


 , Xiaoyun Zhang 2,†, Yanhui Wang 2, Yu Zeng 2, Hongliang Sun 2,*
, Xiaoyun Zhang 2,†, Yanhui Wang 2, Yu Zeng 2, Hongliang Sun 2,* , Xiaodong Cui 2,*
, Xiaodong Cui 2,*1 Department of Anesthesiology, Weifang Hospital of Traditional Chinese Medicine, Shandong Second Medical University, 261061 Weifang, Shandong, China
2 School of Basic Medicine Sciences, Shandong Second Medical University, 261053 Weifang, Shandong, China
†These authors contributed equally.
Abstract
Mesenchymal stem cells (MSCs) are widely utilized in tissue repair, anti-inflammatory treatment, and cell therapy due to their remarkable multidirectional differentiation potential, immunosuppressive capabilities, and low immunogenicity. However, the regulatory mechanisms underlying their functions are intricate, and epigenetic modifications are a significant contributing factor. N6-methyladenosine (m6A) modification affects the proliferation, differentiation, and immunomodulation of MSCs by regulating the stability, transport, and translation of RNA. Studies have shown that m6A modification promotes osteogenic differentiation through the bone morphogehetic protein/small mothers against decapentaplegic (BMP/Smad) and wingless-related integration site/β-catenin (Wnt/β-catenin) pathways. It also enhances the anti-inflammatory effect of MSCs by modulating immune cell polarization and the release of inflammatory mediators. Moreover, exosomes secreted by MSCs contribute to immunomodulation and the response to cancer treatment by regulating the m6A modification of genes in target cells. “Writers” of m6A, such as methyltransferase-like 3 (METTL3) and METTL14, and “erasers”, such as fat mass and obesity-associated protein (FTO) and alkB homolog 5 (ALKBH5), are crucial in regulating the functions of MSCs. Targeting m6A modification via the clinical application of MSCs may represent a new cancer treatment strategy. Therefore, a comprehensive investigation of the m6A regulatory mechanism is essential. This review provides theoretical and technical support for the clinical use of MSCs, facilitating the development of more effective therapeutic strategies.
Keywords
- m6A
- mesenchymal stem cells
- differentiation
- immunity
Mesenchymal stem cells (MSCs) are adult stem cells. They have multidirectional differentiation potential and self-renewal ability and are widely distributed in various tissues such as bone marrow, adipose tissue, and the umbilical cord [1, 2, 3]. MSCs show promising applications in tissue regeneration, immunomodulation, and cell therapy due to their excellent immunomodulatory ability, low immunogenicity, and multilineage differentiation potential [4]. However, the maintenance of the medicinal effects of MSCs represents a highly intricate and arduous subject area, and epigenetic modification may be a significant regulatory factor in this context.
N6-methyladenosine (m6A) modification is crucial in posttranscriptional
regulation [5]. It affects RNA stability, transport, splicing, and translation
efficiency [6]. Additionally, many scientific studies have shown its importance
in the proliferation, differentiation, and immunomodulation of MSCs. For
instance, m6A modulates the osteogenic potential of MSCs by regulating key
signaling pathways, such as phosphoinositide 3-kinase/protein kinase B (PI3K/AKT)
and wingless-related integration site/
m6A modification is a dynamic and reversible process coordinated by multiple proteins. “Writers” like methyltransferase-like 3 (METTL3) and METTL14 recognize specific RNA sequences and catalyze adenine methylation. “Readers” such as YTH domain-containing family protein 1 (YTHDF1) and YTHDF2 bind to m6A sites, regulating RNA stability, splicing, and translation. “Erasers” including fat mass and obesity-associated protein (FTO) and alkB homolog 5 (ALKBH5) remove methyl groups via demethylation, maintaining m6A modification balance and fine-tuning gene expression [15]. METTL3 has been demonstrated to promote osteogenic differentiation and inhibit adipogenic differentiation. Conversely, the deletion of the demethylase ALKBH5 has been shown to significantly enhance self-renewal in MSCs [16, 17]. These research findings suggest that elevating the level of m6A modification by modulating the m6A regulatory mechanism can efficaciously regulate the multidirectional differentiation potential of MSCs, thereby offering novel insights and strategies for regenerative therapies leveraging MSCs. A thorough exploration of the molecular mechanisms underlying m6A modification, as well as the methodologies for enhancing the efficacy of MSC-based clinical treatments through the targeting of m6A regulatory factors, will represent a significant research avenue. This study is designed to systematically investigate the role of m6A modifications in regulating MSC functions, thereby providing valuable theoretical understandings and technical assistance for the application of MSCs in the field of regenerative medicine.
Owing to their strong osteogenic potential, MSCs hold significant promise as a treatment for orthopedic conditions such as osteoporosis, periodontitis, bone defects, and fractures [18, 19, 20]. Accordingly, exploring therapeutic targets by targeting the mechanisms that promote and inhibit the osteogenic differentiation capacity of MSCs can effectively enhance the therapeutic efficacy of MSCs [21]. m6A modifications are common post-transcriptional modifications that not only affect a variety of biological processes such as cellular inflammation, apoptosis, autophagy, senescence, and iron death but also regulate cellular differentiation and transdifferentiation, which in turn are involved in disease progression [22, 23, 24, 25]. Consequently, targeting m6A regulators enables the modulation of the level of epigenetic m6A modifications in vivo and in vitro through the utilization of agonists, inhibitors, or gene silencing and overexpression techniques, thereby influencing cellular function and disease progression [26, 27]. Recent study has found that the osteogenic differentiation process in MSCs is associated with m6A modification. m6A modification promotes osteogenic differentiation and inhibits adipogenic differentiation in MSCs [9]. These findings provide new ideas for enhancing the potential value of MSCs in treating skeletal diseases at the level of m6A modification.
