



 , Najah T. Nassif 1, Ann M. Simpson 1,*
, Najah T. Nassif 1, Ann M. Simpson 1,*
1 School of Life Sciences, University of Technology Sydney, Ultimo, NSW 2007, Australia
Abstract
Transcription factors are significant regulators of gene expression in most biological processes related to diabetes, including beta cell (β-cell) development, insulin secretion and glucose metabolism. Dysregulation of transcription factor expression or abundance has been closely associated with the pathogenesis of type 1 and type 2 diabetes, including pancreatic and duodenal homeobox 1 (PDX1), neurogenic differentiation 1 (NEUROD1), and forkhead box protein O1 (FOXO1). Gene expression is regulated at the transcriptional level by transcription factor binding, epigenetically by DNA methylation and chromatin remodelling, and post-transcriptional mechanisms, including alternative splicing and microRNA (miRNA). Recent data indicate a central role for transcription factors in pancreatic β-cell failure in the context of systemic insulin resistance and chronic inflammation. Therapeutic modulation of transcription factor abundance via gene therapy, small-molecule pharmacology, and epigenetic therapies holds great promise for β-cell restoration and metabolic normalisation. However, further clinical translation will require targeted delivery to appropriate tissues, minimising off-target effects and ensuring long-term safety. This review focuses on the involvement of pancreatic β-cells and transcription factors in diabetes development and their therapeutic implications, intending to develop and consolidate a basis for further research in this area and for the treatment of diabetes in the future.
Graphical Abstract
Keywords
- diabetes
- pancreatic transcription factors
- epigenetic therapy
- chromatin remodeling
- metabolic dysfunction
- insulin resistance
- hepatic gluconeogenesis
- lipid metabolism
- inflammatory responses
- islet cell types
Diabetes is a chronic metabolic disorder characterised by persistently high blood glucose levels over a relatively long period [1]. Due to a paradigm shift in lifestyle, aging of the population, and increasing rates of obesity, the global prevalence of the disease has increased phenomenally within the last two decades [1]. In 2021, there were 529 million (95% uncertainty interval [UI] 500–564) people living with diabetes worldwide, and the global age-standardised total diabetes prevalence was 6.1% (5.8–6.5). Prevalence has been rising more rapidly in low- and middle-income countries than in high-income countries. The study in the Lancet predicted that between 2021 and 2050, the global age-standardised total diabetes prevalence is expected to increase by 59.7% (95% UI 54.7–66.0), from 6.1% (5.8–6.5) to 9.8% (9.4–10.2), resulting in 1.31 billion (1.22–1.39) people living with diabetes in 2050 [2]. Diabetes leads to serious long-term complications, which are broadly categorised as macrovascular or microvascular. Macrovascular complications affect large vessels and result in cardiovascular events such as heart attack and stroke [3]. Microvascular complications affect smaller vessels and cause diabetic retinopathy, which affects the eyes, diabetic nephropathy, and diabetic neuropathy, which affects the nerves [4]. These complications reduce the quality of life of patients with diabetes and contribute to other health conditions, including an increased risk of infection and foot conditions [4, 5]. Diabetes is a life-threatening condition requiring constant management and treatment [5].
Transcription factors (TFs) are a class of proteins that bind specific DNA sequences to modulate the transcriptional activity of genes. They regulate the expression of genes at the transcription stage and, therefore, play a critical role in switching genes on and off and controlling the extent of expression. Transcription factors may function as activators that stimulate transcription or repressors that block this process [6]. This regulatory system is essential for many cellular processes, including development, differentiation, and environmental responses. The binding of transcription factors to DNA at the promoter or enhancer regions is a crucial way RNA polymerase is either enabled or prevented from transcribing a gene to produce the corresponding mRNA [3]. TFs are therefore central to maintaining cellular identity and function, with consequences of aberrant function observed in diseases such as cancer and some genetic disorders such as diabetes, congenital hypothyroidism, and syndromes such as Rett syndrome and Waardenburg syndrome, all considered to be linked with mutations that affect specific transcription factor genes [7].
TFs connect cellular signalling pathways to major metabolic functions such as
insulin secretion and beta cell (
TFs play important roles in diabetes management by regulating metabolic pathways that influence metabolic processes, such as lipid metabolism and insulin sensitivity. For instance, forkhead box protein O1 (FOXO1) is a major transcription factor that controls hepatic gluconeogenesis; inhibition of this factor reduces glucose overproduction and increases sensitivity to insulin. In contrast, sterol regulatory element-binding protein 1c (SREBP1c) controls lipogenesis in the liver and its overactivity contributes to hepatic steatosis and hyperlipidaemia. Thus, the specific targeting of these factors by selective inhibitors or modulators may offer alternative valid therapeutic approaches to metabolic dysfunction with fewer side effects. The complex mechanisms developed in relation to these TFs interact with signalling pathways such as PI3K/protein kinase B (Akt) and AMP-activated protein kinase (AMPK). A deeper understanding of these factors will lead to the development of safer and more effective therapies against diabetes [1].
