





 , Gujun Cong 3,†, Xiaowen Meng 1,4, Yang Zhang 1,4, Haiyan Sun 1,4, Lei Wang 1,4, Chao Zhang 1,4, Junyi Ma 5, Tong Liu 5, Fuhai Ji 1,4,*
, Gujun Cong 3,†, Xiaowen Meng 1,4, Yang Zhang 1,4, Haiyan Sun 1,4, Lei Wang 1,4, Chao Zhang 1,4, Junyi Ma 5, Tong Liu 5, Fuhai Ji 1,4,*
1 Department of Anesthesiology, The First Affiliated Hospital of Soochow University, 215006 Suzhou, Jiangsu, China
2 Department of Anesthesiology, Affiliated Tumor Hospital of Nantong University & Nantong Tumor Hospital, 226001 Nantong, Jiangsu, China
3 Department of Clinical Laboratory Medicine, Nantong Fourth People’s Hospital, 226007 Nantong, Jiangsu, China
4 Institute of Anesthesiology, Soochow University, 215006 Suzhou, Jiangsu, China
5 Institute of Pain Medicine and Special Environmental Medicine, Nantong University, 226019 Nantong, Jiangsu, China
†These authors contributed equally.
Abstract
Bone cancer pain (BCP) related neuronal damage is associated with ferroptosis, a regulated cell death dependent on iron. The noble gas argon is known to have neuroprotective effects, reducing neuroinflammation and enhancing neuronal recovery. However, its potential role in alleviating BCP through the modulation of neuronal ferroptosis remains unexplored.
Ferroptosis was induced by Erastin in SH-SY5Y human neuroblastoma cells. Plasmids were used to overexpress or knock down toll-like receptor (TLR) 2 and cyclooxygenase-2 (COX-2). The effects of argon treatment were evaluated in SH-SY5Y cells in which TLR2 and COX-2 expression was manipulated using viability assays, oxidative stress markers (reactive oxygen species (ROS), malondialdehyde (MDA), and glutathione (GSH)), and ferroptosis-related proteins (acyl-CoA synthetase long-chain family member 4 (ACSL4), glutathione peroxidase 4 (GPX4), and solute carrier family 7 member 11 (SLC7A11)). In vivo, a murine BCP model was developed by injecting Lewis lung carcinoma cells into the femoral cavity. Pain behaviors were analyzed, and spinal cord ferroptosis features were evaluated using histology, immunofluorescence, and transmission electron microscopy (TEM).
In vitro experiments showed that argon treatment restored SH-SY5Y cell viability after Erastin exposure, suppressed ROS and MDA production, and boosted GSH levels. It also downregulated ACSL4 and upregulated GPX4 and SLC7A11. In vivo, argon improved pain behaviors, reduced tumor burden, preserved neuronal integrity, and mitigated ferroptosis-induced damage to the spinal cords of BCP model mice. Argon also significantly suppressed TLR2 and COX-2 expression, disrupting the ferroptosis and inflammation cascades. However, overexpression of TLR2 or COX-2 reversed these protective effects, confirming the pivotal role of the TLR2-COX-2 axis in neuronal ferroptosis and pain modulation.
These findings demonstrate that argon effectively mitigates neuronal ferroptosis and alleviates BCP by downregulating the TLR2-COX-2 pathway, highlighting its therapeutic potential for conditions involving ferroptosis, such as cancer-related pain and neurodegenerative diseases.
Keywords
- argon
- bone neoplasms
- pain
- ferroptosis
- TLR2
- COX-2
A substantial number of patients with cancer, estimated to be between 30 and 50%, experience chronic pain during their treatment [1]. Among these, bone cancer pain (BCP) stands out as one of the most intense and prevalent forms of pain, frequently affecting patients in the advanced stages of cancer [2, 3]. The clinical presentation of BCP includes persistent pain, heightened pain sensitivity, and hyperalgesia, which significantly reduce the quality of life for individuals with bone metastases originating from prostate, breast, or lung cancers [4, 5]. Beyond physical discomfort, BCP often exacerbates psychological distress, contributing to depression, anxiety, appetite loss, sleep disturbances, and reduced social engagement, further diminishing patients’ well-being [3, 5, 6]. Current therapeutic options, including opioids, radiotherapy, and chemotherapy, often fail to provide sufficient relief and are accompanied by substantial side effects, resulting in approximately one-third of patients being inadequately treated [3, 4, 6, 7]. This highlights the urgent need to elucidate the pathogenic mechanisms underlying BCP and to develop more effective and targeted therapeutic strategies to improve patient outcomes.
Ferroptosis is an iron-mediated regulated cell death, notable for lipid peroxides and reactive oxygen species (ROS) accumulation [8, 9]. Mechanistically, it diverges from other cell death pathways, such as apoptosis, as it mainly results from redox disturbances linked to glutathione (GSH) depletion and glutathione peroxidase 4 (GPX4) inactivation [8, 10]. Uncontrolled oxidative stress damages cellular membranes and triggers a cascade of events leading to cell death [10, 11]. Emerging evidence has demonstrated that ferroptosis is a key contributor to both neuropathic and inflammatory pain [10, 12, 13, 14, 15]. For instance, ferroptosis-like cell death in the spine contributes to the progression of neuropathic pain following peripheral nerve injury. Administering liproxstatin-1, a ferroptosis inhibitor, effectively reduces mechanical and thermal hypersensitivities [14]. Another study also revealed that pharmacological inhibition of ferroptosis using ferrostatin-1 attenuated ferroptosis and alleviated BCP by inhibiting the pain-associated activation of extracellular signal-regulated kinase 1/2 (ERK1/2) and cyclooxygenase-2 (COX-2) expression [16]. Therefore, targeting ferroptosis could be a promising therapeutic strategy for improving outcomes in patients with BCP.
Argon, a noble gas first identified in 1885, has emerged as a focus of
scientific interest because of its organ protection properties [17]. Recently,
argon has been shown to have neuroprotective effects in many in vivo and
ex vivo experiments [18, 19, 20]. Argon protects against ischemia/reperfusion
injury-induced loss of nerve cell apoptosis through the inhibition of nuclear
factor kappa-B (NF-
In this study, we hypothesized that argon alleviates pain and inflammation by inhibiting ferroptosis via the TLR2-COX-2 axis in BCP. We demonstrate that argon effectively mitigates neuronal ferroptosis and alleviates BCP by downregulating the TLR2-COX-2 pathway, highlighting its therapeutic potential for conditions involving ferroptosis, such as cancer-related pain and neurodegenerative diseases.
For this study, male C57BL/6J mice weighing 20–25 g were obtained from Hunan SJA Laboratory Animal Co., Ltd. (Changsha, China). They were housed in standard cage under a 12-h light/dark cycle with unrestricted food and water. All experiments were approved by the institutional Ethics Committee of Nantong Tumor Hospital and conducted in accordance with the guidelines of the International Association for the Study of Pain, ensuring ethical and humane treatment of the animals throughout the study (2023-052-02). The experiments were conducted according to the National Institutes of Health (NIH) Guidelines for the Care and Use of Laboratory Animals.