Age-associated osteoporosis (OP) is a disorder typified by the cellular senescence of bone marrow mesenchymal stem cells (BMSCs) resulting from the accrual of oxidative stress, DNA damage, and mitochondrial dysfunction [28]. The senescence of BMSCs not only exacerbates local inflammation but also disrupts the balance of osteogenic and adipogenic differentiation of BMSCs, leading to the progression of age-related skeletal diseases [29]. The current therapeutic approach for OP is primarily focused on the utilization of antidepressant and anabolic agents. The current therapeutic approach for OP is focused primarily on the utilization of antidepressant and anabolic agents. Although these drugs promote bone formation, they also present a number of potential adverse side effects, such as fever, nausea, bone pain, and even the possibility of carcinogenesis [30]. Consequently, the osteogenic potential of BMSCs can be augmented by alleviating their senescence. Research has demonstrated that the antioxidant quercetin mitigates oxidative stress in orofacial mesenchymal stem cells (OMSCs) through the promotion of Per1 m6A modification, thus reducing cellular aging, enhancing the expression of osteogenic genes, and facilitating periodontal tissue regeneration [31]. Knockdown of METTL3 reduces the m6A modification of the critical cell cycle regulator Polo-like kinase 1 (PLK1) in dental pulp stem cells (DPSCs), leading to senescence and apoptosis [32]. Thus, targeting METTL3 in DPSCs may inhibit cellular aging and serve as a therapeutic strategy for periodontitis. Additionally, MSC senescence is closely associated with the onset and progression of osteoarthritis (OA) [33]. In MSCs, ALKBH5 promotes the degradation of cytochrome P450 family 1 subfamily B member 1 (CYP1B1) mRNA via m6A demethylation, thereby alleviating mitochondrial dysfunction and attenuating mesenchymal stem cell senescence and osteoarthritis progression [17].
Autophagy is a protective mechanism that prevents apoptosis [34]. The development of skeletal diseases is often accompanied by impaired autophagy, resulting in defective osteogenic differentiation of BMSCs [35]. Research has shown that METTL14 promotes the m6A modification of beclin-1 mRNA; insulin-like growth factor 2 binding protein (IGF2BP) reader proteins mediate the increase in beclin-1 expression, thereby inducing autophagy and promoting the osteogenic differentiation of BMSCs [36]. Another reader protein, YTHDF1, is highly expressed during the osteogenic differentiation of BMSCs, promoting autophagy and cell proliferation and differentiation, thereby accelerating fracture healing [37]. However, the underlying mechanisms remain to be elucidated. Moreover, MSCs can promote autophagy through paracrine regulation of m6A modification. The initiation of autophagy begins with the activation of the unc-51-like kinase 1 (ULK1) complex, consisting of ULK1, focal adhesion kinase family-interacting protein of 200 kDa (FIP200), and autophagy-related protein 13 (ATG13) [38]. Loss of FIP200 disrupts ULK1 phosphorylation and stability, thereby inhibiting autophagy induction [39, 40]. In cocultured nucleus pulposus cells (NPCs) with BMSCs, ALKBH5 facilitates the demethylation of FIP200 mRNA and, through the reader protein YTHDF2, suppresses FIP200 mRNA degradation. This increase in autophagy helps prevent apoptosis, indicating that BMSC injections can be used as a therapeutic strategy for intervertebral disc degeneration (IVDD) [41].
During bone formation, osteogenesis is closely intertwined with angiogenesis, a process essential for bone tissue regeneration [42]. BMSCs contribute to neovascularization by differentiating into endothelial cells and promoting local angiogenesis via the secretion of factors such as vascular endothelial growth factor (VEGF) and angiopoietin [43]. Knockdown of METTL3 in BMSCs reduces VEGF expression, impairing the angiogenic potential of these cells [44]. Conversely, overexpression of METTL3 and METTL14 upregulates VEGF-A via m6A modification, enhancing osteogenesis and angiogenesis [45]. In addition to their role in bone formation, MSCs play pivotal roles in various other tissue repair processes. For example, METTL3, in conjunction with insulin-like growth factor 2 binding protein 2 (IGF2BP2), facilitates the secretion of vascular endothelial growth factor C (VEGF-C) from adipose-derived stem cells (ADSCs), thereby inducing the formation of lymphatic vessels and accelerating wound healing in diabetic mice [46]. Although various miRNAs in MSC-derived exosomes have been identified as regulators of angiogenic factors, the specific role of m6A modification in the formation and secretion of these exosomes is unclear [47].