This review outlines the knowledge gaps related to the role and mechanism of TFs
in pancreatic islet cells, with particular emphasis on pancreatic
TFs are determinants of developmental stages in pancreatic
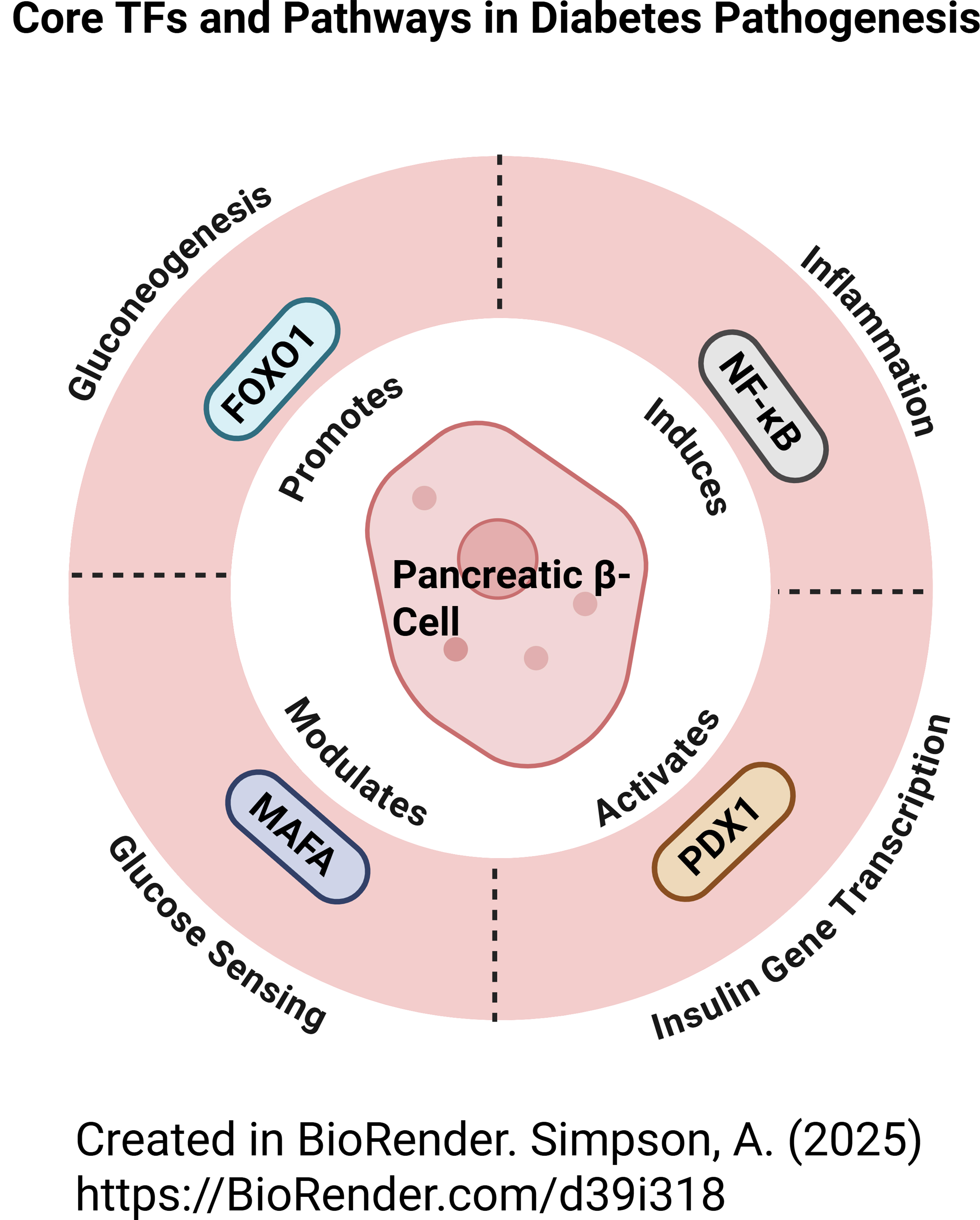
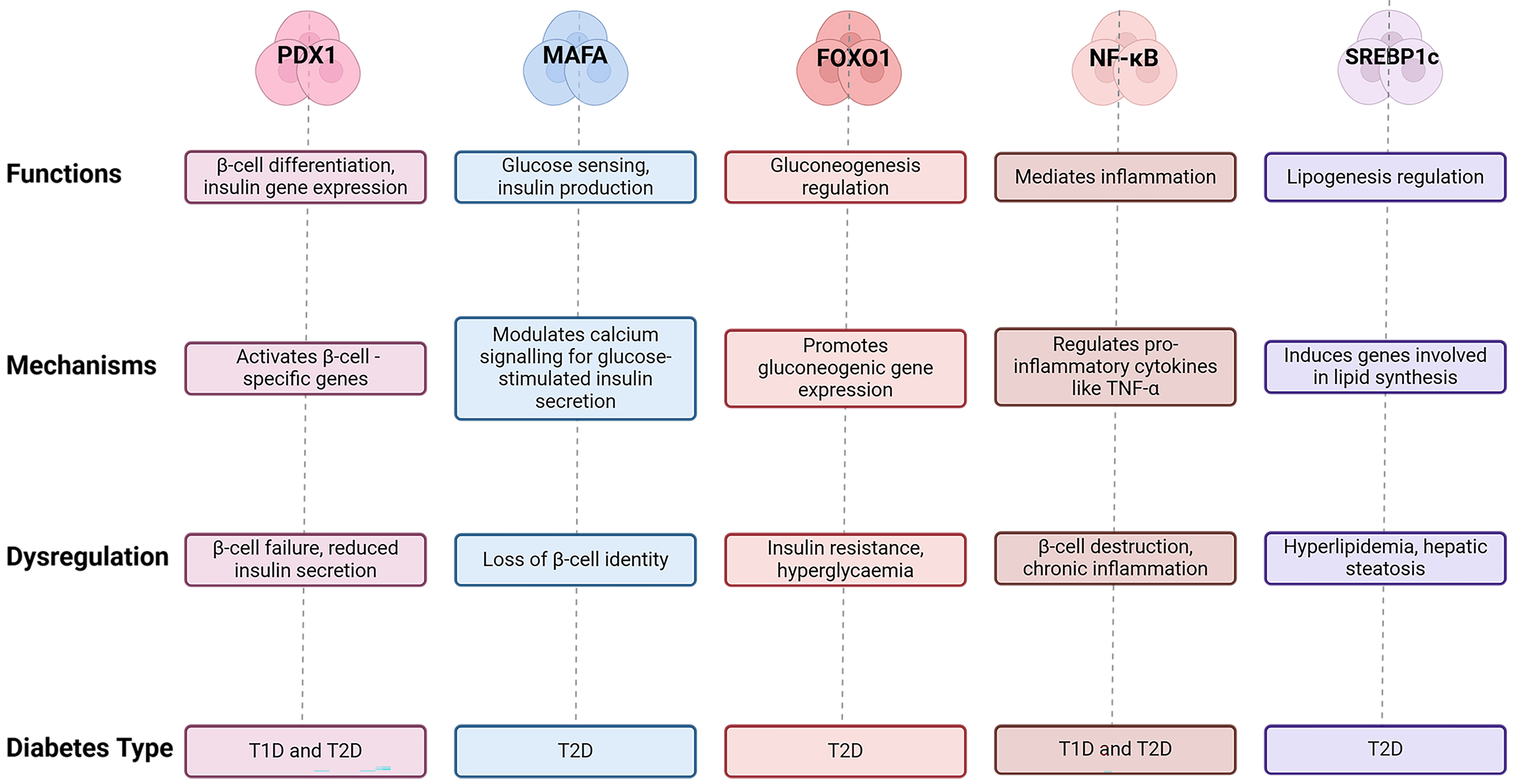 Fig. 1.
Fig. 1.
The role(s) of the major TFs involved in the
pathogenesis of diabetes, including PDX1, MAFA, FOXO1,
NF-
| Transcription factor | Function | Mechanism | Diabetes type | Dysregulation outcome | References |
| PDX1 | Activates |
T1D, T2D | [11, 12] | ||
| MAFA | Glucose sensing, insulin production | Modulates calcium signalling for glucose-stimulated insulin secretion | T2D | Loss of |
[12] |
| FOXO1 | Gluconeogenesis regulation | Promotes gluconeogenic gene expression | T2D | Insulin resistance, hyperglycemia | [12] |
| NF- |
Mediates inflammation | Regulates pro-inflammatory cytokines | T1D, T2D | [13] | |
| SREBP1c | Lipogenesis regulation | Induces genes involved in lipid synthesis | T2D | Hyperlipidemia, hepatic steatosis | [14] |
| HNF4 |
Regulates glucose-responsive genes | Activates SLC2A2, GCK | T1D, T2D, MODY1 | Impaired insulin secretion | [15] |
| KLF11 | Suppresses |