To establish BCP, we utilized luciferase-expressing Lewis lung carcinoma
(LLC-Luc) cells from Hunan Fenghui Biotechnology Co., Ltd. [16]. The cells were
cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% FBS
and 1% penicillin‒streptomycin. For injection, a cell suspension was prepared in
PBS at a density of 2
Mechanical sensitivity was evaluated by measuring the paw withdrawal threshold (PWT) with von Frey filaments applied to the plantar surface, and 50% withdrawal threshold was calculated using the up-down method. Thermal sensitivity was assessed by recording the paw withdrawal latency (PWL) in response to t radiant heat, with a 20 s cutoff time to avoid injury. All behavioral tests were performed by an investigator blinded to group allocation.
For argon therapy, the mice were placed in a custom-sealed animal chamber and
exposed to a mixture of 75% Argon, 21% O2, and 4% N2 for 1 hour daily for 20
consecutive days. The control group received a normoxic mixture of 79% N2 and
21% O2 for the same duration. All treatments were administered between 9:00 and
10:00 AM daily to ensure consistency. For TLR2 overexpression, an
adeno-associated virus (AAV) vector (AAV-oe-TLR2) purchased from Gemma Gene
(Shanghai, China) was administered via tail vein injection. Injections were
performed weekly for a total of three times (on Day 1, Day 7, and Day 14) with 2
Neuroblastoma cells (SH-SY5Y), purchased from American Type Culture Collection (ATCC, Manassas, VA, USA), were grown in DMEM/F12 supplemented with 10% fetal calf serum. All the cell lines were validated by STR profiling and tested negative for mycoplasma. The cells were maintained at 37 °C in a 5% carbon dioxide humidified incubator until they reached 80% confluence.
For the experiments, the cells were exposed to either a control gas blend (74% N2, 21% O2, and 5% CO2) or a treatment gas blend (74% argon, 21% O2, and 5% CO2) for 2 h in a controlled atmosphere. The gas concentrations were adjusted automatically, reaching the preset target levels within 1 min, with the temperature maintained at 37 °C throughout. After exposure, the cell culture medium was replaced with medium containing 1% fetal calf serum to reduce protein interference. Ferroptosis was subsequently induced by the addition of 10 µM Erastin for 24 hours.
SH-SY5Y cells transfected with an overexpression negative control plasmid (oe-NC), knockdown negative control plasmid (sh-NC) or TLR2 plasmid (oe-TLR2), COX-2 plasmid (oe-COX-2), or TLR2 plasmid (sh-TLR2) were treated with or without 10 µM Erastin for 24 h under normal conditions, followed by treatment with or without argon (in the gas mixture described above) for 2 h. To overexpress or knockdown TLR2 or COX-2 in SH-SY5Y cells, TLR2 or COX-2 cDNA was amplified by PCR and then cloned and inserted into the vector pcDND3.1 (RiboBio, Guangzhou, China). The transfection of plasmids was conducted using Lipofectamine® LTX Reagent (Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s protocol.
Cell proliferation was assessed using the CCK-8 assay (Cat. No. K1018; Beyotime, China). After treatment in 96-well plates, 10 µL of the CCK-8 solution was added to each well, followed by a 2 h incubation at room temperature. Absorbance at 450 nm was recorded with a multimode microplate reader to determine cell viability.
We measured the levels of key oxidative stress and ferroptosis markers using a panel of commercial assay kits following the instructions provided by each manufacturer. Specifically, we quantified reactive oxygen species (ROS) with a kit from Abcam (ab186027, Cambridge, MA, USA). Iron levels were determined using an assay from Sigma‒Aldrich (MAK025, St. Louis, MO, USA), which also supplied a kit for measuring glutathione (GSH) activity (CS0260, St. Louis, MO, USA). Malondialdehyde (MDA) concentrations were assessed using a kit from the Nanjing Jiancheng Bioengineering Institute (A003-2, Nanjing, Jiangsu, China).
To observe mitochondrial morphology in the spinal cord, transmission electron microscopy (TEM) was utilized. The mice were deeply anesthetized with 2% pentobarbital sodium (50 mg/kg, intraperitoneally) and perfused with PBS, followed by a fixative solution containing 4% PFA and 2% glutaraldehyde in PBS. The lumbar spinal cords were excised and cut into 100-nm-thick ultrathin sections. The sections were subsequently stained with uranyl acetate and lead citrate to enhance contrast. The prepared grids were observed under a transmission electron microscope (Thermo Fisher Scientific, Eindhoven, Netherlands) operated at 80–120 kV. Digital images were acquired for analysis of mitochondrial morphology, including structure, crista integrity, and vacuolization, in spinal cord neurons.
Approximately 2
Total cellular proteins were extracted using RIPA lysis buffer containing
protease and phosphatase inhibitors on ice for 30 min, then centrifuged at 12,000
Mouse spinal cords were collected following euthanasia and fixed in 4%
paraformaldehyde (PFA) overnight. After fixation, the tissues were cryoprotected
in 30% sucrose solution until they fully sunk, embedded in optimal cutting
temperature (OCT) compound, and sectioned at a thickness of 20 µm using a
cryostat (CM1950, Leica, Wetzlar, Germany). The sections were mounted on glass
slides and air-dried for 1 h at room temperature. To assess cancer cell
proliferation in the femur, conventional hematoxylin and eosin (H&E) staining
was performed. After being postfixed with 4% paraformaldehyde (PFA), the spinal
cord tissues were cryoprotected in 30% sucrose at 4 °C overnight and
then dissected into 10-µm thick sections. The sections were stained with
1% thionine. Nissl-stained sections were analyzed by counting the number of
positive cells in random high-magnification (1
Cell viability and cytotoxicity were assessed using a dual staining approach with calcein-acetoxymethyl ester (AM)/propidium iodide (PI). Following treatment, the SH-SY5Y cells were washed with PBS and incubated for 20 min in the dark with a staining solution containing 2 µM calcein-AM and 5 µM PIr. Live cells fluoresced green due to calcein-AM, while dead cells exhibited red from PI uptake. Five random fields per well were imaged with a fluorescence microscope, and the percentage of live and dead cells was quantified using ImageJ software.
Mouse spinal cords were fixed in 4% PFA overnight, cryoprotected in 30% sucrose, embedded in OCT, and sectioned at 20 µm. Sections were air-dried, permeabilized with 0.1% Triton-100 for 10 min, and blocked with 1% BSA for 30 min at room temperature. The tissues were incubated with primary antibodies against TLR2 (1:200; Cat. No. Ab209216; Abcam, Cambridge, MA, USA) and COX-2 (1:200; Cat. No. Ab179800; Abcam, Cambridge, MA, USA) overnight at 4 °C. Goat anti-rabbit IgG (1:250; Cat. No. A21244; Invitrogen) and donkey anti-goat IgG (1:250; Cat. No. A11058; Invitrogen, Carlsbad, CA, USA) were used as secondary antibodies. Nuclei were stained with Hoechst 33342 (Cat. No. R37605; Invitrogen, Carlsbad, CA, USA). The slides were observed using a fluorescence microscope and analyzed by ImageJ (Image Pro-Plus 6.0; Media Cybernetics, Silver Spring, MD, USA).