BMSCs can differentiate into osteoblastic (OB) cells under certain conditions
[21]. Osteogenic differentiation of BMSCs involves a variety of signaling
pathways, mainly including bone morphogehetic protein/small mothers against
decapentaplegic (BMP/Smad) [48], Wnt/
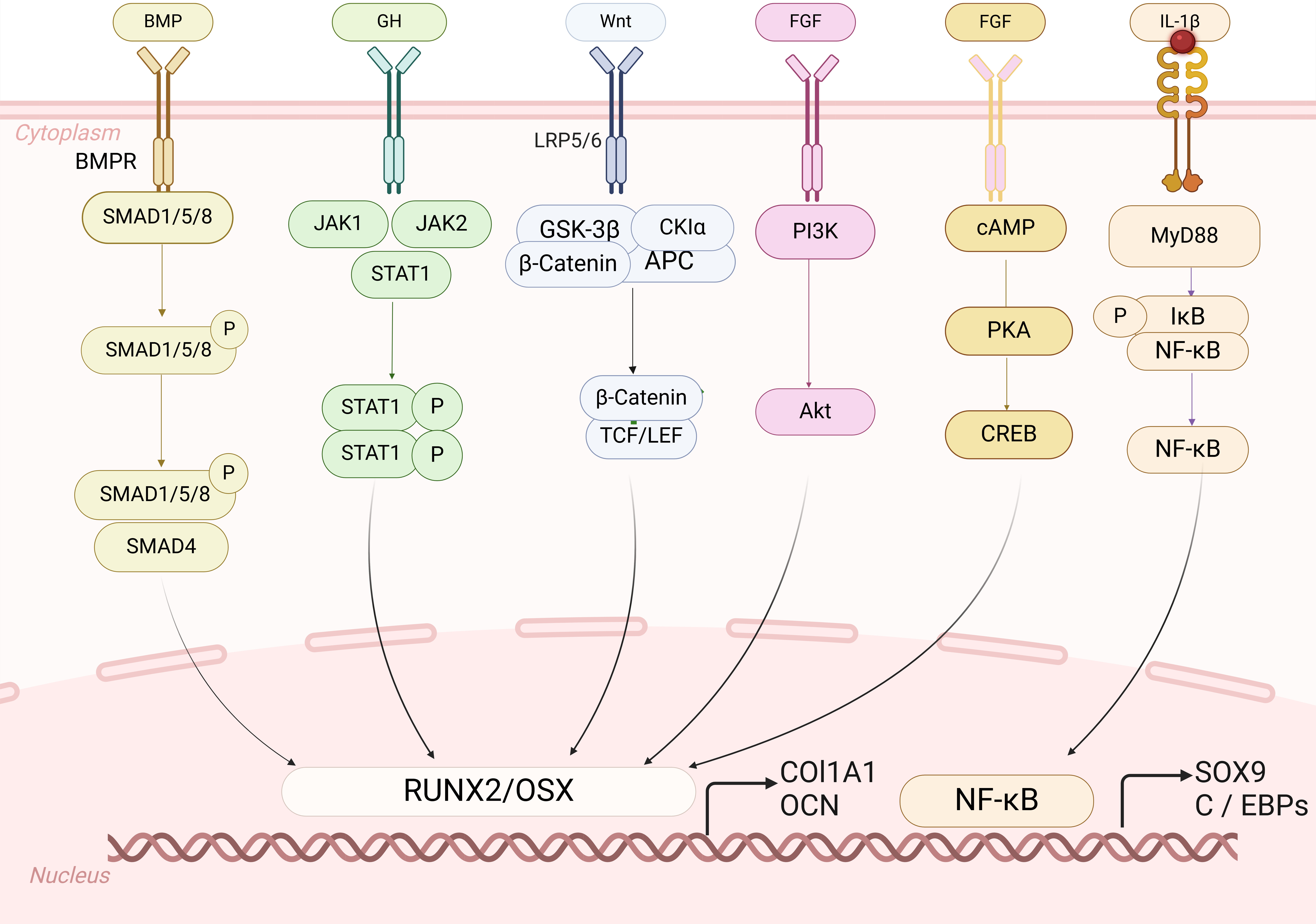 Fig. 1.
Fig. 1.
Osteogenic signaling pathways of mesenchymal stem cells (MSCs).
The osteogenic differentiation of bone marrow mesenchymal stem cells (BMSCs)
involves multiple signaling pathways, including the BMP-Smad,
Wnt/
MicroRNAs (miRNAs), small noncoding RNAs, play crucial roles in the osteogenic
differentiation of BMSCs by regulating gene expression [58]. Different miRNAs
exert varying effects on MSC osteogenic differentiation. For example, miR-21
targets the PI3K/
m6A modification not only acts directly on miRNAs but also regulates MSC osteogenesis by influencing the processing of primary miRNAs (pri-miR) [62]. For instance, WTAP promotes the maturation of pri-miR-181a and pri-miR-181c, while METTL3 inhibits the maturation of pri-miR-320, promoting BMSC osteogenesis [63, 64]. Additionally, METTL14 fosters the maturation of pri-miR-873, inhibiting BMSC proliferation [62]. Furthermore, METTL3 in stem cells of the apical papilla (SCAPs) has been demonstrated to promote osteogenic differentiation of SCAPs by inhibiting the maturation of miR-196b-5p through binding to DiGeorge syndrome critical region 8 protein (DGCR8) [65].