Represses pro-apoptotic BAX | T2D | [16] |
TFs, Transcription factors; PDX1, pancreatic and duodenal homeobox 1;
MAFA, v-Maf musculoaponeurotic fibrosarcoma oncogene homolog A;
FOXO1, forkhead box protein O1; NF-
Additionally, Hepatocyte Nuclear Factor 4
As well as the TFs involved in pancreatic development and differentiation, other TFs have been implicated in diabetic complications. These include CCAAT enhancer binding protein beta (CEBPB), Jun proto-oncogene (JUN), and Fos proto-oncogene (FOS), which have all been implicated in vascular calcification (VC). High-glucose-induced activation of CEBPB upregulates miR-32–5p [20], downregulates the protective regulator of vascular smooth muscle cell differentiation GATA binding protein 6 (GATA6), and promotes VC in T2D [20]. This could provide an example of how TFs interact with diabetic vascular pathology. Other TFs implicated in hepatic gluconeogenesis are FOXO1 and phosphoenolpyruvate carboxykinase (PEPCK) [18, 20]. Dysregulation of FOXO1 promotes gluconeogenic gene expression and perpetuates a state of insulin resistance and hyperglycaemia, with PEPCK, a transcriptional target of FOXO1, being a key mediator of glucose overproduction [20].