All the statistical analyses in the current study were performed by using
GraphPad Prism 8.0 software (GraphPad Software, Inc. La Jolla, CA, USA).
Comparisons between two groups were conducted using a two-tailed Student’s
t test. One-way analysis of variance (ANOVA) followed by an appropriate
post hoc test was applied for comparisons among multiple groups. The data are
shown as the means
To investigate the role of argon in ferroptosis in SH-SY5Y cells, ferroptosis was induced by treating the cells with Erastin. Erastin treatment significantly reduced cell viability (Fig. 1A), as confirmed by calcein-AM and PI staining, which revealed increased cell death (Fig. 1B). Argon exposure effectively restored cell viability and reduced cell death in erastin-treated SH-SY5Y cells (Fig. 1A,B). Erastin treatment also increased the levels of oxidative stress markers, including ROS and MDA, and decreased the level of GSH, whereas argon exposure significantly reversed these changes, reducing the levels of ROS and MDA and restoring the level of GSH (Fig. 1C). Similarly, the iron accumulation induced by Erastin was markedly decreased by argon treatment (Fig. 1C). At the molecular level, argon treatment downregulated ACSL4 but upregulated GPX4, FTL, and SLC7A11 in Erastin-treated SH-SY5Y cells, further confirming the inhibition of ferroptosis (Fig. 1D). Argon alone did not affect the expression of these proteins in SH-SY5Y cells (Fig. 1D). Additionally, TLR2 mRNA levels, which were elevated in Erastin-treated cells, were significantly reduced in the argon-treated group (Fig. 1E). These findings demonstrate that argon mitigates ferroptosis by restoring redox balance, reducing oxidative stress, and regulating the TLR2 pathway.
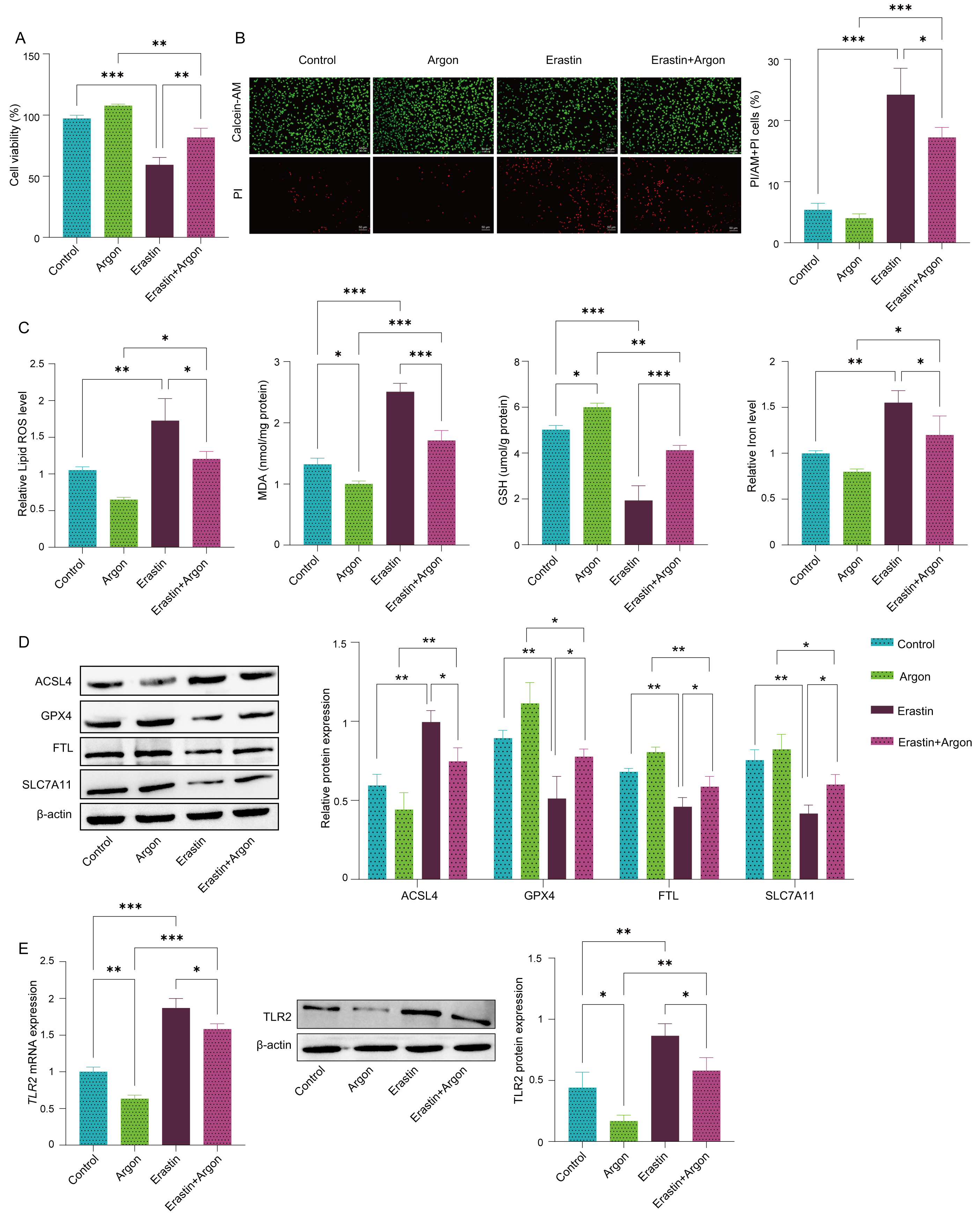 Fig. 1.
Fig. 1.
Argon alleviates ferroptosis in SH-SY5Y cells. SH-SY5Y cells
were pretreated with or without argon (74% argon, 21% O2, and 5%
CO2) for 2 h and then treated with or without 10 µM Erastin for 24 h.
(A) Cell viability was measured by the CCK-8 assay. (B) Live and dead cells were
visualized with calcein-AM and PI staining. Scale bar: 50 µm. (C)
Levels of ROS, MDA, GSH and iron were measured by specific kits. (D) The protein
expression levels of ACSL4, GPX4, FTL and SLC7A11 were detected via Western blot.