Recent studies have also revealed interactions between miRNAs
and long noncoding RNAs (lncRNAs). LncRNAs have been demonstrated to impact miRNA
regulation of osteogenic differentiation in MSCs. For example, the lncRNA H19
acts as a sponge to absorb miR-141, weakening its ability to inhibit
Thus, m6A modification plays a critical role in regulating ncRNA function, inducing diverse osteogenic effects by modulating the expression and activity of ncRNAs. Compared with traditional osteogenic therapies, targeting ncRNAs offers greater specificity, enabling precise bone formation or repair regulation while minimizing systemic side effects [68]. Furthermore, the multitarget nature of ncRNAs, including miRNAs, can increase the therapeutic potential of MSCs [69]. Therefore, studying the impact of m6A modification on miRNA activity may facilitate its application in osteogenic therapies and expand its clinical value in treating bone diseases.
OA is a degenerative joint disease marked by joint deformity, pain, loss of
function, and excessive inflammation, with the knee being the most commonly
affected area [70, 71, 72]. Inflammation plays a central role in the onset and
progression of OA. Macrophages activate the nucleotide-binding domain, leucine-rich–containing family, pyrin domain–dontaining-3 (NLRP3) inflammasome, thereby releasing pro-inflammatory
mediators and cytokines (such as IL-1
BMSCs are progressively being recognized as an innovative therapeutic modality for OA. Their principal biological functionalities encompass: (1) The capacity to differentiate into chondrocyte-like cells within the joint milieu, thereby compensating for the impaired chondrocyte function observed during the progression of OA [79]. (2) The ability to mediate therapeutic effects through the secretion of Extracellular Vesicles (EVs), which play a pivotal role in the management of OA [80]. Consequently, a deeper understanding of the mechanisms by which m6A modifications regulate these biological functions of BMSCs could provide new strategies for OA treatment.
BMSCs have the potential to differentiate into chondrocytes, and the key to
treating cartilage defects is to enhance their proliferation and differentiation
[81]. An investigation has demonstrated that the m6A modification, facilitated by
METTL3 and collaborating with the 5-methylcytosine (m5C) modification mediated by NOP2/Sun RNA
methyltransferase 4 (Nsun4), promotes the translation of SOX9 by recruiting
YTHDF2 and eukaryotic translation elongation factor 1 alpha 1 (eEF1
Furthermore, research findings have indicated that collagen hydrogels, which bear resemblance to the natural cartilage matrix, offer a malleable environment conducive to cell adhesion, proliferation, and differentiation. By downregulating FTO and reducing the expression of cytoskeletal recombinant proteins actinin-alpha 1 (ACTN1) and ACTN4, which regulate adhesion and the actin cytoskeletal signaling pathways, collagen hydrogels facilitate the chondrogenic differentiation of BMSCs [84]. These findings indicate new opportunities for the use of collagen hydrogels in OA treatment.
While mesenchymal stem cell therapy (MSCT) has shown great potential in treating various diseases, challenges such as poor MSC survival, low permeability, and limited blood circulation in bone tissue have hindered its broader application [19]. Consequently, MSCs primarily exert their therapeutic effects through the secretion of cytokines and exosomes rather than through the direct action of the cells themselves [85, 86, 87]. MSC-EVs exhibit inherent targeting properties towards injured tissues and display minimal immunogenicity, thereby mitigating the likelihood of transplant rejection. This renders them exceptionally safe for clinical applications, and they have been investigated as drug-delivery vectors for targeted tissue repair [88].
MSC-EVs are extracellular vesicles that are approximately 40–150 nm in size and contain various bioactive molecules, such as proteins, lipids and RNA, which are key to the therapeutic effects of MSCs [89]. Recent research indicates that m6A modification serves as a downstream effector of MSC-EVs. In this context, MSC-EVs modulate the activity of methyltransferases within recipient cells, subsequently impacting cellular functionalities. For example, human umbilical cord-derived MSC-EVs (hucMSC-EVs) carry miR-1208, which inhibits METTL3 activity in macrophages, reducing the m6A levels of NLRP3 mRNA, alleviating inflammation, and suppressing MMP13 expression in chondrocytes, ultimately slowing the progression of OA in mice [10]. However, BMSC-EVs downregulate METTL3 activity in chondrocytes, promote ACSL4 expression, inhibit ferroptosis, and protect cartilage [90].
Furthermore, MSC-EVs can transport methylation enzymes such as FTO, directly regulating chondrocyte function in an m6A-dependent manner. For example, FTO-overexpressing MSC-derived EVs (FTO-EVs) regulate chondrocyte autophagy and apoptosis in a YTHDF2-dependent manner by upregulating autophagy-related proteins ATG5 and ATG7 while downregulating the pro-apoptotic gene BNIP3, thus triggering autophagy and inhibiting apoptosis, providing a novel therapeutic target for OA treatment [91].
While MSCT involves transplantation, MSC-EVs can be administered via injection, avoiding the complex procedures involved in cell culture and expansion, thereby simplifying the treatment process [92]. Furthermore, MSC-EVs exhibit natural biocompatibility and lower toxicity, with fewer side effects than traditional drug therapies, making them a promising strategy for arthritis treatment and offering new therapeutic options for patients [93, 94]. However, current research on the impact of m6A modification on the formation and secretion of MSC-EVs remains limited, and further development of techniques to regulate the specific cargo within MSC-EVs is needed. Table 1 (Ref. [10, 90, 91]) shows a summary of the therapeutic effects of MSCs-EVs on osteoarthritis.
| EVs content | m6A regulatory factors | Research subjects | Route of administration | Publication date | References |
| hucMSCs-EVs | METTL3 | C57BL/6 NLRP3-/- | Intra-articular injection | 07/2022 | [10] |
| rat BMSC-Exos | METTL3 | SD Rat model of OA | Intra-articular injection | 02/2024 | [90] |
| ratBMSC-Exos | FTO | C57BL/6J | Not specified | 8/2024 | [91] |
MSCs-EVs, mesenchymal stem cells-derived extracellular vesicles; EVs, extracellular vesicles; hucMSC-EVs, human umbilical cord-derived MSC-EVs; m6A, N6-methyladenosine; METTL3, methyltransferase-like 3; FTO, fat mass and obesity-associated protein; OA, osteoarthritis; C57BL/6J, C57 black 6 Jackson laboratory mouse; NLRP3, nucleotide-binding domain, leucine-rich–containing family, pyrin domain–dontaining-3.