Some TFs have opposing functions in diabetes. For example, mothers against
decapentaplegic homolog 3 (SMAD3) and PDX1 are representative
of the bifunctional nature of TFs in diabetes, with SMAD3 inhibiting
insulin transcription and promoting
TFs contribute to T1D and T2D through shared but distinct mechanisms, suggesting
that their roles in the pathogenesis of diabetes are multi-layered. While
PDX1 and NEUROD1 are essential in both T1D and T2D because of
their essential roles in
Pancreatic islet TFs are among the most important regulators of
Dysregulation of the action of the TFs NKX6.1 and the regulatory factor
X (RFX) family is highly implicated in diabetes pathogenesis. NKX6.1
maintains
Specific TFs control insulin biosynthesis and secretion at both levels. For
example, Hasnain et al. (2014) [17] showed that interleukin (IL)-22
could rejuvenate glucose-induced insulin secretion by inhibiting endoplasmic
reticulum stress caused by proinflammatory cytokines. This illustrates how TFs
maintain
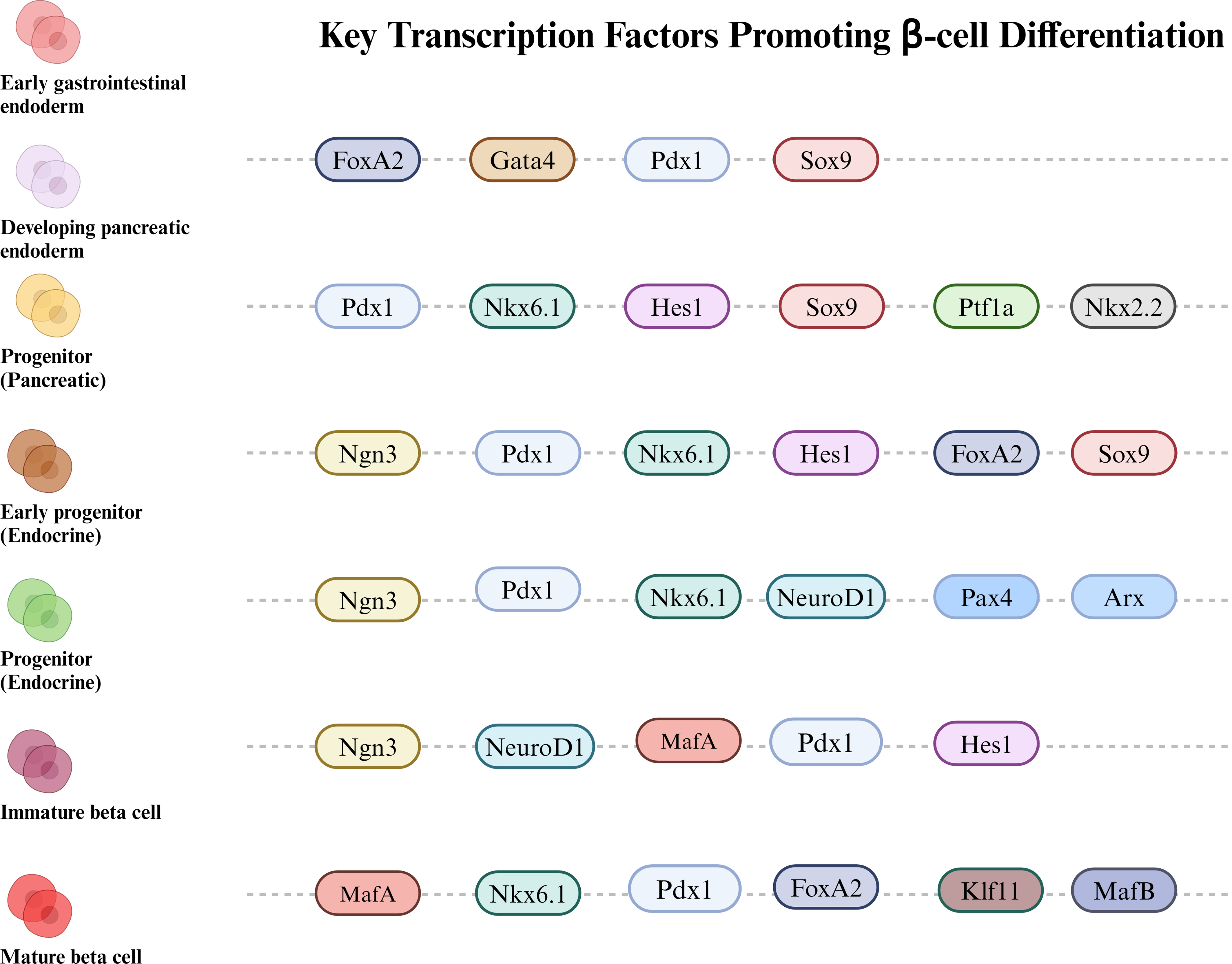 Fig. 2.
Fig. 2.
Schematic representation of how various TFs
sequentially contribute to the
TFs in insulin-responsive tissues, such as the liver, skeletal muscle, and adipose tissue, are critical for maintaining glucose and lipid homeostasis. Dysfunctional activity seriously underlies the development of insulin resistance and T2D [30].
FOXO1 is an essential modifier of hepatic glucose metabolism that mediates gluconeogenesis in response to metabolic and carbohydrate signalling. Stojchevski et al. (2024) [31] demonstrated that in a model of induced insulin resistance, increased FOXO1 activity leads to higher hepatic glucose output through the upregulation of gluconeogenic genes, thereby exacerbating hyperglycaemia. More importantly, the interaction of FOXO1 with a key insulin signalling component, insulin receptor substrate 2 (IRS2), further supports the role of FOXO1 in perpetuating insulin resistance and disturbing glucose metabolism during chronic states [22, 32].
