining and a JC-1 aggregate-forming cationic dye assay to measure the mitochondrial membrane potential [17]. Several independent studies analyzing the function of NGLY1 in human fibroblasts, mouse embryonic fibroblasts (MEFs) and human neurons supported that observation, reporting that NGLY1 deficiency led to severe mitochondrial fragmentation, reduced mitochondrial respiration and altered mitochondrial membrane potential [2, 17, 23]. Finally, the excessive disruption of mitochondria in combination with their impaired clearance in NGLY1 deficient MEFs induced chronically activated cytosolic nucleic-acid sensing pathways, causing an overall elevated cellular stress response [23].
These findings show the importance of NGLY1 in the regulation of cellular homeostasis and how its reduction or complete loss of function can shape diseases.
A functional immune system is our main protection against infections and uncontrolled cellular mutagenesis or cancer. A tight regulation is indispensable to prevent inadequate or excessive responses that lead to potentially fatal damage through uncontrolled infection, tumor growth, autoimmune responses or excessive inflammation. More often than not, it is only through the precise fine-tuning of immune cells that the appropriate immune response is achieved, protecting the organism against foreign structures (antigens) without causing unnecessary damage [43]. The cells of the innate immune system provide the body with a rapid, non-specific primary defense, offering immediate protection against a wide range of pathogens through physical barriers and cells such as macrophages, dendritic cells and natural killer cells. In contrast, the adaptive immune system takes more time to properly react to antigens, but it provides a highly specific defense against pathogens and establishes an immunological memory. This enables the body to recognize re-encountered pathogens, even decades after initial contact, for example through vaccination [44]. Adaptive immune cells are lymphocytes: T cells and B cells, which recognize specific antigens via their respective receptors (T cell receptors and B cell receptors). B- and T cell activation leads to a targeted, highly efficient and effective immune response upon current and future pathogen encounters [44]. After their activation, B cells can differentiate into antibody-secreting plasmablasts and plasma cells, with or without the help of T cells. Therefore, they mount an acute humoral immune response and provide long-term titers of protective antibodies [45, 46]. Thus, the innate immune system initiates a rapid response to pathogens, while the adaptive immune system provides a long-lasting, antigen-specific defense. In order to function properly, activated immune cells require constant protein translation and turnover [44, 45].
As previously outlined, NGLY1 plays a regulatory role in numerous intracellular pathways and is involved in vital transcriptional and metabolic processes, including protein processing. The significance of mitochondria and metabolism in the functions of immune cells has been recognized more in the past decade. They impact T cell and macrophage function via oxidative phosphorylation (OXPHOS), regulate inflammation, and maintain epigenetic modifications [47]. Consequently, substantial alterations to the mitochondrial compartment are likely to impact immune functions through diverse mechanisms. The mitochondrial dysfunction seen in NGLY1-deficient neurons suggests that NGLY1 plays a significant role in immunity [2, 17, 23]. This review aims to shed light on the potential role of NGLY1 in immune cell regulation and function.
A lack of NGLY1 led to increased transcription of interferon-stimulated genes (ISGs) due to impaired NFE2L1 signaling in patient-derived lymphoblastoid cells [23]. The type I Interferon response is a vital defense mechanism in viral infections and can modulate immune responses [48]. It can stimulate the proliferation of cytotoxic CD8+ T cells by providing costimulatory effects [49]. Yang and colleagues [23] suggest that an increased interferon-type I response in NGLY1-deficient cells is linked to changes in mitochondrial function, due to impaired mitophagy and subsequent activation of innate immune sensors for free mitochondrial-derived cytosolic nucleic acids (mtDNA), Cyclic GMP-AMP-Synthase (cGAS), inducing stimulator of IFN genes (STING), and Retinoic Acid Inducible Gene 1 (RIG-1). Another study describes a mechanism of reciprocal regulation between STING and T cell receptor signaling [50]. Using T cells from wild-type and STING-deficient mice, the authors demonstrated that STING activation only inhibits mechanistic target of rapamycin complex 1 (mTORC1) signaling in wild-type T cells [50]. mTORC1 is a well-described protein complex that plays a crucial role in orchestrating growth and metabolism in a wide variety of tissues [50, 51]. It promotes anabolic processes, such as protein and organelle synthesis, while suppressing catabolic processes, such as autophagy (cellular self-degradation) [52]. Thus, mTORC1 acts as a sensor for growth signals, such as nutrients, hormones, and growth factors, linking cellular energy availability to cell growth and proliferation [50, 53]. Imanishi and colleagues demonstrated that activating mTORC1 through T cell receptor stimulation is essential for STING-mediated type I interferon (IFN-I) production, while partial inhibition of mTORC1 through STING activation results in impaired proliferation of naïve CD4+ T cells upon anti-CD3/CD28 stimulation [50]. This balanced coordination allows IFN-I production from antigen-specific T cells without promoting their excessive proliferation. Based on these data, we propose that NGLY1 activity could tip this coordinated balance and may regulate the production of inflammatory cytokines in immune cells, such as T cells. Thereby, NGLY1 could control even further immune responses downstream of the cytokine induced signaling cascades (Fig. 2).
