
 , Arnaud Chevrollier 1,*
, Arnaud Chevrollier 1,*
1 University of Angers, MitoLab, Unité MITOVASC, UMR CNRS 6015, INSERM U1083, SFR ICAT, 49330 Angers, France
2 Departments of Biochemistry and Molecular Biology, University Hospital Angers, 49933 Angers, France
Abstract
The bioenergetic machinery of the cell is protected and structured within two layers of mitochondrial membranes. The mitochondrial inner membrane is extremely rich in proteins, including respiratory chain complexes, substrate transport proteins, ion exchangers, and structural fusion proteins. These proteins participate directly or indirectly in shaping the membrane’s curvature and facilitating its folding, as well as promoting the formation of nanotubes, and proton-rich pockets known as cristae. Recent fluorescent super-resolution images have demonstrated the strong dynamics of these events, with constant remodeling processes. The mitochondrial outer membrane itself is also highly dynamic, interacting with the endoplasmic reticulum and its environment to ensure a rapid diffusion of surface components throughout the mitochondrial networks. All these movements occur besides migration, fusion, and fission of the mitochondria themselves. These dynamic events at the level of mitochondrial membranes are primarily dependent on their unique lipid composition. In this review, we discuss the latest advances in phospholipid research, focusing on their metabolism and role in mitochondrial dynamics. This process emphasizes the importance of interactions with the endoplasmic reticulum and mitochondrial matrix enzymes, extending its relevance to lipid sources, in particular, cardiolipins and phosphatidylethanolamines at the cellular, tissue and even whole-organism level. Given the expanding array of characterized mitochondrial functions, ranging from calcium homeostasis to inflammation and cellular senescence, research in the field of mitochondrial lipids is particularly significant. As mitochondria play a central role in various pathological processes, including cancer and neurodegenerative disorders, lipid metabolism may offer promising therapeutic approaches.