MSCs are a vital source of adipocytes, and their adipogenic
differentiation is also regulated by m6A modification [95], primarily through the
ERK1/2 and peroxisome proliferator-activated receptor (PPAR) signaling pathways.
PPAR-
FTO has been demonstrated to promote osteogenic differentiation of BMSCs by
reducing PPAR-
In the tumor microenvironment, adipocytes derived from MSCs contribute to tumor growth and drug resistance by accumulating free fatty acids and releasing adipokines [103, 104, 105]. Study has shown that loss of METTL3 promotes MSC adipogenic differentiation by inhibiting the AKT1 signaling pathway, accelerating cancer progression, and increasing resistance to cytarabine in acute myeloid leukemia (AML) [106]. Therefore, targeting m6A modification to regulate the AKT signaling pathway presents a potential strategy for overcoming chemotherapy resistance in AML [107].
In the context of endocrine diseases, obesity exacerbates insulin resistance, worsening glucose metabolism in diabetic patients [108]. Thus, targeting the regulation of adipogenic differentiation in adipose-derived MSCs may offer a novel approach to alleviate insulin resistance in obese diabetic patients [101]. m6A modification has demonstrated significant therapeutic potential in inhibiting MSC adipogenic differentiation, providing promising new targets for treatment. Further exploring the specific regulatory mechanisms of m6A modification in this process will offer crucial theoretical insights and therapeutic strategies for addressing adipose-related diseases and cancer drug resistance.
MSCs communicate information with the surrounding microenvironment and cells
through paracrine effects, thereby regulating inflammatory responses, influencing
microenvironmental status, and exerting immunomodulatory functions. Research
indicates that MSCs can modulate cell functions and the microenvironment by
regulating the activity of m6A-modifying proteins in nearby cells. The exchange
of information is of significant importance for the processes of tissue repair,
immune homeostasis, and the improvement of pathological states. For example, MSCs
reduce inflammation and improve kidney injury in diabetic nephropathy (DN) mice
by inhibiting WTAP expression in renal tubular epithelial cells (HK-2), which
leads to decreased Enolase 1 expression and reduced secretion of inflammatory
factors [109]. Conversely, research has demonstrated that the microenvironment is
capable of regulating the pluripotency of MSCs at the post-transcriptional level.
For example, TNF-
MSCs are vital in maintaining immune and inflammatory homeostasis [111, 112]. As key immune cells, monocytes can be recruited by MSCs from peripheral blood to inflamed tissues, where they differentiate into either M1 pro-inflammatory or M2 anti-inflammatory macrophages [113]. This procedure is of paramount importance for the preservation of immune homeostasis. The recruitment of monocytes induced by MSCs and the polarization of M1 macrophages play significant roles in the pathogenesis of a diverse range of diseases, such as ankylosing spondylitis (AS), spinal cord injury, and inflammatory bowel disease (IBD) [114].
MSC-EVs facilitate the polarization of macrophages towards an anti-inflammatory phenotype by regulating m6A modification, thereby exerting immunoregulatory effects [115]. For instance, hucMSC-EVs can upregulate and enhance the binding of METTL3 to Slc37a2 mRNA, increasing Slc37a2 expression and promoting the polarization of macrophages towards the M2 anti-inflammatory phenotype, which alleviates IBD [12]. Study has also shown that specific treatments of MSCs can regulate the contents of MSC-EVs via m6A modification. For example, melatonin administration results in a reduction in METTL3 expression in MSCs. This leads to an increase in the levels of the deubiquitinase ubiquitin specific peptidase 29 (USP29) in extracellular vesicles derived from MSCs. Extracellular vesicles containing USP29 interact with nuclear factor-like 2 (NRF2) in macrophages, thereby promoting the deubiquitination of NRF2 and increasing its stability. This, in turn, induces the anti-inflammatory M2 macrophage phenotype. This intricate process facilitates functional recovery in mice following spinal cord injury (SCI) [116].