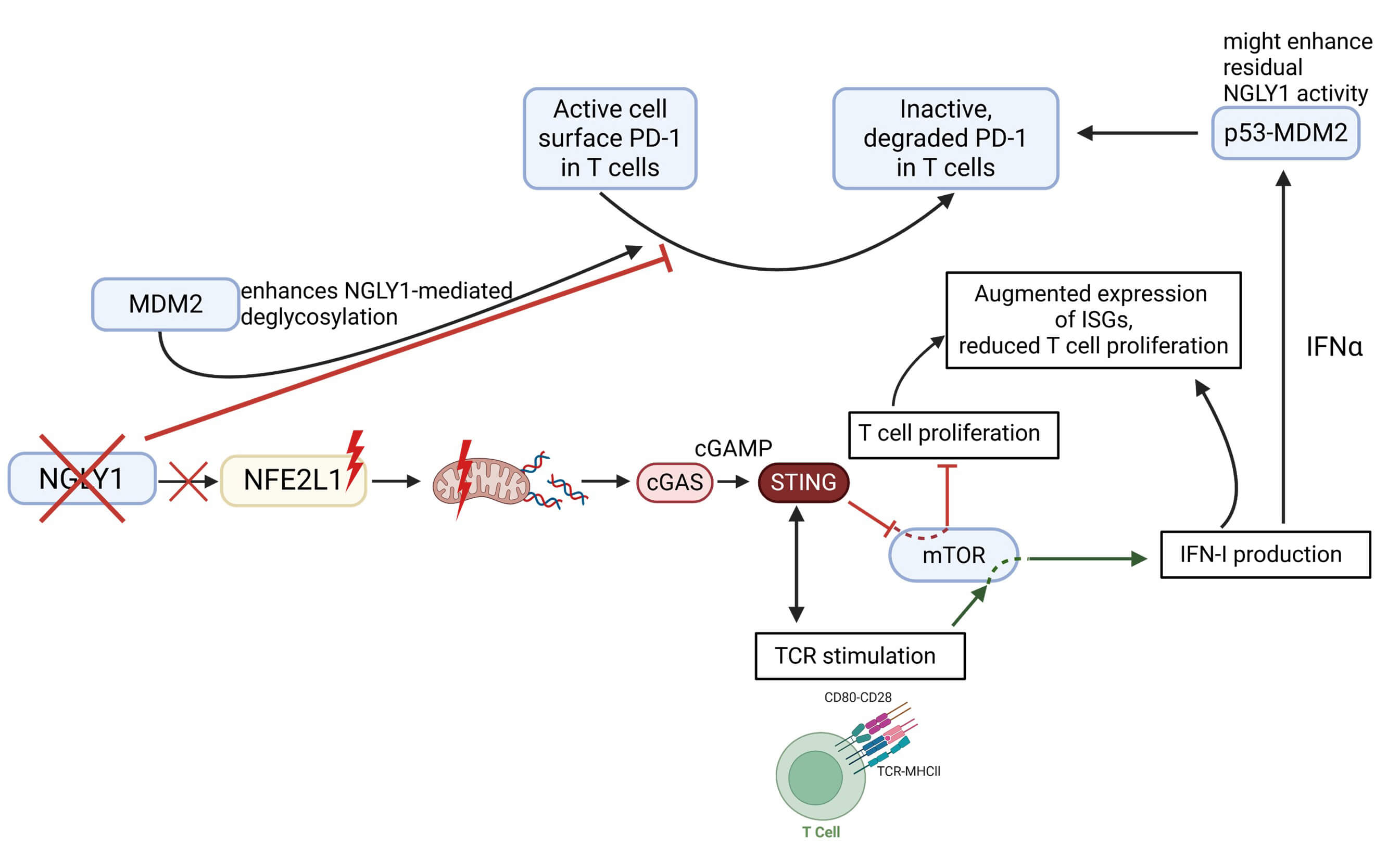 Fig. 2.
Fig. 2.
Proposed model of potential impact of NGLY1 deficiency on T
cells based on known mechanisms. When N-Glycanase 1 (NGLY1) activity is lost,
the transcription factor nuclear factor (erythroid-derived 2)-like 1 (NFE2L1)
remains inactive. This leads to disturbed mitochondrial homeostasis and the
release of mitochondrial DNA (mtDNA). The activation of the cyclic GMP-AMP
synthase (cGAS)-stimulator of interferon genes (STING) pathway may result in
reduced T cell proliferation through STING-mediated inhibition of mechanistic
target of rapamycin (mTOR), as well as interferon (IFN)-I production via T cell
receptor (TCR)-mediated stimulation of mTOR. Release of IFN
Interestingly, in a case study analyzing blood samples from a NGLY1-CDDG patient, elevated antibody titers against rubella and/or rubeola following the administration of the measles, mumps and rubella triple vaccination were described [31]. Based on these observations, we speculate that there may be alterations to the B cell compartment and populations of antibody-secreting cells in NGLY1-CDDG patients. This could potentially be caused by NGLY1’s regulatory role in the B cell compartment or antibody production. However, an altered B cell response could also be a secondary effect of the aforementioned alterations in NGLY1-deficient T cells and their activity. Nevertheless, no studies have yet investigated the role of NGLY1 in B cells or B cell-related disorders.
A recent study identified a connection between protein deglycosylation and T cells, implicating a potential role of NGLY1 in the context of cancer [54]. Analyzing the mechanisms involved in programmed cell death protein 1 (PD-1) degradation, mouse double minute 2 homolog (MDM2) was identified as a key regulator of T cell activation and a link between deglycosylation processes and ubiquitination [54]. PD-1 is upregulated after T cell activation. In fact, it inhibits activated T cells and prevents them from becoming overactive and attacking the body’s own tissues. PD-1 therefore functions as an important immune checkpoint, maintaining homeostasis in the immune system [55]. Indeed, mice that are deficient in PD-1 have developed accelerated autoimmunity [56, 57, 58, 59, 60]. Importantly, the transmembrane protein PD-1 must be glycosylated to be functional and its deglycosylation and ubiquitination are essential for its efficient degradation on T cells [54]. Unfortunately, tumor cells have learned to exploit the PD-1 axis by upregulating PD-1 ligand 1 (PD-L1), thereby inhibiting the anti-tumor response of T cells [61]. Thus, not only PD-1 abundance but also its glycosylation has to be tightly regulated to maintain immune cell homeostasis. We therefore suggest a potential role of NGLY1 in the mechanisms controlling T cell activity via PD-1 (Fig. 2).