Peroxisome proliferator-activated receptor gamma (PPAR
SREBP1c, through Akt signalling, is an effective inducer of lipogenic genes in the insulin response, which requires nuclear trafficking in the endoplasmic reticulum followed by proteolytic processing for nuclear translocation to activate target gene expression [33, 34]. In insulin-resistant states, however, where this is impaired, apart from disturbing glucose homeostasis, SREBP1c unleashes uncontrolled lipogenesis, leading to hyperglycaemia, hyperlipidaemia, and hepatic steatosis. Insulin resistance is characterised by metabolic dysfunction and further drives the development of diabetes.
Carbohydrate-responsive element-binding protein (ChREBP) is a glucose-responsive transcription factor that interacts specifically with carbohydrate response elements, which mediate the primary transcriptional activity of genes encoding glycolytic enzymes in response to high glucose availability [33]. This transcription factor induces glycolytic and lipogenic genes in response to a high intake of carbohydrates in the liver, including liver pyruvate kinase (LPK), which is required for the appropriate use of glucose and deposition of triglycerides [33, 35]. The state of its phosphorylation controls its activity, that is, its DNA-binding ability and subsequent transcriptional activity. This marks the final role attributed to TFs, such as ChREBP, in maintaining blood glucose levels.
These interactions in insulin-responsive tissues are integrated into complex
networks that regulate glucose and lipid metabolisms. For example, insulin,
through Akt, activates SREBP1c but inhibits FOXO1 in the liver,
and this reciprocal regulation maintains the balance between glucose production
and lipogenesis [32, 35]. Insulin signalling in skeletal muscle regulates the
expression of TFs responsible for glucose uptake and fatty acid oxidation, thus
playing a critical role in maintaining insulin sensitivity. Thus, TFs such as
PPAR
Recent studies have focused on how some TFs, including anterior open (Drosophila
ETS transcription factor) (AOP), FOXO, and pointed (Drosophila ETS transcription
factor) (PNT), interact in hepatic and adipose tissues to change the metabolic
rate and, as a result, longevity [10, 36]. In the skeletal muscle, factors such
as fibroblast growth factor 19 (FGF19) stimulate glucose uptake and lipid
metabolism through signalling pathways initiated by AMPK and sirtuin 1 (SIRT1),
which control PGC-1
Insight into such interactions may provide a general understanding of possible therapeutic approaches that target TFs to improve insulin resistance and T2D. Such tissue-specific actions and systemic interactions of regulators may reveal unique ways to restore metabolic imbalance.
TFs are critical regulators of gene expression related to the inflammatory and
immune responses. They are molecular switches that drive the expression of genes
essential for various immune processes [13].
Nuclear factor kappa-light-chain-enhancer of activated B cells (NF-
The STAT family of TFs, particularly STAT4, have emerged as
critical regulators in immune responses implicated in the pathogenesis of
diabetes [8, 21]. Induction by cytokines, such as IL-12, triggers
STAT4-dependent transactivation of IFN-
Interferon Regulatory Factors (IRFs) modulate the expression of chemokines in
the context of chronic inflammation associated with diabetes and, by doing so,
prolong the infiltration of immune cells into the sites of inflammation, creating