(E) The mRNA levels and protein levels of TLR2 in cells were quantified
via RT‒PCR and Western blot. n = 3. Data represent means
To determine whether argon inhibits ferroptosis through the regulation of TLR2, we overexpressed TLR2 in SH-SY5Y cells using a TLR2 overexpression plasmid. Compared with erastin treatment, Argon treatment significantly decreased both the mRNA and protein levels of TLR2 (Fig. 2A,B). However, TLR2 overexpression reversed the reduction in TLR2 expression caused by argon in Erastin-treated cells, indicating that the effects of argon on TLR2 expression are dose- and expression-dependent. We also assessed cell viability using CCK-8 assays and observed that argon treatment significantly increased cell viability in Erastin-treated control cells (oe-NC), but no protective effect was observed in Erastin-treated TLR2-overexpressing cells (Fig. 2C). Calcein-AM/PI staining further confirmed these results, revealing increased cell death in TLR2-overexpressing cells despite argon treatment (Fig. 2D). Moreover, TLR2 overexpression blocked the protective effects of argon on oxidative stress markers. The ability of Argon to reduce ROS and MDA levels and restore GSH and iron homeostasis was significantly impaired in TLR2-overexpressing cells (Fig. 2E). Finally, argon failed to reduce ACSL4 expression and increase GPX4, FTL, and SLC7A11 expression in Erastin-treated TLR2-overexpressing cells, further supporting the role of TLR2 in mediating the protective effects of argon (Fig. 2F). Collectively, these data indicate that argon inhibits neuronal ferroptosis in SH-SY5Y cells by regulating the TLR2 pathway.
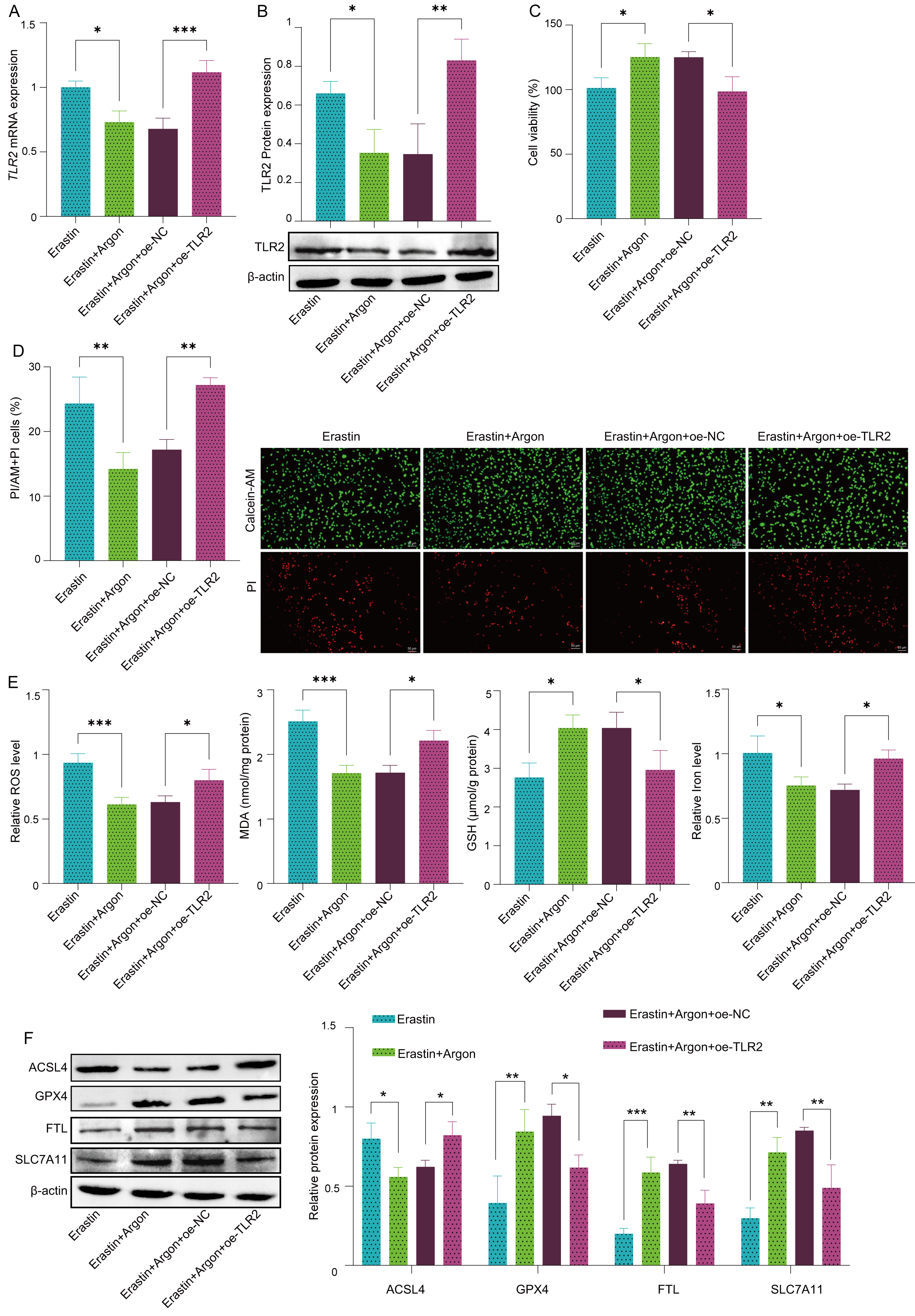 Fig. 2.
Fig. 2.
TLR2 overexpression reverses the neuroprotective effect
of argon in SH-SY5Y cells. SH-SY5Y cells were transfected with a TLR2
overexpression plasmid (oe-TLR2) or negative control plasmid (oe-NC), pretreated
with or without argon (74% argon, 21% O2, and 5% CO2) for 2 h, and
then treated with or without 10 µM Erastin for 24 h. (A) TLR2 mRNA
levels were measured via RT‒PCR. (B) TLR2 protein expression was assessed by
Western blot. (C) Cell viability was measured by CCK-8 assay. (D) Live/dead cells
were stained with calcein-AM/PI. Scale bar: 50 µm. (E) The levels of
oxidative stress markers, including ROS, MDA, GSH, and iron, were quantified via
biochemical kits. (F) The protein expression levels of ACSL4, GPX4, FTL and
SLC7A11 were detected by Western blot. n = 3 per group. Data represent means
In SH-SY5Y cells, overexpression of TLR2 resulted in marked increases in the mRNA and protein levels of both TLR2 and COX-2 (Fig. 3A,B), and downregulation of TLR2 expression significantly reduced the mRNA and protein levels of both TLR2 and COX-2 (Fig. 3A,B). These data suggest that TLR2 regulates COX-2 expression in SH-SY5Y cells. Furthermore, to investigate the role of the TLR2-COX-2 axis in ferroptosis, SH-SY5Y cells were transfected with a TLR2 downregulation plasmid and a COX-2 overexpression plasmid. In the Erastin-treated cells, TLR2 downregulation mitigated ferroptosis, as reflected by decreased cell death (Fig. 3C,D) and reduced levels of oxidative stress markers, including ROS and MDA, and restored GSH levels and iron homeostasis (Fig. 3E). However, overexpression of COX-2 in TLR2-knockdown cells partially reversed this protection and exacerbated ferroptosis (Fig. 3C,D). TLR2 downregulation also reduced ACSL4 expression while increasing GPX4, FTL, and SLC7A11 expression (Fig. 3F). These protective effects were reversed upon COX-2 overexpression in TLR2-knockdown cells (Fig. 3E,F). To further verify that TLR2 mediates ferroptosis via COX-2, we performed a rescue experiment. Overexpression of TLR2 (oe-TLR2) significantly increased the expression of the proferroptotic protein ACSL4 and reduced the expression of the anti-ferroptotic protein GPX4. However, when COX-2 was silenced (sh-COX-2) in TLR2-overexpressing cells, the expression levels of ACSL4 and GPX4 returned to baseline, indicating that silencing COX-2 rescues cells from the proferroptotic effects of TLR2 activation (Supplementary Fig. 1). Together, these results indicated that TLR2 activates neuronal ferroptosis by regulating COX-2.