Regarding MDM2, it is crucial to highlight that it displays no deglycosylase
activity on its own. Potential deglycosylases involved in the process were
identified by investigating the binding partners of the PD-1 protein complex
[54]. Using HEK293T cells for protein complex purification followed by mass
spectrometry analysis and deglycosylation assays assessed by Western Blot
analysis, NGLY1 was identified as the enzyme responsible for PD-1 deglycosylation
in vitro [54]. As deglycosylated PD-1 is easily ubiquitinated and
degraded, we hypothesize that NGLY1-mediated destabilization of PD-1 might be an
important regulator of its function. Our suggestion is supported by a study of
the human osteosarcoma cell line U2OS and its interactions with the immune
system. The study revealed that loss or overexpression of NGLY1
increased or decreased PD-1 levels, respectively, which was associated with a
change in PD-1 deglycosylation [54]. Conversely, neither the knockdown nor the
overexpression of NGLY1 initiated a substantial alteration in the amount
of glycosylated PD-L1. This observation suggests that, in this context, NGLY1
specifically targets PD-1, while other enzymes regulate PD-L1 glycosylation and
activity. NGLY1-mediated deglycosylation of PD-1 was found to be enhanced by MDM2
and reduced in its absence, indicating MDM2’s role in potentiating the
interaction between NGLY1 and PD-1 [54]. Interestingly, MDM2 is mainly known as
an oncogene that is overexpressed in cancer cells, where it downregulates the
tumor suppressor protein p53 and thereby promotes tumor cell growth [62]. By
contrast, analysis of primary T cells revealed that stimulation with type I
interferon (IFN-
Based on these results and previous studies, we propose that the induction of type I interferons is associated with NGLY1 function and connected to both MDM2 and PD-1 (Fig. 2). Upon partial or complete loss of NGLY1, PD-1 cannot be deglycosylated and degraded properly in T cells [54]. However, at the same time and controlled by the cGAS/STING pathway, reduced NGLY1 abundance elevates type I interferon expression [23]. We propose that this mechanism could ultimately lead to the activation of the p53/MDM2 axis, creating a feedback loop that destabilizes PD-1 by enhancing residual NGLY1 activity.
A study of doxorubicin, a popular chemotherapy drug, supports the connection between NGLY1 activity and PD-1 function in disease settings [61]. A proximal ligation assay using U2OS cells revealed that doxorubicin induces PD-1 destabilization through its enhanced interaction with NGLY1, resulting in increased NGLY1-mediated deglycosylation and subsequent PD-1 degradation. As cancer cell-intrinsic PD-1 sensitizes tumor cells to doxorubicin, NGLY1-mediated degradation directly counteracts the drug’s anti-proliferative effect, potentially diminishing its anti-tumor efficacy [61]. Thus, the authors discussed that patients may benefit from a combined approach of targeting NGLY1 specifically in tumor cells while using doxorubicin to restore and even enhance the drug’s anti-tumor effects. It should be noted that PD-1 is also expressed on B cells, natural killer cells and myeloid lineage cells [59]. However, this fact is not widely known and the function of PD-1 in these cells has not yet been fully elucidated. This leaves room for further potential functions of NGLY1 in these immune cells.
We discussed whether NGLY1 could play a pivotal role in T cell activity by regulating the deglycosylation-facilitated degradation of PD-1. This process has been investigated in cancer cells, but not in T cells. However, we hypothesize that NGLY1 deficiency could result in a diminished T cell response through stabilized PD-1. This PD-1 regulation could be beneficial for the treatment of cancer patients if tumor cells themselves are NGLY1-negative. Conversely, NGLY1 upregulation might induce a heightened state of T cell activity, which may contribute to an elevated risk of autoimmunity.
Another potential mechanism by which NGLY1 may influence T cell signaling is via
the AMP-activated protein kinase (AMPK). By inducing catabolic pathways, such as
autophagy or fatty acid oxidation, AMPK plays a pivotal role in the regulation of