a continuous inflammatory environment that contributes to insulin resistance and
metabolic dysregulation [10, 37]. Abnormal IRF function disrupts the balance
between pro- and anti-inflammatory mediators, characterising chronic inflammation
in T1D and 2 diabetes [37, 38]. Therefore, the regulation of chemokines and
inflammatory responses places IRFs in a critical role in the development of
diabetes. IRF5 and AP-1 further exacerbate diabetic inflammation. The
activation of IRF5 leads to M1 macrophage polarisation, which results in
enhanced TNF-
Chronic inflammation mediates a positive feedback loop by inducing several TFs
that in turn promote
Anti-inflammatory treatments decrease
CHOP is an essential mediator of apoptosis after ER stress, and while necessary
to maintain cellular homeostasis, it becomes deleterious after stress if it is
highly activated [9]. This further complicates the understanding of
Another key player is represented by JNK and its corresponding JNK pathway,
which is implicated in the
By its very nature, hyperglycaemia impairs the activity of key TFs, further
promoting
Other critical oxidative stress pathways, induced by activating certain TFs,
also induce
Through interacting and regulatory networks, these TFs determine the balance
between survival and apoptosis of
The connection between glucose metabolism (Fig. 3A), ER
stress (Fig. 3B), reactive oxygen species (ROS) (Fig. 3C), NF-
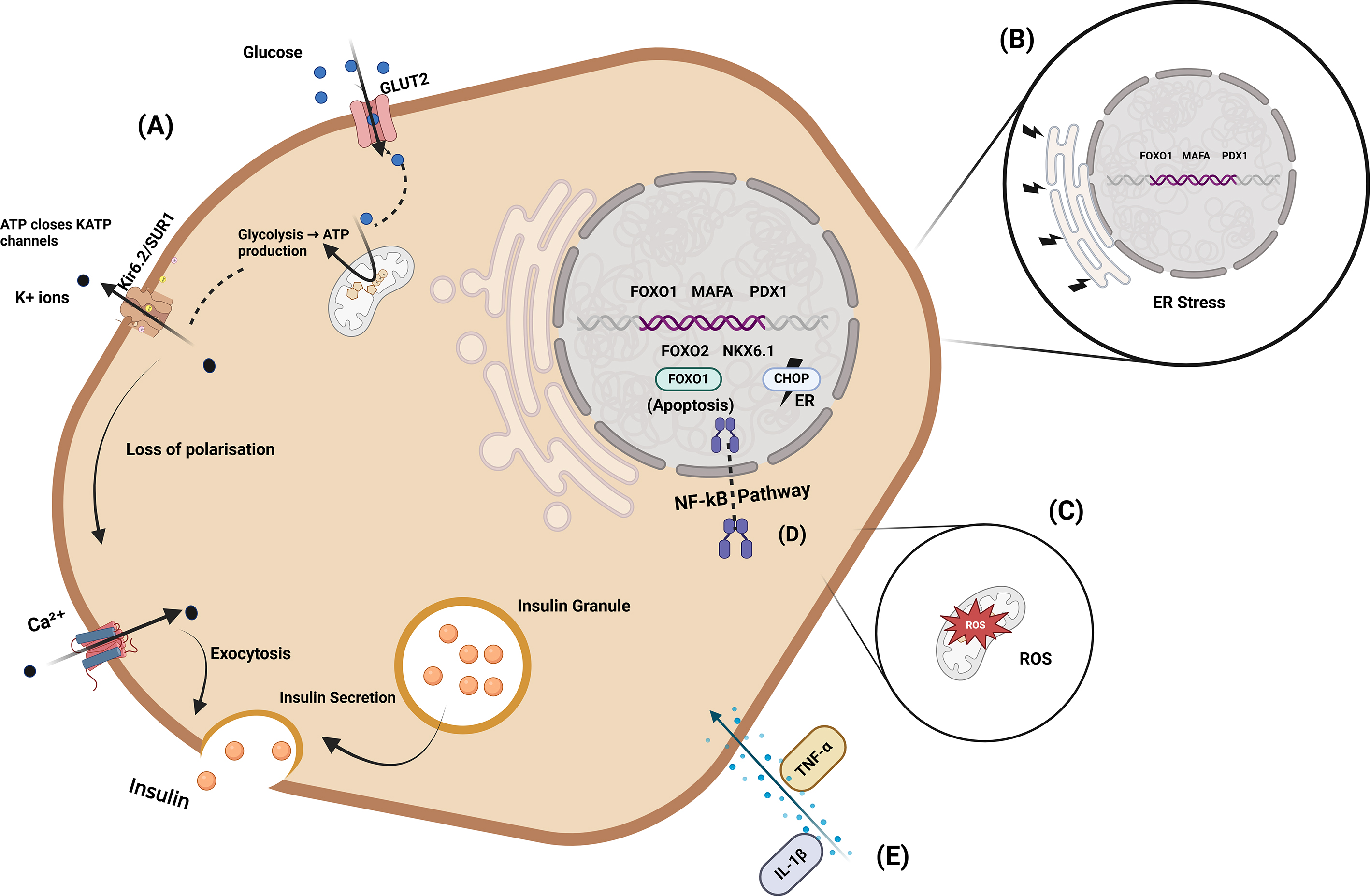 Fig. 3.
Fig. 3.
Integrated pathways regulating insulin production and
The development of diabetes involves contributions from non-beta islet cells
through the action of transcription factors that include aristaless-related
homeobox (ARX) in
Specific TF pathways are implicated in the development of diabetic complications
including retinopathy, nephropathy, and cardiovascular diseases [4]. Increased
vascular endothelial growth factor (VEGF) levels due to HIF-1
TFs represent one of the most essential classes of gene expression regulators
and are significant players in cellular homeostasis in response to metabolic and
environmental signals. Their mode of action involves direct interaction with DNA
at specific promoter or enhancer regions, recruitment of coactivators and
corepressor proteins, and modification of chromatin structures [49]. TFs are
targets of highly sophisticated regulation at multiple levels, including
transcriptional regulation (including DNA methylation and histone remodelling),
post-transcriptional regulation (such as microRNA (miRNA)-mediated control), and
post-translational modifications (including phosphorylation and ubiquitination)
[50]. These regulatory mechanisms dynamically control the critical pathways
essential for
| Modification types | Target TFs | Effect on activity | Relevance to diabetes | References |
| Phosphorylation | FOXO1 | Suppresses nuclear activity, promotes cytoplasmic retention | Reduces |
[14] |
| Ubiquitination | EZH2 | Targeted for degradation | Reduces inflammation, enhances |
[51] |
| Acetylation | PDX1 | Enhances DNA binding and transcription | Promotes |
[34, 52] |
| Sumoylation | FOXO1, NF- |
Stabilises protein and enhances inflammatory gene expression | Exacerbates chronic inflammation in diabetes | [13, 53] |
| Deacetylation | SIRT1 | Promotes anti-inflammatory response | Reduces oxidative stress and insulin resistance | [36] |
EZH2, enhancer of zeste homolog 2; SIRT1, sirtuin 1.