Keywords
- mitochondria
- dynamic
- lipids
- membrane
- mitochondrial diseases
Mitochondria are organelles present in the majority of our cells, and are the
site of many essential metabolic processes, including the Krebs cycle (citric
acid cycle) and the oxidative phosphorylation (OXPHOS) [1]. OXPHOS efficiently
generates adenosine triphosphate (ATP) from energy substrates via the respiratory
chain complexes and the ATP synthase, thus supplying cells with their primary
energy source. The origin of these organelles can be traced back to an ancestral
bacterium that established a symbiotic relationship with a primitive eukaryotic
cell, progressively leading to the formation of mitochondria through a process
known as endosymbiosis [2]. A direct consequence of this evolutionary process is
the persistence of a small circular genome (mtDNA) and of two membranes, known as
outer and inner mitochondrial membranes (OMM and IMM, respectively), which
delimit two compartments, the intermembrane space and the mitochondrial matrix
[3, 4]. The IMM forms closed invaginations called cristae that extend into the
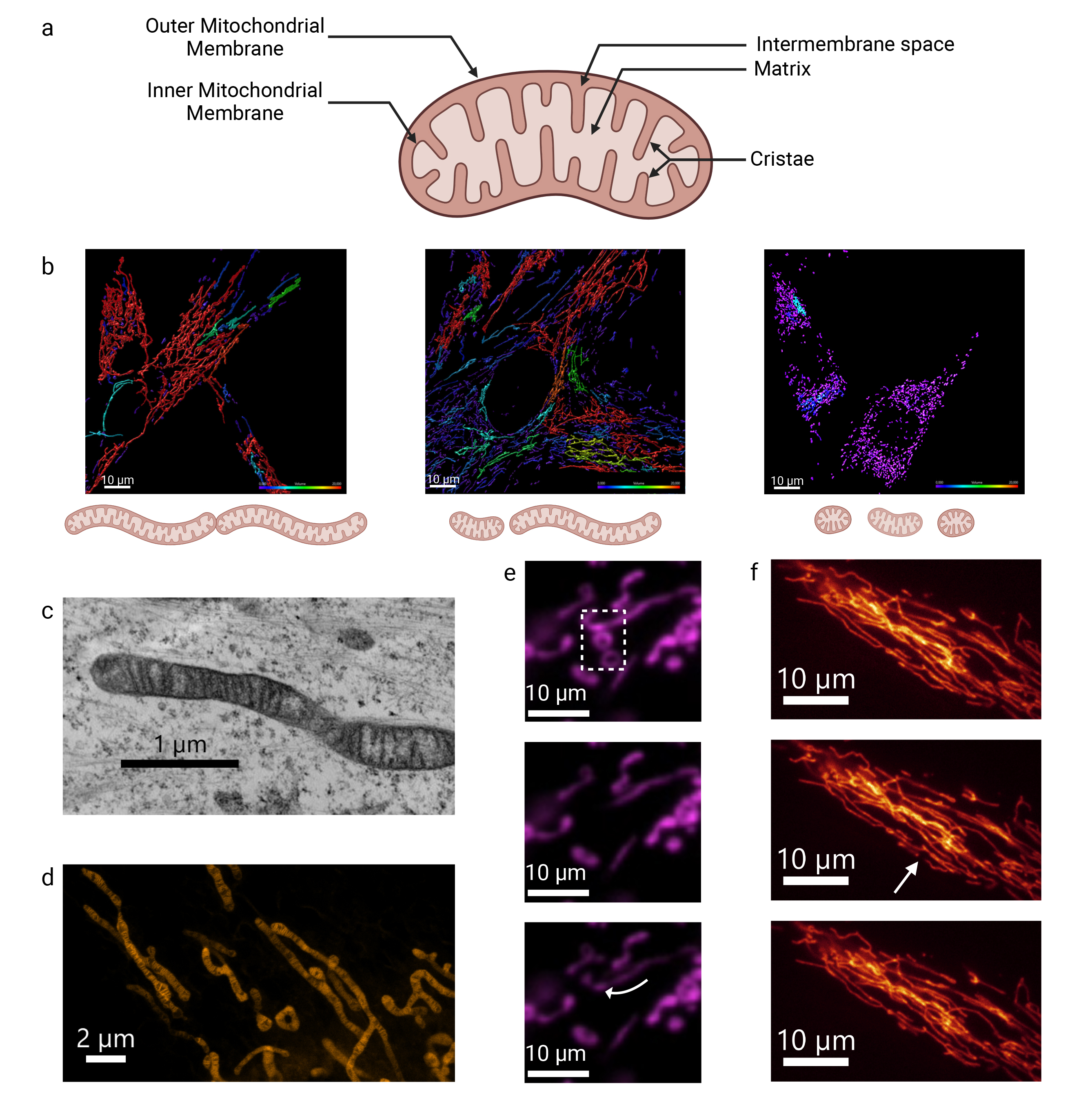
mitochondrial matrix, giving rise to a third mitochondrial compartment (Fig. 1a).
The mitochondrial matrix contains key metabolites such as nicotinamide adenine
dinucleotide (NADH) and ions, including calcium. It is also the site where fatty
acyl-CoA undergoes
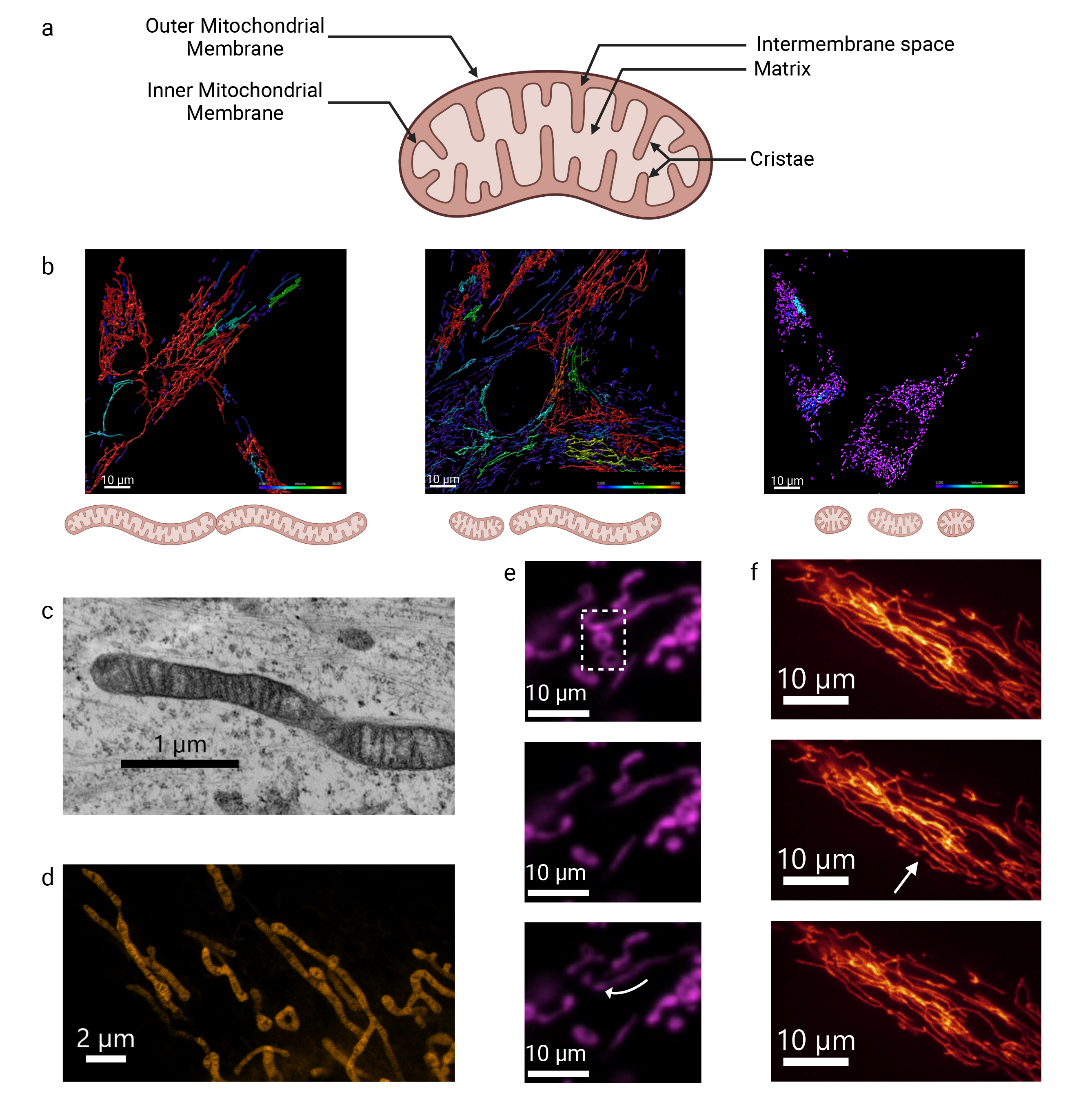 Fig. 1.
Fig. 1.
Mitochondrial membrane structure and dynamics analyzed by fluorescent and electron microscopy. (a) Schematic representation of the structure of mitochondria. Created with BioRender.com. (b) Representative image of the mitochondrial network in control conditions and under mitochondrial stress. An increase in fusion and/or a decrease in fission leads to hyperconnectivity, as shown on the left panel. Conversely, a decrease in fusion and/or