cellular energy homeostasis [63]. Importantly, a recent study analyzing
NGLY1-deficient murine and human fibroblasts revealed that NGLY1 plays a role in
decreasing the protein abundance of an AMPK catalytic subunit, AMPK
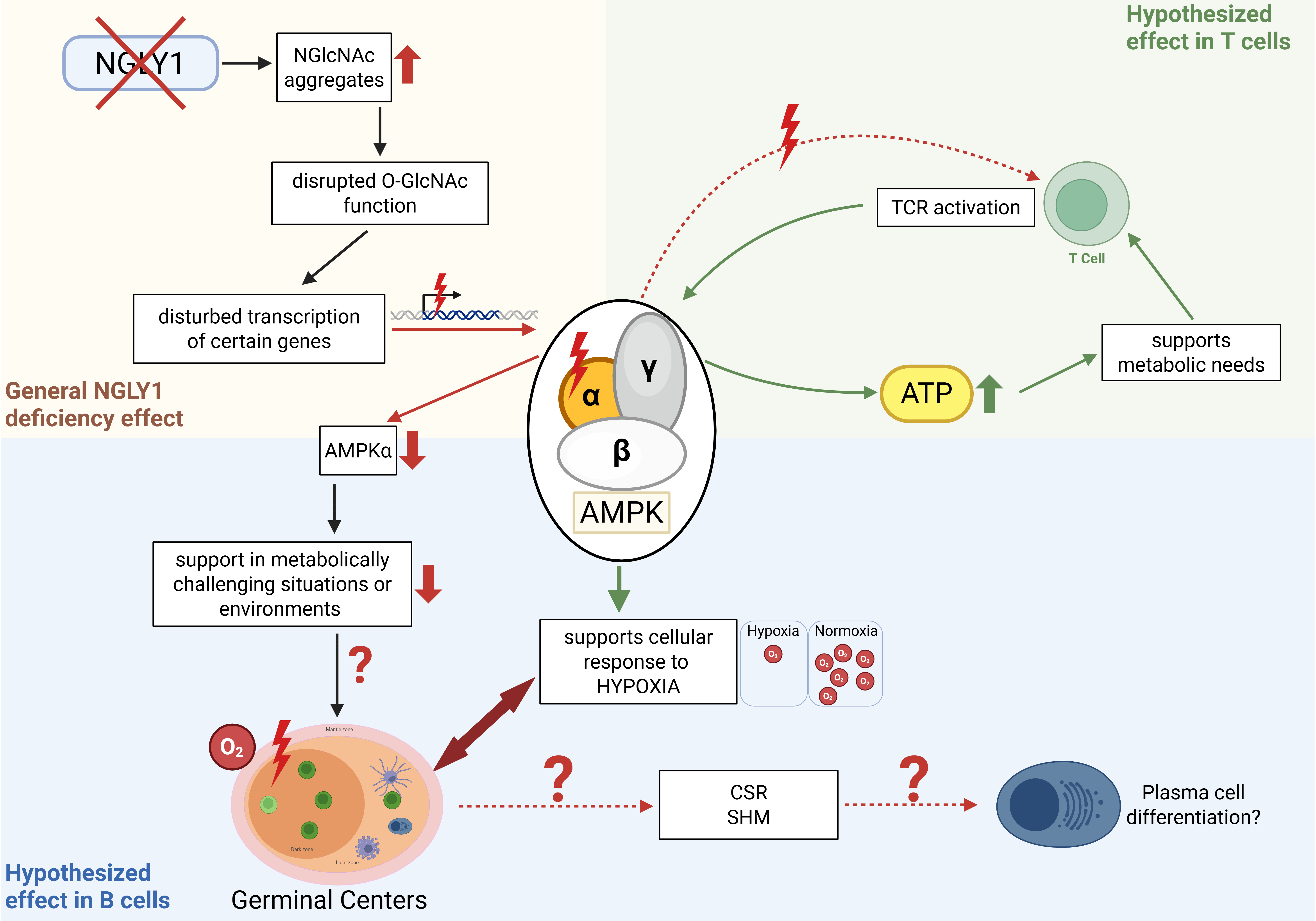 Fig. 3.
Fig. 3.
Described function of NGLY1 in AMPK
In an immunological context, a well-studied hypoxic environment that could be
affected by NGLY1 deficiency and subsequent AMPK
These studies demonstrate that NGLY1 plays a pivotal role in a number of processes related to (de)glycosylation and glycan modifications, thus underscoring its critical function in regulating comprehensive cellular processes and mechanisms. As NGLY1 deficiency impairs AMPK function and consequently an essential regulatory mechanism in hypoxic conditions, we hypothesize that T and B cells lacking NGLY1 may face challenges in establishing an adequate germinal center response. This could result in reduced antigen-specific humoral immunity (Fig. 3).
NGLY1’s deglycosylating function not only removes glycans from proteins, but also hydrolytically cleaves the N-Glycan bond between the asparagine residue and the first N-acetylglucosamine sugar. The amide group of asparagine (Asn) is broken off in the process, which results in its conversion to aspartic acid (Asp) [72]. The deamidation of Asn to Asp within a glycosylation motif (NX(S/T)) resulting from the deglycosylation of an immunogenic tyrosinase peptide has been described as an important regulatory mechanism since tyrosinase-specific cytotoxic T cell receptors favor the deamidated peptide [73]. Thus, NGLY1 could play an important role in activating cytotoxic T cells, which are essential for clearing infected and cancerous cells. However, they can also contribute to the development of autoimmune diseases [74]. Importantly, studies using DM331 melanoma cells and the molecule Z-VAD-FMK, which is an inhibitor of caspases but was shown to block PNGase activity in yeast and mammalian cells [75], revealed that this post-translational modification from Asn to Asp requires the glycosylation step in the endoplasmic reticulum (ER) prior to its deglycosylation by NGLY1 and cannot occur otherwise [76].
In 1999, two independent studies showed that epitopes of viral antigens presented by the major histocompatibility complex (MHC) class I exhibited deamidation of Asn residues [77, 78]. MHC-I molecules bind to and present proteins or peptides that are generated within the cell on the cell’s surface. This activates cytotoxic T cells, which destroy the infected cell and restrict the pathogen’s spread in the body. Thus, MHC-I plays an essential role in identifying foreign intracellular structures, such as viral particles. However, endogenous antigen presentation is also crucial in preventing autoimmunity by presenting self-antigens to T cells in the thymus. T cells that bind to self-antigens with their T cell receptor are eliminated, establishing central tolerance and restricting autoimmune reactions [79]. Ferris and colleagues’ study [77] from the end of the last century aimed to better characterize the transport, processing and loading of HIV envelope proteins onto MHC-I via Transporter Associated with antigen Processing 1/2 (TAP1/2). They used B-lymphoblastoid cell lines infected with a virus expressing the complete HIV envelope protein (gp120 and gp41) to serve as target cells presenting env-derived antigens to envelope-specific cytotoxic T cells. They found that after the retro-translocation from the ER to the cytosol, certain Asn residues within the epitopes of both envelope subunits undergo deglycosylation mediated by NGLY1, resulting in their conversion to Asp. Crucially, recognition of these Asp-containing epitopes on MHC-I by the specific cytotoxic CD8+ T cells was considerably enhanced, resulting in significantly increased lysis of the infected cells [77]. A second study from the same year found a high level of mechanistic consistency when analyzing the loading of hepatitis C virus envelope protein E1 to MHC-I in a chimpanzee cell line, indicating that this mechanism is not restricted to a single virus strain or organism [78].