While MSCs influence monocyte recruitment and polarization, macrophages also
affect them. For example, in diabetic peri-implantitis, M1 macrophage
polarization is also present, and an “information exchange” between
inflammatory cells and alveolar bone MSCs inhibits MSC osteogenic differentiation
[117]. Recent study suggests that reduced ALKBH5 expression may be how
macrophages inhibit MSC osteogenic differentiation in diabetic peri-implantitis
[118]. The interaction between MSCs and monocytes is also evident in AS. AS-MSCs
secrete monocyte chemoattractant protein 1 (MCP1) and C-C motif chemokine ligand
2 (CCL2), leading to monocyte infiltration and M1 polarization, exacerbating
chronic inflammation [119]. Furthermore, TNF-
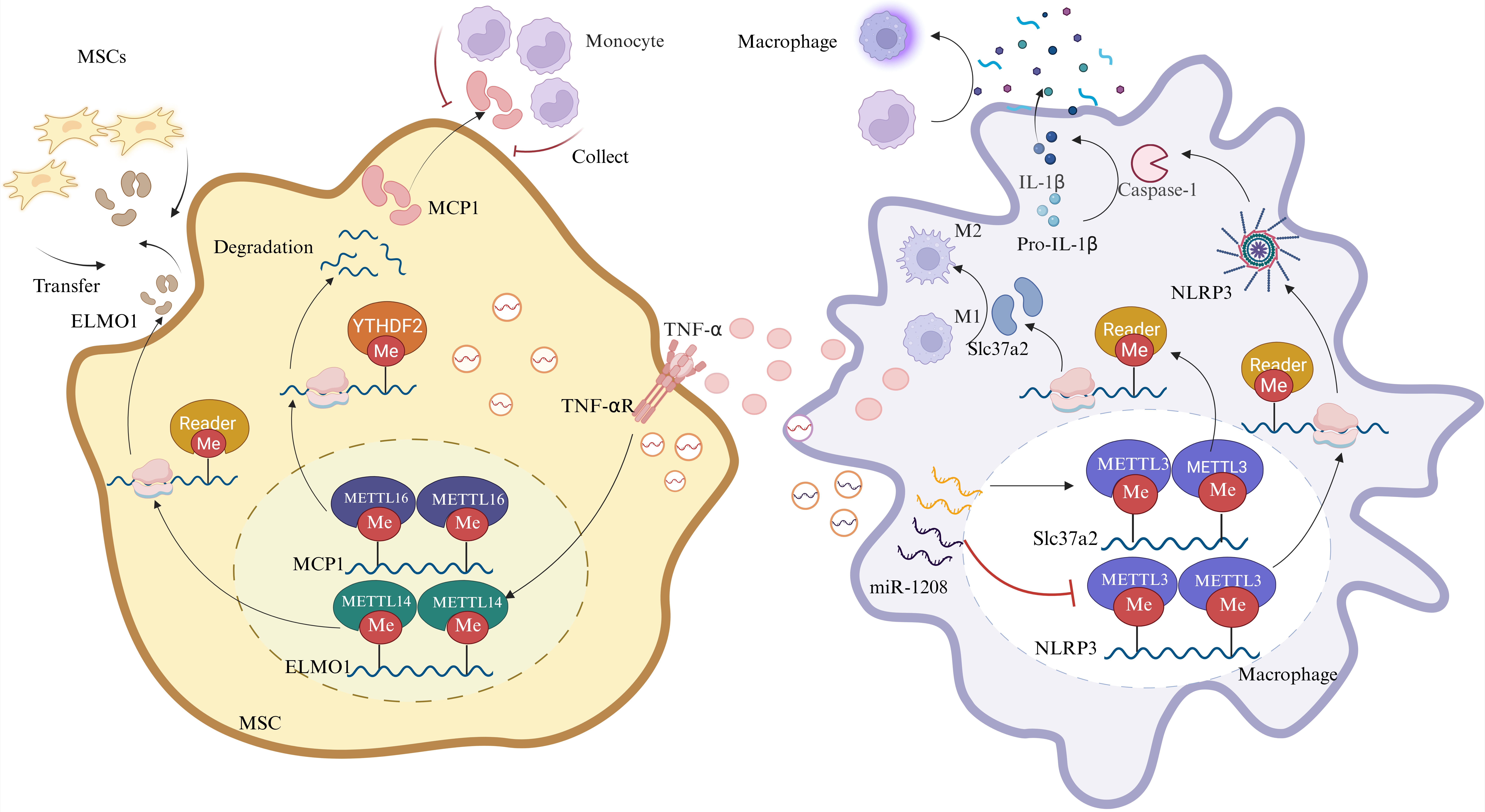 Fig. 2.
Fig. 2.
The role of m6A modification in the Interaction between
MSCs and macrophages. MSCs recruit monocytes to inflammatory sites, where they
differentiate into M1 proinflammatory or M2 anti-inflammatory macrophages.
METTL16 in MSCs inhibits monocyte recruitment by degrading monocyte
chemoattractant protein 1 (MCP1) mRNA via YTH domain-containing family protein 2
(YTHDF2). hucMSCs-EVs suppress METTL3 in macrophages through miR-1208, decreasing
NLRP3 m6A levels and proinflammatory factor levels. Macrophage-secreted
TNF-
Research has also shown that pro-inflammatory factors in the microenvironment
can induce MSCs to express immunosuppressive molecules, enhancing their
immunosuppressive effects. For instance, IFN-
In summary, m6A modification plays a crucial role in the interaction between MSCs and immune cells and in immunomodulation, influencing the progression of various diseases. MSCs also play a key role in the tumor microenvironment, and m6A modification is involved and has significant impacts. Next, we will delve into the specific mechanisms of m6A modification in the tumor microenvironment and related research progress.