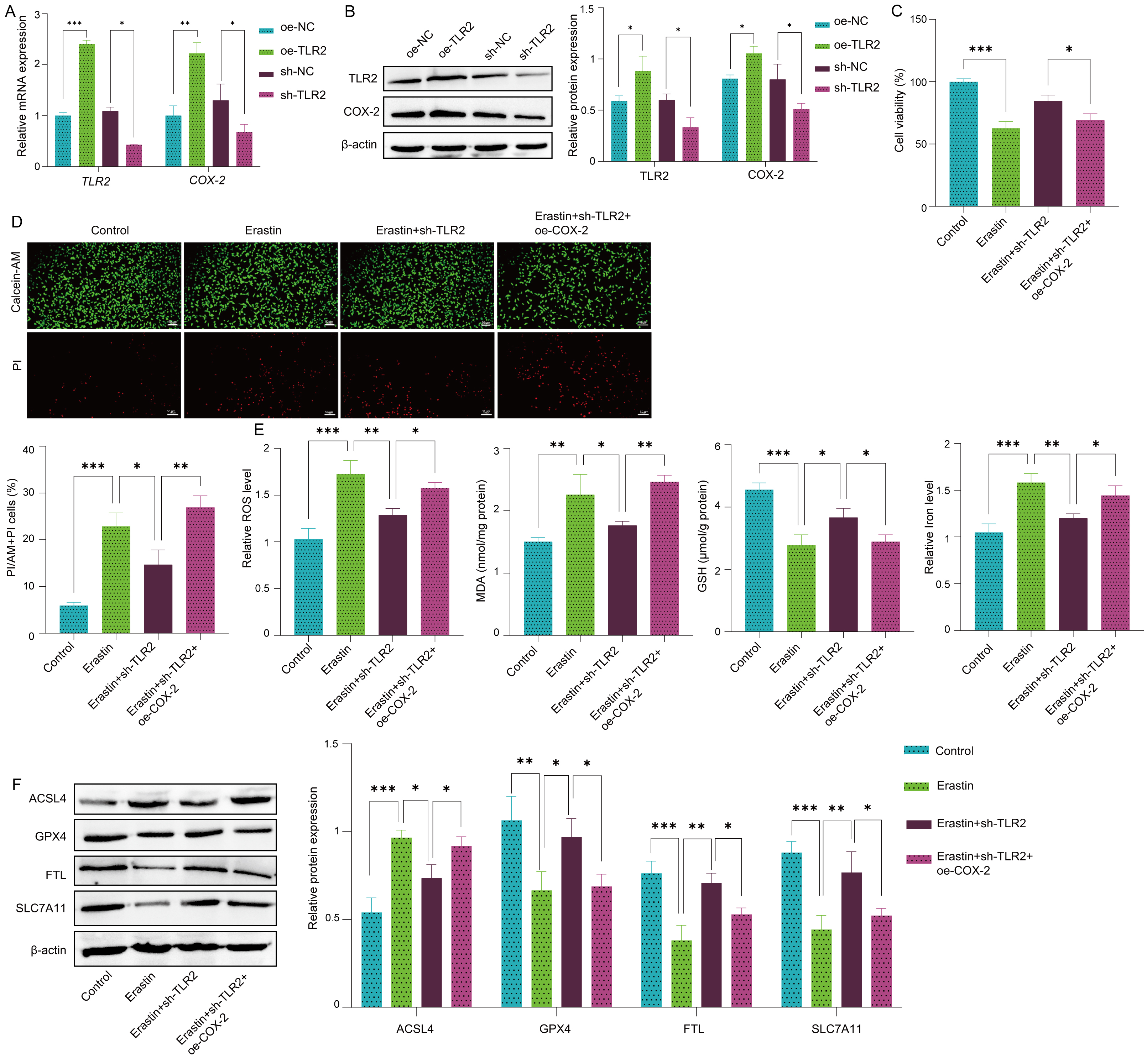 Fig. 3.
Fig. 3.
TLR2 regulates neuronal ferroptosis through COX-2 activation.
SH-SY5Y cells were transfected with a COX-2 overexpression plasmid
(oe-COX-2), a negative overexpression control plasmid (oe-NC), a TLR2
knockdown shRNA plasmid (sh-TLR2) or a negative knockdown control plasmid
(sh-NC). After transfection, the cells were treated with or without 10 µM
Erastin for 24 h. (A) The mRNA levels of TLR2 and COX-2 in
SH-SY5Y cells were measured by RT‒PCR. (B) The protein expression levels of TLR2
and COX-2 were detected via Western blot. (C) The viability of SH-SY5Y cells was
measured by CCK-8 assay. (D) Live/dead SH-SY5Y cells were stained with
calcein-AM/PI. Scale bar: 50 µm. (E) The levels of oxidative stress
markers, including ROS, MDA, GSH, and iron, were quantified using biochemical
kits. (F) The protein expression levels of ACSL4, GPX4, FTL and SLC7A11 were
detected by Western blot. n = 3. Data represent means
To investigate the role of COX-2 in the neuroprotective effects of argon, SH-SY5Y cells were transfected with a COX-2 overexpression plasmid. Compared with Erastin treatment, Argon treatment significantly reduced TLR2 and COX-2 mRNA and protein levels. However, COX-2 overexpression partially reversed the argon-induced reduction in COX-2 expression in the Erastin-treated SH-SY5Y cells (Fig. 4A,B). In addition, COX-2 overexpression did not affect TLR2 expression in erastin- or argon-treated SH-SY5Y cells (Fig. 4A,B). In Erastin-treated cells, COX-2 overexpression negated the protective effects of argon against neuronal ferroptosis. This was evidenced by increased cell death, as shown by the CCK-8 assay and calcein-AM/PI staining results (Fig. 4C,D), as well as elevated levels of oxidative stress markers, including ROS and MDA, and disrupted GSH and iron homeostasis (Fig. 4E). Furthermore, COX-2 overexpression reversed the effects of argon on the expression of ferroptosis markers, restoring ACSL4 expression and reducing GPX4, FTL, and SLC7A11 levels (Fig. 4F). These findings demonstrate that argon inhibits neuronal ferroptosis by downregulating COX2 expression in SH-SY5Y cells.