A more recent study that examined posttranslational modifications of MHC-I-presented peptides in more detail found that 2.5–7% of peptides contained deamidated residues, with the deamidation of Asn being far more prevalent than the deamidation of glutamine [80, 81]. Furthermore, a significant proportion of those peptides with deamidated Asn also exhibited the aforementioned NX(S/T) glycosylation motif described for CD8+ cytotoxic T cell mediated cell lysis. Finally, in vitro inhibition of NGLY1 by Z-VAD-FMK in the Epstein-Barr virus-transformed B-lymphoblastoid cell line CIR1 and subsequent isolation of peptides from MHC-I complexes revealed a drastic reduction in deamidated NX(S/T)-containing peptides, but no other Asn-containing motifs [80]. These findings suggest that NGLY1 significantly contributes to the antiviral response of cytotoxic immune cells by regulating the loading of viral peptides to MHC-I complexes. In addition, it is described that MHC-I usually strictly incorporates peptides that are non-glycosylated [81]. Therefore, regardless of their origin, the deglycosylation of proteins or peptides is essential for their correct presentation by MHC-I, and consequently for the efficient activation of the immune response against intracellular pathogens or for the central tolerance in the thymus.
Based on these studies, we suggest that a full protection against certain viruses by vaccination may not be guaranteed in NGLY1-CDDG patients, potentially also depending on their residual NGLY1 activity. Furthermore, a lack of proper deglycosylation of self-antigens in their thymus could lead to inadequate MHC-I-mediated presentation during central tolerance. This may result in an elevated number of auto-reactive T cells and therefore, hypothetically, in the patients’ increased risk of developing autoimmune diseases, such as rheumatoid arthritis (RA).
Another mechanism how NGLY1 could impact antigen recognition by immune cells is via MHC-II-mediated presentation of peptides. MHC-II binds and presents proteins or peptides that were internalized by phagocytosis or pinocytosis, so molecules that do not derive from the cell itself [82]. Thus, it is vital for recognizing pathogens and developing tolerance to self. However, it is also crucial for peripheral tolerance, which prevents autoimmunity. T cells bind to MHC-II-presented self-antigens via their T cell receptor. If they do not receive co-stimulatory or inhibitory signals, the T cell is deleted by apoptosis or becomes anergic or exhausted. The co-stimulatory signal is induced by a pathogen-associated molecular pattern (PAMP, e.g., LPS) or a damage-associated molecular pattern (DAMP, released by stressed, damaged, or dying cells) recognized by the pathogen recognition receptor (PRR) of the antigen-presenting cell. MHC-II is also essential for the development and function of regulatory T cells, which in turn inhibit the activation of cytotoxic or T effector cells and B cells [83]. In contrast to MHC-I, MHC-II binding pockets are three-dimensionally less confined and, therefore, allow a less restricted repertoire of peptides to bind. In addition, described molecular models suggest that glycosylated proteins can be incorporated into the MHC-II complex [81]. Importantly, DAMPs and PAMPs both can be glycosylated, too [84, 85]. Thus, NGLY1 may not only play a significant role in the deglycosylation of individual proteins presented by MHC-II, but also in the induction of the co-stimulatory signal to T cells, by regulating the glycosylation of DAMPs. The presentation of foreign antigens by B cells via MHC-II is also essential for their T-dependent activation, since T helper cells only support them when they recognize the presented antigen via their own T cell receptor.
These studies suggest that NGLY1 deficiency in antigen-presenting cells may disrupt the recognition and presentation of pathogens and self-antigens by MHC molecules, potentially leading to immune system imbalances or autoimmune reactions. We suggest that NGLY1 might play a pivotal role in integrating innate and adaptive immunity by augmenting the variety of the immunopeptidome.
A study on B cell precursor acute lymphoblastic leukemia with mixed-lineage leukemia gene rearrangement (MLL-r) found drastic remodeling of the leukemia cells’ glycome compared to healthy B cell progenitors by integrating transcriptomics, proteomics and glycomics on primary patient-derived cells [86]. This approach revealed extensive changes in the glycocalyx, which is a solid layer of carbohydrates including glycoproteins, proteoglycans and glycolipids arranged in a dense network on the cell surface. Such profound glycome remodeling impacts leukemia cell survival. The study identified NGLY1 as being required for the survival of leukemia cells, suggesting that proper glycoprotein quality control is critical for maintaining leukemia cell viability. Thus, we suggest that NGLY1 could indirectly affect the extensive glycome remodeling in MLL-r leukemia and further contribute to cell survival by providing a functional processing and clearance machinery for misfolded glycoproteins. A strengthened glycocalyx protects cancer cells from immune cells, enabling evasion and metastasis [87, 88]. Furthermore, the glycocalyx influences a vast number of biological processes also impacting immune cell activity. Its functions include protecting cells against mechanical or chemical stress as a physical layer and controlling the permeability of endothelial cells in blood vessels to allow proper blood flow. In addition, it binds soluble molecules such as growth factors, influencing their binding to surface receptors and intracellular signaling, as well as intercellular recognition, adhesion and migration [89]. These functions are all essential for B and T cells.