MSCs are critical in shaping the tumor microenvironment and regulating tumor cell functions, influencing tumor proliferation, metastasis, angiogenesis, and immune evasion [122, 123]. MSCs can be reprogrammed within different microenvironments through various mechanisms, inducing epithelial-mesenchymal transition (EMT), promoting cell proliferation, maintaining the tumor stem cell niche, and modulating tumor metabolism [124]. For example, exosomes derived from gastric cancer cells upregulate colony-stimulating factor 2 (CSF2) through the IGF2BP2-dependent m6A pathway, thereby inducing the ubiquitination of Notch1 and inhibiting its signaling cascade. This reprograms MSCs into a pro-tumor phenotype, enhancing gastric cancer cell proliferation, migration, and drug resistance, and providing new therapeutic targets and intervention strategies for gastric cancer [125, 126, 127].
Moreover, MSC-EVs hold significant regulatory and therapeutic potential in the tumor microenvironment. MSC-EVs can transport proteins, lipids, nucleic acids, and RNA demethylases [128, 129]. For instance, the exosomes derived from bone marrow mesenchymal stem cells (FTO-EVs) can demethylate long non-coding RNA long non-coding RNA of colorectal cancer (GLCC1), thereby activating the c-Myc signaling pathway and augmenting the invasiveness and drug resistance of acute myeloid leukemia cells [130]. In vivo study have also shown that BMSC-EVs loaded with shRNA-ALKBH5 can inhibit the expression of the ubiquitin-conjugating enzyme ubiquitin-conjugating enzyme E2C (UBE2C), reduce the ubiquitination and degradation of p53, and suppress the stemness, proliferation, and metastasis of triple-negative breast cancer (TNBC) cells [131].
The anti-inflammatory and anti-cancer effects of m6A modification in MSCs provide new opportunities for identifying therapeutic targets. m6A modification can regulate paracrine functions, modulating immune responses and inflammation to improve disease microenvironments. In cancer therapy, m6A modification inhibits tumor proliferation and metastasis by controlling signaling between MSCs and tumor cells. Targeting m6A modification can enhance the anti-inflammatory effects of MSCs, reducing inflammatory factors through gene regulation and offering new therapeutic avenues for inflammation-related diseases and cancer. Therefore, further understanding of its specific mechanisms will be crucial for developing novel treatment strategies.
MSCs are adult stem cells found in a wide range of tissues, including bone marrow, heart, lungs, and adipose tissue [2]. m6A modification plays a crucial role in regulating MSC differentiation, but abnormal differentiation can lead to the progression of various diseases. For instance, METTL3 promotes the aberrant differentiation of MSCs into myofibroblasts in lung tissue, contributing to pulmonary fibrosis [132]. In aortic valve interstitial cells, METTL3 enhances osteogenic differentiation, leading to aortic valve calcification (AVC) [133, 134]. Additionally, under hypoxic conditions, METTL3 drives the differentiation of ADSCs into vascular smooth muscle cells (VSMCs), accelerating the progression of myocardial infarction (MI) [135]. These findings highlight the critical role of m6A modification in controlling MSC differentiation and disease progression, offering new potential therapeutic targets for related conditions.
MSC-EVs can also regulate various cellular functions by modulating m6A
modification. For example, MSC-EVs play a neuroprotective role in neurological
disorders by reducing neuronal damage and improving recovery after ischemic
stroke. Kruppel-like factor 4 (KLF4) in BMSC-EVs targets lncRNA-ZFAS1 in neurons
damaged by stroke, upregulating FTO, reducing dynamin-related protein 1 (Drp1)
expression, increasing translocase of outer mitochondrial membrane 20 (TOM20)
levels, mitigating mitochondrial dysfunction, and alleviating neuronal injury
[136]. Additionally, MSC-EVs loaded with si-FTO can be delivered to the brains of
animal models, where they stabilize ataxia-telangiectasia mutated (ATM) mRNA,
suppress
| EVs sources | EVs contents | Acting cell | m6A regulatory factors | m6A-modified molecule | Molecular function | Reference |
| huMSCs | miR-1208 | Macrophage | Inhibit METTL3 | NLRP3 | Inhibiting NLRP3 inflammasome activation and the release of inflammatory factors helps alleviate OA. | [10] |
| huMSCs | / | Macrophage | Upregulated METTL3 | Slc37a2 | Upregulating Slc37a2 expression promotes the polarization of anti-inflammatory M2 macrophages and alleviates inflammatory bowel disease. | [12] |
| huMSCs | miR-335-5P | AEC | Inhibit METTL14 | ITG |
Inhibiting ITG |
[138] |
| hucMSCs | miR-26a-5p | NPCs | Inhibit METTL14 | NLRP3 | IGFBP2-dependent reduction of NLRP3 expression prevents focal cell death and alleviates IVDD. | [141] |
| huMSCs | si-FTO | Dopamine neuron | / | ATM | Increased expression of the apoptosis-related factor ATM promotes |
[137] |
| BMSCs | FTO | Acute leukemia cell | / | LncRNA GLCC1 | Activating the GLCC1-IGF2BP-Cmyc complex enhances drug resistance and aggressiveness in AML. | [130] |
| BMSCs | shRNA-ALKBH5 | Triple-negative breast cancer cells | Inhibition of ALKBH5 | UBE2C | Reduced expression of UBE2C decreases p53 ubiquitination and degradation, thereby inhibiting cell stemness and metastasis. | [131] |
| BMSCs | FTO | Chondrocyte | / | Atg5/7 | The YTHDF2-dependent promotion of autophagy proteins ATG5 and ATG7 triggers autophagy, which inhibits apoptosis. | [16] |
| BMSCs | / | Chondrocyte | Inhibit METTL3 | ACSL4 | Reducing m6A modification of ACSL4 prevents cellular ferroptosis and alleviates OA. | [90] |
| BMSCs | HIF-1 |
Islet |
Upregulated YTHDF1 | Atg5/2/14 | Promoting the expression of autophagic proteins ATG5, ATG2, and ATG14B to counteract hypoxia-induced cell apoptosis and senescence. | [140] |
OA, osteoarthritis; ALI, acute lung injury; IVDD, intervertebral disc
degeneration; PD, Parkinson’s disease; AML, acute myeloid leukemia; AEC, alveolar
epithelial cell; NPCs, nucleus pulposus cells; LncRNA, long noncoding RNA;
IGF2BP, insulin-like growth factor 2 binding protein; ITG
In recent years, the roles of m6A modification in MSCs, especially in regulating osteogenic differentiation, antiaging, autophagy, and immunomodulation, have drawn much attention. MSCs can differentiate into various cell types and self-renew; thus, they have great potential in treating bone diseases and immune disorders and in regenerative medicine. However, the functions of MSCs and their regulatory mechanisms are complex. As a critical epigenetic regulatory mechanism, m6A modification profoundly influences MSC biological functions through various signaling pathways and targets.