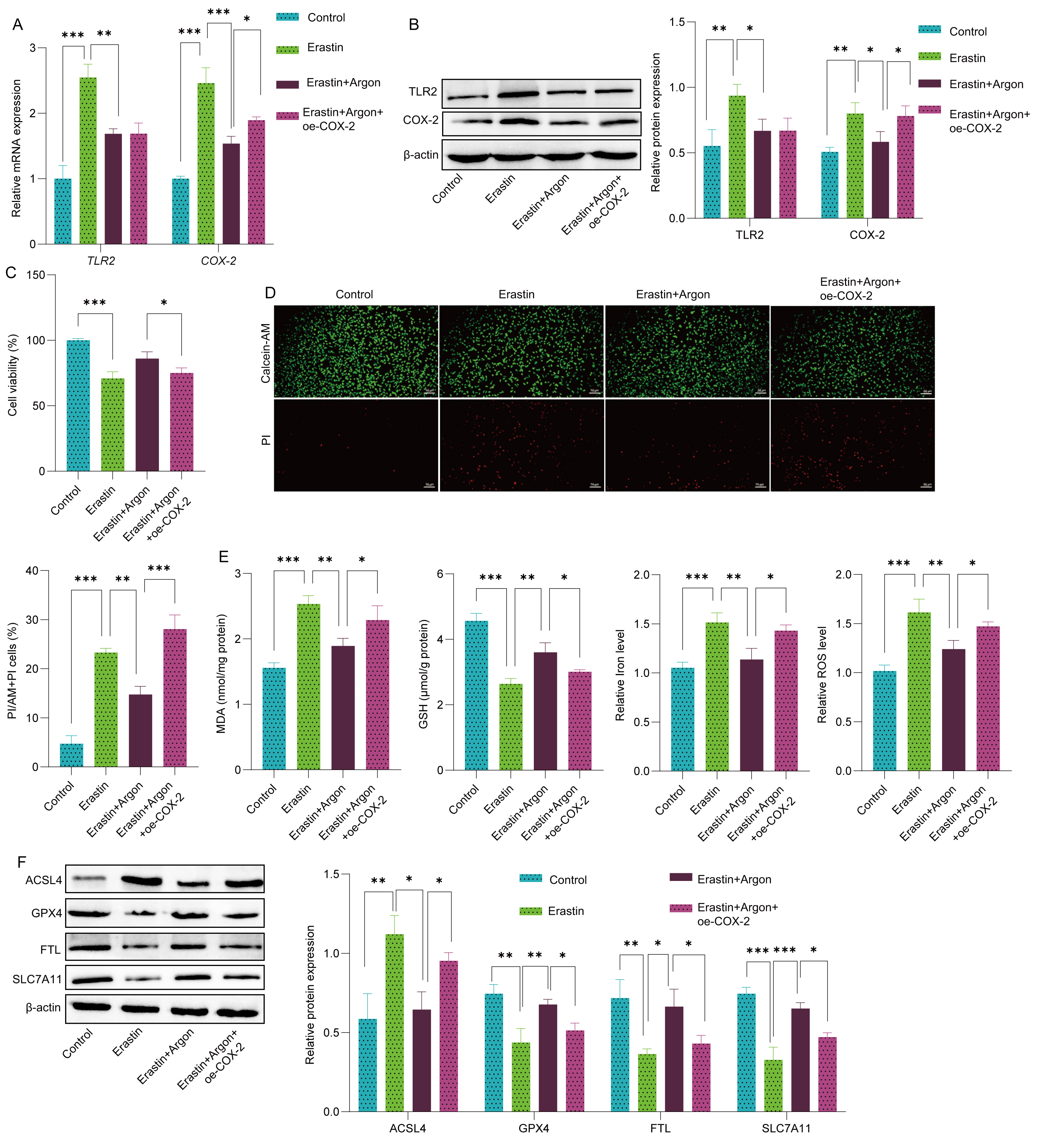 Fig. 4.
Fig. 4.
Argon suppresses neuronal ferroptosis via TLR2-regulated COX-2. SH-SY5Y cells were transfected with either a COX-2 overexpression
plasmid (oe-COX-2) or a control plasmid (oe-NC). Following transfection, the
cells were pretreated with or without argon (74% argon, 21% O2, and 5% CO2)
for 2 h, after which they were treated with or without erastin (10 µM, 24
h) followed by argon exposure. (A) TLR2 and COX-2 mRNA levels
in SH-SY5Y cells were quantified by RT‒PCR. (B) The protein levels of TLR2 and
COX-2 in SH-SY5Y cells were analyzed by Western blot. (C) The viability of
SH-SY5Y cells was assessed via CCK-8 assay. (D) Live/dead SH-SY5Y cells were
stained with calcein-AM/PI. Scale bar: 50 µm. (E) The levels of
oxidative stress markers, including ROS, MDA, GSH, and iron, were quantified
using biochemical kits. (F) The protein expression levels of ACSL4, GPX4, FTL and
SLC7A11 were detected by Western blot. n = 3. Data represent means
To investigate the therapeutic effects of argon on BCP, we established a mouse model of BCP in TLR2-overexpressing mice. In the BCP model, argon treatment significantly improved pain behaviors, as evidenced by increased paw withdrawal thresholds and paw withdrawal latency and reduced tumor growth, as indicated by bioluminescence imaging (Fig. 5A,B). Histological analysis revealed less bone damage in the femur tissue of argon-treated mice (Fig. 5C). Nissl staining revealed improved neuronal integrity in the spinal cord after argon treatment compared with that in the BCP group (Fig. 5D). TEM further confirmed that ferroptosis damage was reduced in the spinal cord neurons of argon-treated mice (Fig. 5E). To confirm that the effects of argon were specific to disease pathology, we administered argon to healthy, naïve mice and found no significant changes in the paw withdrawal threshold (PWT), paw withdrawal latency (PWL), or expression of ferroptosis markers (ACSL4, GPX4, FTL, SLC7A11) in the spinal cord (Supplementary Fig. 2A–C), suggesting that argon does not alter basal ferroptosis pathways in a healthy state. However, TLR2 overexpression in the BCP mouse model reversed these neuroprotective effects of argon. In TLR2-overexpressing BCP mice, argon failed to improve the PWT, PWL, tumor growth, bone integrity, or neuronal preservation (Fig. 5A–E). Immunofluorescence staining revealed significantly lower TLR2 and COX-2 expression in the spinal cord dorsal horns of argon-treated mice than in those of BCP-treated mice, indicating inhibition of the TLR2-COX-2 axis (Fig. 5F). Conversely, TLR2 overexpression in BCP mice increased TLR2 and COX-2 expression, negating the inhibitory effects of argon on this pathway (Fig. 5F). Additionally, TLR2 overexpression reversed the suppression of neuronal ferroptosis by argon, as indicated by elevated levels of the oxidative stress marker MDA, disrupted GSH levels, and iron homeostasis in the spinal cord dorsal horn (Fig. 5G). Furthermore, the ability of argon to reduce the expression of ferroptosis marker ACSL4 and increase the expression of GPX4, FTL, and SLC7A11 was blocked by TLR2 overexpression (Fig. 5H). These findings suggest that argon alleviates BCP and neuronal ferroptosis primarily by downregulating the TLR2-COX-2 axis, positioning this pathway as a critical target for its therapeutic effects.