Based on these studies we suggest that NGLY1 plays a vital role in the healthy functioning of adaptive immune cells and the regulation of autoimmune responses. Intense studies on pre-clinical models and translational systems are necessary to determine NGLY1’s function in adaptive immune cells and understand the mechanisms by which it controls them. These studies may provide further insight into the regulation of the adaptive immune response. This knowledge is crucial for a significant proportion of patients, including those with rare NGLY1-CDDG, but may also benefit cancer patients or those with autoimmune or autoinflammatory diseases.
An extensive recent N-glycoproteomics study of NGLY1-CDDG patient-derived dermal fibroblasts identified a variety of proteins with altered glycosylation patterns compared with those derived from healthy individuals [90]. Among the top 20 glycoproteins which exhibit frequent alterations in glycosylation, two have known functions in the immune system.
The C-type mannose receptor 2 (MRC2) presented a reduced abundance of glycopeptides in NGLY1-deficient cells. Mannose receptors are an essential component of the antigen presentation machinery in dendritic cells, which patrol the body’s tissues to detect foreign invaders such as viruses and bacteria [91]. Their primary function is to capture antigens and present them to T cells, thereby activating the adaptive immune response. In dendritic cells, MRC2 is rapidly recycled between the plasma membrane and early endocytic compartments, functioning as a high-capacity, broad-specificity antigen receptor. The uptake of antigens by dendritic cells via MRC2 is approximately 100 times more efficient than the internalization of antigens by the fluid phase [92]. We already described the potential role of NGLY1 in antigen-presentation by MHC molecules as an interface between innate and adaptive immunity. In addition, MRC2-mediated internalization of antigens strongly enhances human leukocyte antigen (HLA) class II-restricted antigen presentation on dendritic cells. This is the human equivalent of mouse MHC-II molecules [91]. Although the precise implications of downregulating MRC2 glycopeptides in NGLY1-CDDG remain unclear, these findings provide further support for the potential involvement of NGLY1 in MHC/HLA class II-mediated antigen presentation.
As a second interesting protein originating from the study by Budhraja and colleagues, glycopeptides from the interleukin-17 receptor A (IL-17RA) were more abundant in NGLY1 deficient cells [90]. Compared to other cell types in the body, IL-17RA shows an essentially higher expression in several immune cells, especially in granulocytes and monocytes [93]. IL-17A is a proinflammatory cytokine predominantly secreted by T helper 17 cells. It induces cytokine and chemokine release in dendritic cells, lymphocytes and monocytes, leading to the recruitment of additional immune cells, especially neutrophils, and the production of antimicrobial proteins at the site of infection [94]. As reviewed extensively elsewhere, IL-17 is also described to be involved in the development of RA [95]. Finally, it is still unclear how altered glycosylation of IL-17RA could impact the binding of secreted IL-17A or how this might affect signal transduction. However, these findings indicate a link between NGLY1 activity and the function of the innate and adaptive immune system through antigen presentation and cytokine activity. We therefore suggest NGLY1 as a potential fine-tuner of inflammatory responses.
Besides these two clearly immune system-associated proteins, the glycosylation of several extracellular matrix proteins was also altered in NGLY1 deficient fibroblasts [90]. This observation is tightly associated with the suggested role of NGLY1 in influencing the reshaping of the glycocalyx on the MLL-r B-cell precursor acute lymphoblastic leukemia cells described before. A co-culture study of fibroblasts and endothelial cells showed that the composition and density of the extracellular matrix of endothelial cells is altered by fibroblast activity [96]. Thus, changes in the extracellular matrix proteins produced by fibroblasts have an essential influence on the migration, adhesion and extravasation of innate immune cells, which transmigrate along cytokine gradients through the basement membrane after successfully crossing the vascular endothelium to reach adjacent tissues [97]. Importantly, fighting pathogens or detecting antigens in muscles post-vaccination depends on innate immune cells invading and crossing the vascular endothelium. These cells transport pathogens into the lymphatic system and activate adaptive immune cells [98]. This mechanism is essential for an effective tissue-specific immune response, but also associated with inflammatory diseases such as RA [99, 100]. Thus, by shaping the features of vascular endothelial cells we hypothesize that NGLY1 affects the innate and adaptive immune cell function by controlling tissue specific pathogen recognition and local immune cell infiltration.
As previously discussed, increased activation of stimulator of IFN genes (STING)
is a clearly described phenotype in NGLY1 deficient cells [23]. Further analysis
of MEFs demonstrated that the increase in STING activation dependent on NGLY1
also relies on the function of cGAS upstream (Fig. 2). The same mechanism could
be identified in murine macrophages and in lymphoblastoid cell lines [23]. This
is of particular interest for innate immune cells as STING activation induced by
the release of nucleic acids from acute myocardial infarction was described to
repolarize anti-inflammatory M2 macrophages into pro-inflammatory M1 macrophages
[101]. Another inflammatory mechanism that was described in murine mast cells
showed that NFE2L1, that is activated by functional NGLY1, induces TNF
Based on the described mechanisms, we hypothesize that NGLY1 is not only involved in adaptive immune regulation, but can also affect innate immunity. This could occur through the regulation of immune receptor glycosylation and, consequently, activity, or through inflammatory signals via NFE2L1 function. Finally, we hypothesize that NGLY1 may impact the homeostasis of innate immune cells and the initiation of inflammatory responses by regulating the activity of the cGAS-STING pathway.