Firstly, m6A modification plays a crucial role in promoting MSC differentiation. Additionally, m6A modification mitigates MSC aging by modulating autophagy, oxidative stress, and mitochondrial function, thus enhancing their potential for use in bone repair and anti-aging therapies. Targeted regulation of m6A factors like METTL3 and ALKBH5 has been recognized as an effective strategy for treating osteoporosis, fractures, and arthritis. Moreover, m6A modification helps alleviate obesity and insulin resistance by inhibiting adipogenic differentiation pathways, improving the tumor microenvironment, and reducing cancer cell stemness, metastasis, and drug resistance. Secondly, in the realm of immunomodulation, m6A modification significantly reduces inflammation by regulating MSC recruitment of monocytes, inhibiting inflammasome formation, and controlling the polarization of M1 macrophages. Furthermore, the interaction between immune cells and the microenvironment also impacts MSC function. Research indicates that MSC paracrine signaling modulates immune function by influencing the activity of m6A-modifying proteins [135].
MSCs hold significant potential for treating cardiovascular diseases (CVD) by promoting angiogenesis, reducing inflammation, and improving heart function [47, 142]. The knockout of ALKBH5 has been shown to enhance the viability of AMSCs and improve heart function in mice [141]. Also, microRNAs from MSC-EVs offer a new approach to treating CVD. Research suggests that miRNAs within MSC-EVs can regulate the activity of m6A-modifying proteins in target cells, thereby modulating cellular functions [136]. However, the impact of m6A modification on the generation and secretion of MSC-EVs warrants further investigation.
Future research should focus on several key areas: First, a deeper analysis of the specific mechanisms by which various m6A regulatory factors (such as METTL3, FTO, etc.) influence MSC differentiation and immune regulation will aid in developing precision treatment strategies for specific diseases. Second, integrating high-throughput sequencing technology with gene editing tools will further elucidate the global role of m6A modification in regulating MSC function and its potential targets. These approaches are expected to optimize the therapeutic effects of MSCs in orthopedic diseases, immune-related disorders, and regenerative medicine.
Notably, m6A modification research has revealed multiple new avenues for the clinical application of MSCs. In the treatment of bone diseases, further research on enhancement of the osteogenic differentiation ability of MSCs by regulating m6A modification may lead to the development of novel bone repair materials or cell-based treatment regimens. For example, gene-editing techniques can be used to upregulate the expression of METTL3 in MSCs, enhancing their osteogenic differentiation for the treatment of bone defects or osteoporosis. In the treatment of immune-related diseases, regulating m6A modification can optimize the immunomodulatory function of MSCs. In inflammatory bowel disease, by modulating m6A-related molecules in MSC-EVs, it is possible to promote the polarization of macrophages into the anti-inflammatory M2 type, thereby reducing the inflammatory response. In the field of cancer treatment, targeting m6A modification to affect the interaction between MSCs and tumor cells can inhibit tumor growth and metastasis. For example, by regulating m6A modification, the supportive effect of MSCs on tumor cells can be reduced, and the sensitivity of tumor cells to chemotherapeutic drugs can be increased, providing new ideas and methods for cancer treatment.
In conclusion, this review comprehensively elucidates the role of m6A modification in MSCs. It significantly impacts MSC differentiation, including promoting osteogenic and chondrogenic while inhibiting adipogenic differentiation, offers new strategies for skeletal and metabolic diseases and cancer treatment. In immunomodulation, it’s crucial for the interaction between MSCs and immune cells, potentially optimizing MSC-based therapies for immune-mediated diseases. In the tumor microenvironment, targeting m6A modification in MSCs may develop new cancer therapies. However, the precise mechanisms and translation to clinical applications need further study. Overall, research on m6A modification in MSCs has great potential for advancing regenerative medicine and disease treatment.
JHZ, HLS and XDC designed the study. XYZ, YZ and YHW conducted research. JHZ and HLS polished the language. All authors contributed to manuscript editing, read and approved the final version, and are accountable for all aspects of the work.
Not applicable.
The figures were created with BioRender software (BioRender.com).
This research received no external funding.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.