an increase in fission results in fragmentation, as shown on the right panel. These images were obtained from human primary fibroblasts (HPF) labeled with MitoTracker Green and further analyzed with the Imaris software. Mitochondria colored in red showcase hyperfusion, while those colored in purple showcase fragmentation. The scale bar = 10 μm. (c) The IMM and cristae structure can be observed in fixed HPF using transmission electron microscopy and, (d) more recently, in live HPF using STED microscopy with the fluorescent probe PKmito Orange. c: scale bar = 1 μm. d: scale bar = 2 μm. (e) Fluorescence Recovery After Photobleaching (FRAP) enables to explore diffusion movements within mitochondrial membrane using OMMprotein-fluorescent Tag in the human glioblastoma (SNB 75) cell line. As shown, only connected mitochondria recover signal (arrow) due to membrane fluidity from other parts of the mitochondrial filament, while isolated donut-shaped mitochondria remain unbleached (see Motion 1). (f) Fission occurs spontaneously within the mitochondrial network of HPF (arrow). The scale bar = 10 μm. The transmission electron microscopy images were acquired using a JEOL JEM 1400 at the SFR ICAT Facility, Angers University, France. Super-resolution STED images were acquired using a TCS SP8 gSTED 3X (Leica Microsystems, Wetzlar, Germany) in collaboration with the CECAD imaging facility, University of Cologne, Germany (Unpublished data, Mitolab, Angers University). IMM, inner mitochondrial membranes; STED, Stimulated emission depletion.
In humans, mitochondrial diseases are rare genetic pathologies caused either by pathogenic variants in mitochondrial DNA (mtDNA), or by mutations in numerous nuclear genes coding for subunits of the OXPHOS system, key metabolic enzymes or proteins involved in mitochondrial structure [6, 7, 8, 9, 10]. Mitochondrial dysfunction is also playing a role in a wide range of pathologies, including cancer, cardiovascular disease, and neurodegenerative disorders [11, 12, 13, 14, 15].