Our society is facing an increasing threat from cancer, autoimmune diseases, and
bacterial or viral infections. The Western world, in particular, is witnessing a
consistent rise in the prevalence of autoimmune and autoinflammatory diseases
[107, 108]. Although checkpoint inhibitors (
Evading internal control mechanisms, pathogenic plasma cells secrete self-reactive auto-antibodies such as the anti-citrullinated protein antibody (ACPA) that plays a critical role in the onset of RA [110]. RA patients, who are 3-times more often female than male, suffer from synovial inflammation and joint destruction, which drastically reduces their quality of life [111]. Previous studies demonstrated that plasma cells are enriched in the synovium of many RA patients and that this can be associated with a reduced therapy response rate, indicating a critical role of plasma cells for successful RA treatment [112]. The underlying causes of the onset of RA are not yet fully understood. However, it was suggested that it is most likely the result of an interplay between genetic and environmental factors.
A recent study analyzing single-cell RNA sequencing data from the joints of RA patients using bioinformatics and machine learning techniques identified lactylation as a posttranslational modification associated with RA [113]. During lactylation, a lactyl group is attached to a protein, a process that has been most extensively studied in relation to the amino acid residue lysine [114]. Lactylation is driven by lactate accumulation, which is largely generated during non-oxidative glycolysis. When lactate combines with coenzyme A (CoA), lactyl-CoA is generated, which then transfers a lactyl group to specific proteins. Lactylation of histones can alter chromatin structures and thereby control gene expression [115]. Thus, lactylation links the metabolic state of the cell to gene expression and has been described as a new regulator of biological processes with medical implications. For example, by promoting the expression of anti-inflammatory genes in immune cells such as macrophages, it is postulated to regulate inflammation [115, 116]. However, aberrant lactylation has also been correlated with progression of diseases including cancer and neurological disorders [114].
Single-cell RNA sequencing data from the synovial tissue of RA patients revealed an increase in the expression of core lactylation-promoting genes, identifying NGLY1 as one of them [113]. NGLY1 expression was specifically increased in a group of plasma cells with a high lactylation score found in RA tissues. These cells also showed pathways linked to oxidative phosphorylation, glycolysis and ER stress, indicating enhanced metabolic activity. They also exhibited increased MHC-II expression, which may also be linked to NGLY1 activity. This observation could be a further indication of enhanced antigen presentation by pathogenic plasma cells in RA joints [113]. Indeed, MHC-II-expressing plasma cells have been associated with autoimmune diseases, such as systemic lupus erythematosus [117]. However, MHC-II downregulation has also been linked to the maturation of antibody-secreting cells from early plasmablasts to long-lived plasma cells [118]. Based on these data we could also hypothesize that plasma cells associated with inflammatory or autoimmune diseases have been generated more recently and potentially are a result of chronic B cell activation and differentiation. Thus, analyzing inflamed tissues from RA patients indicates that NGLY1 expression is increased, most likely in antibody-secreting cells, and is associated with the development of the disease.
Interestingly, NGLY1 was also shown to be upregulated in melanoma cell lines and patient-derived tumors compared to human normal melanocytes or human pluripotent stem cells [119]. Suppressing NGLY1 in melanoma causes an anticancer response, suggesting that NGLY1 could be a target for melanoma therapy. However, only a few studies examined the effect of ectopic overexpression of NGLY1 for a broader purpose than just rescuing a NGLY1-deficiency. Further investigation of NGLY1-controlled mechanisms in immune cells may provide new insights and result in novel therapeutic strategies for inflammatory or autoimmune diseases.
Viral infections, including the common cold and the novel virus responsible for the global Coronavirus Disease 2019 (Covid-19) pandemic, are part of our daily lives. They are usually kept at bay by our immune system. When we do become ill after all, our immune system works at full capacity to mount a pathogen-specific immune response. However, some people are more susceptible to infections than others and especially children or the elderly are more prone to viral infections. In a “The New Yorker” article from 2014, Mnookin described that parents of NGLY1-CDDG children often observed that they seem to be resistant to viral infections that are common during childhood [120]. It was suggested that this apparent viral resistance could be due to viruses not being able to spread further after attaching to defective glycoproteins on host cells. This hypothesis was based on a recently published article addressing viral infections in patients with another disorder of deglycosylation [120]. This case report described that children suffering from the mannosyl-oligosaccharide glucosidase congenital disorder of glycosylation (MOGS-CDG) are less susceptible to viral infections, probably through the impaired entry and replication of viruses, both depending on glycoproteins [121]. MOGS is the first enzyme involved in the processing of N-glycans after they are initially attached to a protein. Thus, MOGS-CDG patients have an impairment in the subsequent processing and modification of the protein-bound glycans, inhibiting calnexin/calreticulin dependent quality control. This results in the accumulation of improperly processed glycoproteins [121, 122].
However, although patients with MOGS-CDG and NGLY1-CDDG both show increased resistance to viral infection, the potential underlying mechanisms are fundamentally different: While MOGS-CDG patients’ impairments in glycoprotein processing lead to viral resistance, it was shown that interferon stimulated genes are elevated in NGLY1-deficient mouse embryonic fibroblasts and that loss of NGLY1 enzymatic activity was the critical factor driving that upregulation [23]. Following up on the finding that resistance to viral infections is exhibited by cells upon cell intrinsic activation of these interferon stimulated genes, NGLY1-deficient MEFs were infected with vesicular stomatitis virus. Indeed, reduced virus replication was found in NGLY1-deficient cells. Mechanistically, increased phospho-STING in NGLY1-deficient MEFs correlated with the activation of the STING-pathway. As described before, the authors suggested that the cGAS/STING pathway is the main driver for NGLY1-mediated activation of interferon stimulated genes, resembling an antiviral state [23]. One of the main intracellular sources for endogenous ligands of the cGAS/STING pathway is mtDNA [23, 123, 124, 125]. Thus, the authors also analyzed mitochondrial structure and function in the NGLY1-deficient MEFs [23]. They observed that a loss in NGLY1 resulted in fragmentation of mitochondria and impaired mitophagy, causing a release of free mtDNA into the cytosol, thus activating the cGAS/STING pathway. This research reveals a crucial role of NGLY1 in mitochondrial homeostasis, activation, and shaping of immune responses. Further analysis of this NGLY1-driven mechanism could identify strategies for targeting autoimmune disorders and influencing the course of viral infections.