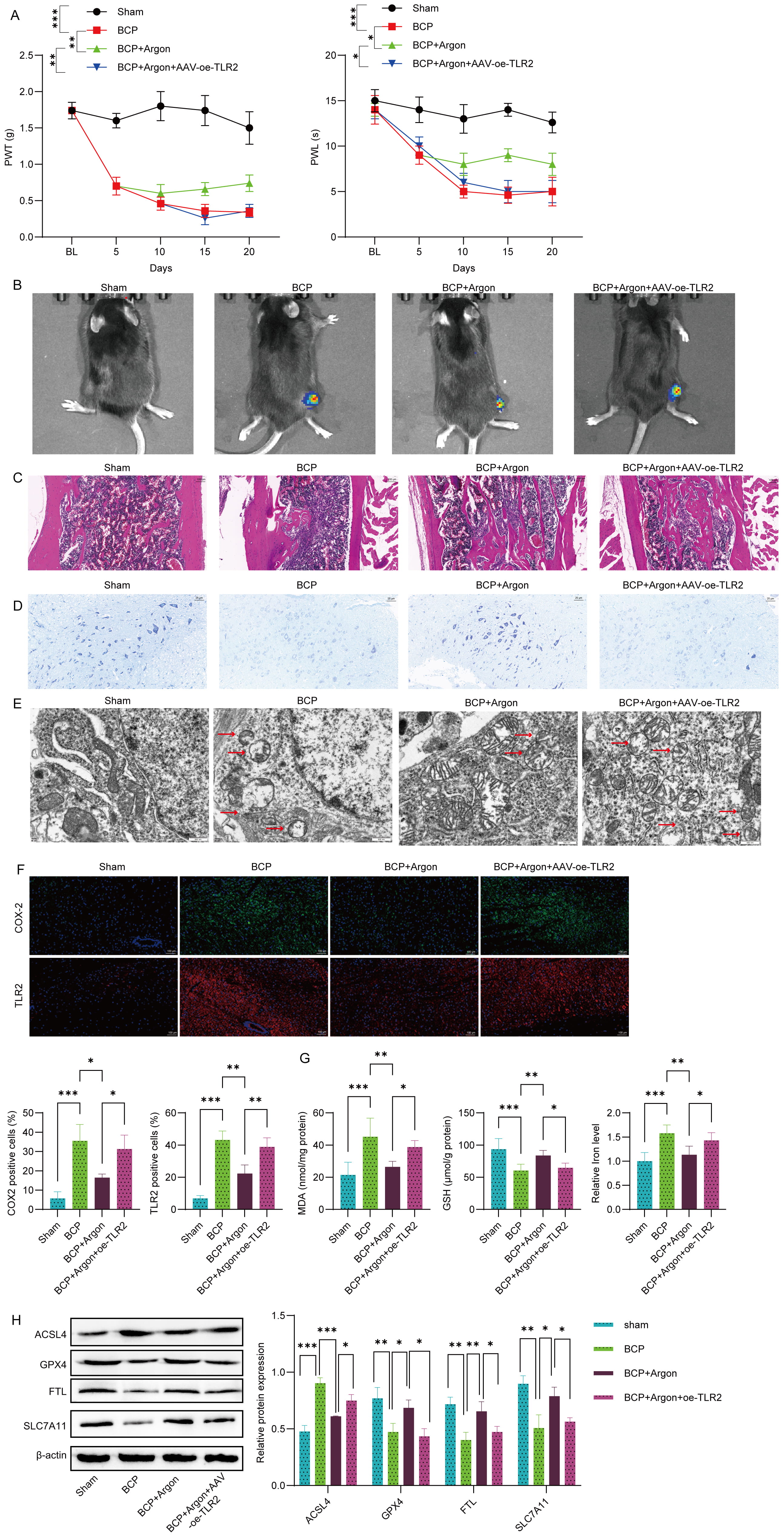 Fig. 5.
Fig. 5.
Argon alleviated BCP and neuronal ferroptosis by downregulating
the TLR2-COX-2 axis in a mouse model. Live luciferase-expressing Lewis lung
carcinoma cells (LL/2-Luc2) were injected into the femoral cavity of C57BL/6 mice
with AAV-induced TLR2 overexpression to establish a bone cancer pain
(BCP) model. Afterward, the mice were treated with or without argon for 20 days.
(A) The paw withdrawal threshold (PWT) and paw withdrawal latency (PWL) of the
mice were measured at specified time points to evaluate pain behaviors. (B) Tumor
growth was assessed via bioluminescence imaging. Representative images and
quantitative analysis of bioluminescence intensity are shown. (C) Hematoxylin and
Eosin staining of the femur. Scale bar: 100 µm. (D) Nissl staining
of the spinal cord dorsal horn of mice and the quantification of Nissl-positive
cells are presented. Scale bar: 25 µm. (E) Transmission electron
microscopy (TEM) revealed reduced mitochondrial damage and ferroptosis features
in the spinal cord neurons of argon-treated mice. The arrow indicates the
mitochondria. Scale bar: 500 nm. (F) The expression levels of TLR2 and COX-2 in
the spinal cord dorsal horn were detected by immunofluorescence staining. Scale
bar: 100 µm. (G) Oxidative stress markers (MDA, GSH, and iron) in
spinal cord tissue were quantified using specific biochemical kits. (H) The
protein expression levels of ACSL4, GPX4, FTL and SLC7A11 were detected by
Western blot. n = 5. Data represent means
BCP or other type of chronic pain is a challenging clinical problem because of its complex pathophysiology and the limited effectiveness of current treatments, such as opioids and radiotherapy, which primarily address symptoms rather than underlying mechanisms [1, 6, 22, 23], Ferroptosis, a form of controlled cell death triggered by lipid peroxidation and oxidative stress, contributes to neuronal damage, amplifying pain sensitivity and neuroinflammation in BCP [12]. The current study explored the potential of argon gas as a therapeutic method to alleviate BCP through the targeting of ferroptosis. We identified the TLR2-COX-2 signaling axis as a pivotal pathway driving neuronal ferroptosis and inflammation in BCP. Using both cell-based and live-animal models, we demonstrated that argon gas, with its established neuroprotective properties, significantly suppressed TLR2 and COX-2 expression, inhibited ferroptosis, and reduced oxidative stress, thereby improving pain behaviors, preserving neuronal integrity, and mitigating ferroptosis-induced spinal cord damage. These findings suggest that argon is a promising noninvasive therapy for BCP and highlight the TLR2-COX-2 axis as a novel target for effective pain management.