TFs linked to diabetes control essential cellular functions by interacting with
their target DNA-binding motifs, including positive regulatory domain
III/positive regulatory domain I (PRDIII/PRDI) and AP-1 [50]. For instance, MafB
activates the AP-1 motif, stimulating the cytokine promoter activity of genes
such as interferon
Generally, post-translational modifications are essential for fine-tuning the activity of the TFs involved in diabetes. Phosphorylation is likely the most important process in post-translational mechanisms. Phosphorylation is expected the most important step in these processes. FOXO1, for instance, undergoes insulin-induced phosphorylation at sites such as Thr-24, Ser-256, and Ser-319, which prevents its translocation into the nucleus and causes its retention in the cytoplasm, thereby hindering its transcriptional activity [14]. Furthermore, the phosphorylation of Ser-256 may be subjected to additional phosphorylation events that allow interaction with nuclear export proteins. Similarly, phosphorylation affects TFs such as SREBP1c, where signalling pathways regulate its expression via factors such as specificity protein 1 (Sp1) and liver X receptor (LXR), modulating the transcription of target genes [14].
Ubiquitination affects the stability of TFs. For example, the E3 ligase F-box/WD
repeat-containing protein 7 (FBW7) targets enhancer of zeste homolog 2 (EZH2) for
proteasomal degradation [14]. Lower levels of FBW7 in T1D favour the stability of
EZH2 and enhance
Under ER stress FOXO1 becomes Sumoylated which maintains its nuclear
localisation and increases the activity of pro-apoptotic genes such as
CHOP [9, 57, 58]. The study reveals that sumoylation of NF-
Acetylation and deacetylation are dynamic epigenetic modifications that regulate the extent of gene expression. Histone acetyltransferases (HATs) catalyse the addition of an acetyl group to histones, maintaining an open configuration of chromatin with active transcription [61]. On the contrary, histone deacetylases remove the acetyl groups from histones, enabling chromatin’s condensation and inhibiting transcription [56, 61]. The process of DNA repair is regulated by histone deacetylase 1 (HDAC1) and HDAC2 histone deacetylases, either directly altering the key histone residues histone H3 lysine 56 (H3K56) and H4K16, which affect the choice of repair pathway [51]. The other family of nicotinamide adenine dinucleotide (NAD+)-dependent deacetylases consists of sirtuins, which regulate stress responses and metabolic processes, and thus participate in maintaining cellular homeostasis under conditions of metabolic stress.
Sumoylation is the post-translational attachment of small ubiquitin-like modifier proteins that links the stability, localisation, and activity of TFs to oxidative or metabolic stress [43, 53]. This modification supports cellular adaptation because it mechanistically influences the expression of stress-related genes that are essential for maintaining homeostasis and evading apoptosis [42, 43].
miRNAs are important post-transcriptional regulators of gene expression through
the specificity of their binding to mRNAs, leading to either the prevention of
translation or the degradation of these mRNAs [62]. For
instance, miR-709 was previously identified to directly target the TFs CCAAT
enhancer-binding protein
Epigenetic modifications determine the activity of transcription factors that control gene expression and influence the development of diabetes. Transcription factor expression shows a connection with DNA methylation which functions as a crucial epigenetic mechanism through changes in the methylation status of cytosine-phosphate-guanine dinucleotide (CpG) sites [65, 66]. Environmental exposure may induce methylation changes in genes that control inflammation, which is a risk factor for the development of diabetes. For example, DNA methylation may target TFs, such as bromodomain-containing protein 4 (BRD4) and basic leucine zipper ATF-like transcription factor 3 (BATF3), which repress the expression of their respective genes or be deposited on their target genes, thus preventing correct target regulation by these factors. The methylation pattern changes impact both immune responses during cytomegalovirus infection and diabetes susceptibility [65]. The mentioned case stands as a single instance demonstrating how methylation changes control diabetes-related biological processes.
Post-transcriptional control by miRNAs functions as a vital mechanism for gene
expression regulation in diabetes beyond DNA methylation. miRNAs are small
molecules of non-coding RNA which attach to specific mRNA targets which results
in mRNA degradation or translational blockage. The microRNA miR-709 targets
transcription factors CEBPA and MYC directly which affect glucose metabolism and
Non-coding RNAs include long non-coding RNAs (lncRNAs) and miRNAs, which are
major regulators of epigenetic modifications of transcription factor genes.
lncRNAs can act as circRNAs (ccRNAs), thus regulating miRNA availability and the
number of TFs [67]. lncRNA myocardial infarction-associated transcript (MIAT)
affects transcription factor expression indirectly by changing intracellular
signalling, which includes TGF-
Histone modifications including acetylation and methylation are essential for
chromatin accessibility and transcriptional activity. High levels of acetylated
histone three (H3) within myoblasts with reduced vacuolar protein sorting
associated protein 39 (VPS39) indicate disturbances in the differentiation
processes associated with diabetes [65]. In a somewhat related scenario, high
glucose levels cause changes in the degree of histone acetylation, for example,
histone 3 lysine 9 and 14 (H3K9/K14), and reduce repressive trimethylation marks
on H3K9, thus allowing for the perpetual activation of inflammatory genes, such
as IL-6 and monocyte chemoattractant protein 1 (MCP-1) [51]. Increased lipid
levels, including oxidised low-density lipoprotein (LDL), trigger active
epigenetic reprogramming and maintain both proinflammatory responses and
metabolic disorders. Hence, environmental stressors such as high glucose and
lipid levels would strongly influence epigenetics, particularly in endothelial
cells and monocytes, resulting in long-lasting changes in gene expression
observed in metabolic and cardiovascular disorders [65]. The lncRNA
metastasis-associated lung adenocarcinoma transcript 1 (MALAT1) maintains
NF-
Epigenetic therapies targeting DNA methyltransferases (DNMTs) and HDACs are promising approaches for restoring transcription factor regulation in diabetes [65]. Dietary extremes of lipid and carbohydrate consumption, as defined by the IDECG Working Group, have been shown to modulate DNA methylation patterns and thereby alter transcription factor accessibility and downstream gene expression [69]. Simultaneously, HDAC inhibitors improve histone acetylation, thereby increasing chromatin openness and transcriptional activity [65]. Such epigenetic therapies could offer a potential approach for correcting dysregulated gene expression and slowing diabetes progression Table 3 (Ref. [31, 50, 56, 70, 71]) summarises epigenetic modifications influencing TFs and their clinical implications in diabetes.