In this review, we highlighted NGLY1’s function and suggested its potential role in the context of several immunologically relevant pathways. Until now, most studies have investigated NGLY1 in the context of the congenital loss of function disorder. In most cells, NGLY1 deficiency generally leads to cytotoxicity through protein accumulation and a higher cellular stress response through mitochondrial damage. It also results in dysfunctional ferroptosis regulation leading to hyperubiquitination and increased cell death. These processes typically trigger the activation of immunological pathways. Table 1 (Ref. [2, 17, 22, 23, 54, 65, 76, 86, 90, 103, 119]) provides a summary of non-immune cells in which NGLY1-related mechanisms have been demonstrated.
| Cell type | Mechanisms shown | Ref. |
| DM331 melanoma cells | Asn to Asp conversion requires the glycosylation step in the ER prior to its deglycosylation by NGLY1 | [76] |
| HEK293T | NGLY1 is responsible for PD-1 deglycosylation in vitro | [54] |
| Human neurons | NGLY1 deficiency leads to severe mitochondrial impairment | [2, 17, 23] |
| (NGLY1-deficient) Human fibroblasts | • NGLY1 deficiency impairs AMPK function | [2, 17, 23, 65, 90] |
| • Altered protein glycosylation patterns upon NGLY1 deficiency | ||
| • Altered protein glycosylation of ECM proteins upon NGLY1 deficiency | ||
| • NGLY1 deficiency leads to severe mitochondrial impairment | ||
| Human umbilical vein endothelial cells | iNOS is negatively regulated by NFE2L1 | [22, 103] |
| (NGLY1-deficient) MEFs | • NGLY1 deficiency impairs AMPK function | [2, 17, 23, 65] |
| • NGLY1 deficiency activates cGAS-STING pathway | ||
| • Reduced virus replication in NGLY1-deficient cells | ||
| • NGLY1 deficiency leads to severe mitochondrial impairment | ||
| Melanoma cell lines & patient derived tumors | NGLY1 is upregulated in individual tumor cell lines and NGLY1 suppression causes anticancer responses | [119] |
| MLL-r | NGLY1 is one of the genes required for the leukemia cell´s survival | [86] |
| U2OS human osteo-sarcoma cell line | A change in NGLY1 levels induces a change in overall PD-1 levels | [54] |
Examining specific cell types that demonstrate NGLY1’s involvement in regulating
the STING signaling pathway suggests NGLY1 may influence T cell proliferation by
regulating STING signaling and T cell activity via PD-1. Studies in cancer cells
show that upregulation of NGLY1 results in lower abundance of PD-1. Applying this
mechanism to T cells suggests that NGLY1 upregulation could hyperactivate T
cells, increasing the risk of autoimmunity. Therefore, analyzing the role of
NGLY1 in T cell homeostasis and cancer treatment could be a promising area of
future research. Furthermore, it appears that NGLY1 affects the presentation of
antigens by MHC/HLA molecules. However, its actual impact on antigen-presenting
cells must be reliably determined. NGLY1 also influences AMPK
Currently, there is no list of NGLY1 substrates. Such a list would demonstrate the extent to which NGLY1 is involved in immune cell function. To understand the role of NGLY1 in the immune system, we first need to improve our understanding of its regulatory capacities in immune cells. This will require further research.
Based on the described studies, we propose that NGLY1 might play a key role in maintaining a healthy immune system and could be exploited as a target for novel therapies aimed at fine-tuning overshooting or underperforming immune responses in the context of autoimmune and auto-inflammatory diseases as well as cancer.
AMPK, AMP-activated protein kinase; Asn, asparagine; Asp, aspartic acid; BCR, B cell receptor; cGAS, cyclic GMP-AMP synthase; CDG, Congenital Disorders of Glycosylation; DDI2, DDI1 homolog 2; ENGase, endo-beta-N-acetylglucosaminidase; ER, endoplasmic reticulum; ERAD, ER-associated degradation; GlcNAc, acetylglucosamine; IL-17RA, interleukin-17 receptor A; iPSC, Induced pluripotent stem cells; JC-1, J-aggregate-forming cationic dye; iNOS, inducible nitric-oxide synthase; MDM2, mouse double minute 2 homolog; mtDNA, Mitochondrial DNA; MEF, Mouse embryonic fibroblasts; MHC, major histocompatibility complex; MTCO2, Mitochondrially Encoded Cytochrome C Oxidase ll; NFE2L1, nuclear factor (erythroid-derived 2)-like 1; NRF1, NF-E2-related factor 1; OXPHOS, oxidative phosphorylation; PBMCs, peripheral blood mononuclear cells; PD-1, programmed cell death protein 1; PD-L1, programmed cell death protein 1 ligand 1; RIG-1, Retinoic Acid Inducible Gene 1; STING, stimulator of IFN genes; TCR, T cell receptor; TNF
KP conceived the review, with MeS and MaS generating the first outline. CBB and KP wrote the manuscript, supported by FK. CBB, MO, FK, MeS and MaS generated the figures. UGD performed literature screening, provided scientific input and revised the manuscript. KP acquired funding. All authors read and approved the final manuscript. All authors contributed to editorial changes in the manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
All figures within the review were designed under a license acquired from biorender.com.
This work was supported by the German Research Foundation (DFG) through the project grant GRK2599 and the “Interdisziplinäre Zentrum für Klinische Forschung (IZKF)” of the FAU Erlangen-Nürnberg.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.