Ferroptosis is a regulated form of cell death characterized by excessive lipid peroxidation and the aggregation of ROS [8, 24]. The resulting oxidative stress damages cellular membranes and triggers a cascade of events leading to cell death [25]. The regulation of ferroptosis involves the lipid repair enzyme GPX4, along with transcription factors such as p53, Nrf2, and COX-2, and the phosphorylation of extracellular signal-regulated kinases (ERKs) [25, 26]. Emerging evidence links ferroptosis to neurological diseases, such as neuropathic pain, Alzheimer’s disease, ischemic injury, and BCP, highlighting its role in neuroinflammation and neurodegeneration [12, 16, 27]. Moreover, suppressing spinal ferroptosis could help alleviate neuropathic pain [14, 28]. In BCP, ineurons in the spinal dorsal horn exhibit elevated ferroptosis, characterized by iron buildup, lipid peroxidation, increased COX-2 expression, and mitochondrial structural changes [16]. In our study, we showed that neuronal ferroptosis is increased in the spinal cord dorsal horn of mice with BCP. These findings are consistent with those of previous studies. The inhibition of ferroptosis by ferrostatin-1 has been proven to prevent pathogenic features linked to ferroptosis and reduces BCP [16]. These findings highlight ferroptosis as a promising therapeutic target that could mitigate oxidative stress and neurodegeneration, improving pain management outcomes.
A noble gas, argon was discovered in 1885 and has emerged as a scientific focus because of its organ protection properties [29, 30]. Recently, argon has been shown to have impressive neuroprotective effects in many in vivo and ex vivo experiments [17, 19, 31, 32]. The results of our study reveal that argon treatment effectively mitigated ferroptosis by reducing the levels of oxidative stress markers, including ROS and MDA, while restoring the levels of GSH and iron homeostasis. These observations align with those of earlier studies highlighting the ability of argon to modulate redox balance in ischemic and inflammatory models [17, 18, 19]. Argon also suppressed the expression of key ferroptosis markers, including ACSL4, whereas it upregulated protective markers such as GPX4 and SLC7A11. These findings underscore the therapeutic potential of argon in addressing ferroptosis-mediated neuronal damage in BCP. The TLR2-COX-2 axis plays a critical role in ferroptosis and pain modulation [33, 34]. Studies have shown that the COX-2 enzyme inhibitor lumiracoxib attenuated bone destruction and pain in a mouse bone cancer pain model [33, 34]. Previous studies have shown that the neuroprotective effects of argon are entirely blocked by the inhibition of TLR2 and TLR4 [21, 35]. Our findings revealed that overexpression of TLR2 and COX2 reversed the neuroprotective effects of argon, exacerbating oxidative stress and ferroptosis markers. Conversely, argon treatment significantly suppressed TLR2 and COX-2 expression, effectively interrupting the ferroptosis cascade. These results align with existing studies that identify the TLR2-COX-2 axis as a pivotal target for therapeutic intervention in BCP, further validating its role in inflammation and pain pathophysiology.
Our current study confirmed these findings in a mouse model of BCP. Argon treatment improved pain behaviors, reduced tumor burden, and preserved bone and neuronal integrity. TEM and Nissl staining revealed less ferroptotic damage, whereas immunostaining revealed reduced TLR2 and COX-2 expression in the spinal cord dorsal horn. Interestingly, studies have also highlighted the role of ferroptosis in neurodegeneration and pain sensitization, with iron accumulation and lipid peroxidation contributing to neuronal death and spinal cord dysfunction [36]. The potential of Argon to reduce ferroptotic damage indicates that targeting this pathway could offer a valuable therapeutic strategy, which is consistent with findings that ferroptosis inhibitors alleviate pain behaviors in animal models of chronic pain [28]. Studies indicate that treating chronic constriction injury rats with liproxstatin-1, via intraperitoneal injection attenuates both mechanical and thermal pain hypersensitivity. Additionally, it reduces neuropathic pain caused by peripheral nerve injury in rats. In contrast, TLR2 overexpression reversed these benefits, highlighting the critical role of the TLR2-COX-2 axis in pain modulation and ferroptosis regulation. These findings align with those of previous studies showing that compared with wild-type mice, TLR2-deficient mice exhibit reduced mechanical allodynia and thermal hyperalgesia following nerve transection, as well as decreased glial activation and lower inflammatory gene expression in the spinal cord [37]. Another in vivo study revealed that nerve injury-induced thermal hyperalgesia was completely abolished in TLR2 knockout mice, highlighting the crucial role of TLR2 in pain hypersensitivity [38]. These findings provide compelling evidence that argon exerts a protective effect by targeting oxidative stress and inflammatory pathways.
Although our findings are promising, we acknowledge several limitations of this study. First, the use of Erastin to induce ferroptosis in vitro comes with the caveat that we did not experimentally rule out its potential off-target effects. Second, our in vivo model utilized young, healthy male mice, which does not fully represent the typically older, comorbid human population affected by BCP. Finally, although the 20-day treatment protocol is standard for this animal model, its direct clinical translation requires further investigation to establish optimal and practical treatment regimens. It is important to note, however, that the gas mixture used (containing 21% O2) was normoxic and did not pose a risk of hypoxia, addressing a potential translational concern.
In summary, this study provides compelling evidence that argon gas effectively alleviates bone cancer pain by inhibiting neuronal ferroptosis. The therapeutic effect of argon is mediated through the downregulation of the TLR2-COX-2 signaling pathway. These findings not only elucidate a novel mechanism underlying the neuroprotective effects of argon but also identify the TLR2-COX-2 axis as a key therapeutic target. These findings position argon as a promising candidate for developing new, noninvasive treatment strategies for BCP and other neurodegenerative conditions associated with ferroptosis.
ACSL4, acyl-CoA synthetase long-chain family member 4; BCP, bone cancer pain; CCK-8, cell counting kit 8; COX-2, cyclooxygenase-2; DMEM, Dulbecco’s modified Eagle’s medium; FTL, ferritin light chain; GPX4, glutathione peroxidase 4; GSH, glutathione; MDA, malondialdehyde; PWL, paw withdrawal latency; PWT, paw withdrawal threshold; ROS, reactive oxygen species; SLC7A11, solute carrier family 7 member 11; TEM, transmission electron microscopy; TLR2, toll-like receptor 2.
The datasets used or analyzed during the current study are available from the corresponding author on reasonable request.
PK, XM, TL, and GC guaranteed the integrity of the entire study. PK, GC, XM, and YZ designed the study and literature research. PK, GC, XM, YZ, and JM defined the intellectual content. HS, LW, and FJ performed experiment. HS, LW, TL, and FJ collected the data. CZ, JM, and FJ analyzed the data. PK, GC, and XM wrote the main manuscript and prepared figures. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
All experimental procedures were approved by the institutional Ethics Committee of Nantong Tumor Hospital and conducted in accordance with the guidelines of the International Association for the Study of Pain, ensuring ethical and humane treatment of the animals throughout the study (2023-052-02). The experiments were conducted according to the National Institutes of Health (NIH) Guidelines for the Care and Use of Laboratory Animals.
Not Applicable.
This work was supported by the Nantong Health and Wellness Commission’s Scientific Research Program (MS2023063), the Natural Science Foundation of Nantong Municipality (JCZ2024001), and the Chinese Anti-Cancer Association’s Research Program (GWRX-CR-2024-3).
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/FBL43761.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.