| Modification | Target gene/factor | Impact on gene expression | Clinical implication | References |
| DNA methylation | NF- |
Reduces transcriptional activity | Suppresses pro-inflammatory responses, reduces |
[50] |
| Histone acetylation | PDPX1 | Enhances chromatin openness | Improves insulin secretion and |
[71] |
| Histone methylation | IL-6 | Increases inflammatory gene expression | Exacerbates chronic inflammation and insulin resistance | [70] |
| Non-coding RNAs | PPAR |
Regulates lipid metabolism gene expression | Reduces lipid accumulation, improves insulin sensitivity | [31] |
| lncRNA-mediated modification | TGF- |
Impacts TFs indirectly | Alters |
[56] |
PPAR
Therapeutic interventions aimed at modulating these transcription factors have,
therefore, become therapeutic targets in antidiabetic treatment approaches,
primarily through the enhancement of
PDX1 is critical for
Similarly, NEUROD1 is essential for
MAFA is a key modulator of the glucose-sensing pathway, supports
insulin secretion and
Other key transcription factors implicated as general regulators of metabolic
pathways, including FOXO1, PPAR
Epigenetic treatments add another layer of complexity to target transcription
factors in diabetes. Inhibitors of HDACs and DNMTs-enzymes that modify chromatin
accessibility can indirectly influence the transcription and activity of
transcription factors [65]. Such methods have shown promise in restoring
appropriate gene expression in
The therapeutic potential associated with the modulation of TFs in diabetes reflects the advances that have been made in understanding their role in disease mechanisms Table 4 (Ref. [7, 11, 12, 37, 41, 73, 78, 79]) outlines therapeutic strategies for targeting transcription factors in diabetes management, including their mechanisms and challenges. Although there have been valuable insights into preclinical studies and early clinical trials, developing safe, selective, and effective treatments is a major barrier to the translation of TFs into diabetes therapeutics.
| Transcription factor | Therapeutic approach | Mechanism of action | Challenges | References |
| PDX1 | Gene therapy | Restores |
Precision targeting of |
[12, 79] |
| NEUROD1 | Pharmacological modulator | Enhances |
Off-target effects, in vivo modulation issues | [7, 11, 73] |
| FOXO1 | Inhibitor | Reduces |
Systemic side effects due to broad activity | [12] |
| SREBP1c | Modulator | Reduces lipogenesis and hepatic steatosis | Difficulties in achieving tissue-specific action | [41, 78] |
| NF-κB | Anti-inflammatory drugs | Suppresses pro-inflammatory pathways | Risk of immunosuppression | [37] |
Transcriptional activators based on the clustered regularly interspaced short
palindromic repeats/deactivated Cas9 system (CRISPR/dCas9) system allow targeted
overexpression of PDX1 in
Transcription factors are instrumental targets for therapeutic intervention in
treating the complex pathogenesis of diabetes. They are relevant to disease
progression, given their key roles in beta-cell function, glucose metabolism, and
inflammation. Therapeutic restoration approaches for transcription factors,
including PDX1, NEUROD1, and MAFA, have shown promise,
whereas modulation of FOXO1, SREBP1c, and PPAR
Future research should focus on delivery systems, such as tissue-specific gene
therapy and advanced drug delivery techniques, to achieve greater therapeutic
specificity. Studying combinatorial approaches using transcription factor
manipulation combined with different metabolic interventions may yield additional
benefits. This calls for increasing insights into transcriptional networks,
especially the mutual influences between transcription factors and epigenetic
regulators when developing new therapeutic approaches. Overcoming these issues
with thorough preclinical and clinical testing will be important to ensure that
transcription factor-based therapies can be safely and effectively applied to
treat diabetes. Also, future research should focus on single-cell multi-omics
techniques to investigate TF networks within human islets from various diabetes
subtypes. Light-activated dCas9 among other inducible CRISPR systems presents a
method for controlling TF activity during specific time periods. Patient-derived
organoid models will help demonstrate the effects of genetic variations in
transcription factors such as HNF4
TFs, transcription factors;
NKP and AMS established the concept, NKP wrote the initial draft, AMS and NTN provided advice on the structure of the review, all authors edited the manuscript, and all authors read and approved the manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not Applicable.
Not Applicable.
This research received no external funding.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.