The first detailed images of mitochondria were obtained by transmission electron microscopy, revealing a small bean-shaped structure with characteristic double membrane and distinctive zebra-like inner membrane folds (Fig. 1c,d) [16]. Advances in fluorescent optical microscopy combined with improved acquisition times have opened up a new field for exploring mitochondrial structure [17, 18, 19, 20]. These innovations have revealed the diversity of mitochondrial shapes and lengths, and allowed the observation of dynamic processes such as mitochondrial fusion and fission, which occur with varying frequency within the cells (Fig. 1) [21, 22].
Mitochondria cannot be generated de novo; instead, they undergo various turnover processes, which include mitochondrial fusion and fission (Fig. 1e,f). Fission results in the division of one mitochondrion into two daughter mitochondria, while fusion merges two mitochondria into one [23]. This turnover is crucial to ensure the proper distribution of mitochondria within cells and to maintain a sufficient number of mitochondria in the daughter cells following cell division [24]. Fission also produces smaller mitochondria that can be selectively degraded via mitophagy, an essential process that removes dysfunctional mitochondria [25, 26]. Fusion, conversely, facilitates the mixing of mitochondrial matrix contents, thus contributing to mitochondrial quality control and the maintenance of mitochondrial function [27]. Several recent reviews summarize the latest findings that led to a better understanding of mitochondrial fusion and fission, and how they are orchestrated at the protein level [28, 29, 30, 31, 32, 33, 34]. For fission, the key protein is Drp1 (Dynamin-related protein 1), a GTPase that is recruited to the OMM to facilitate the fission of both the outer and the inner membranes. For outer membrane fusion, the main proteins are mitofusins 1 and 2, which form homodimers or heterodimers with other mitofusins from a neighboring mitochondrion, to bring them closer together. In the inner membrane, the main player is OPA1 (Optic Atrophy 1), a GTPase that exists in two isoforms: short-OPA1 localized in the matrix and long-OPA1 anchored in the IMM. The proportion of these isoforms regulates inner membrane fusion [35]. Interestingly, as discussed below, these fusion factors are closely linked to membrane lipids and play a significant role in shaping membrane’s curvature [36].
Lipids in the mitochondrial membranes are either imported from the cytosol or synthetized de novo within mitochondrial compartments. Furthermore, lipids can be modified in situ and transported or exchanged between mitochondrial membranes, or even with other organelles such as the endoplasmic reticulum. The composition of mitochondrial membranes plays a critical role in regulating the permeability to substrates, ions, and proteins, thereby facilitating the establishment of ionic gradients that are essential for optimal mitochondrial function. Accordingly, the inner and outer mitochondrial membranes exhibit distinct protein and lipid compositions. This asymmetry suggests that some lipids can be transferred from one leaflet to the other through a “flip-flop” process, passing from one membrane layer to the other [37, 38]. In the process of mitochondrial dynamics, the position and organization of lipids contribute to changes in mitochondrial shape. Recent advances in mass spectrometry, fluorescence chemistry and microscopy have enabled the study of lipid composition in the context of mitochondrial fusion and fission, thus paving the way for a new research field to decipher the dynamic interaction between lipids and mitochondrial function [21, 39].
The mitochondrial membranes are primarily composed of lipids known as glycerophospholipids (GPLs), a class of phospholipids (PLs) derived from a common precursor, phosphatidic acid (PA). PA is chemically described as a glycerol moiety with two fatty acyl chains attached to carbons 1 and 2 and a phosphate group bound to carbon 3. Common GPLs are of five types (phosphatidylcholine (PC), phosphatidylethanolamine (PE), phosphatidylglycerol (PG), phosphatidylinositol (PI) and phosphatidylserine (PS)) depending on the polar molecule, with at least one alcohol function (choline, ethanolamine, glycerol, inositol or serine) linked to the phosphate group. Cardiolipin (CL) is a particular GPL with four acyl chains, found almost exclusively in the IMM and formed by two PA, each linked to carbon 1 and 3 of a glycerol moiety. Each member of a GPL class is uniquely determined by the fatty acyls (length and unsaturation degree of the hydrocarbon chain) attached to glycerol. Most of these lipids are synthesized in the endoplasmic reticulum (ER) and subsequently transported to the mitochondria [40]. This transport occurs at specialized regions known as Mitochondria-Associated Membranes (MAMs), where the ER and mitochondria are closely aligned, facilitating the exchange of lipids, calcium, and proteins [41, 42, 43].
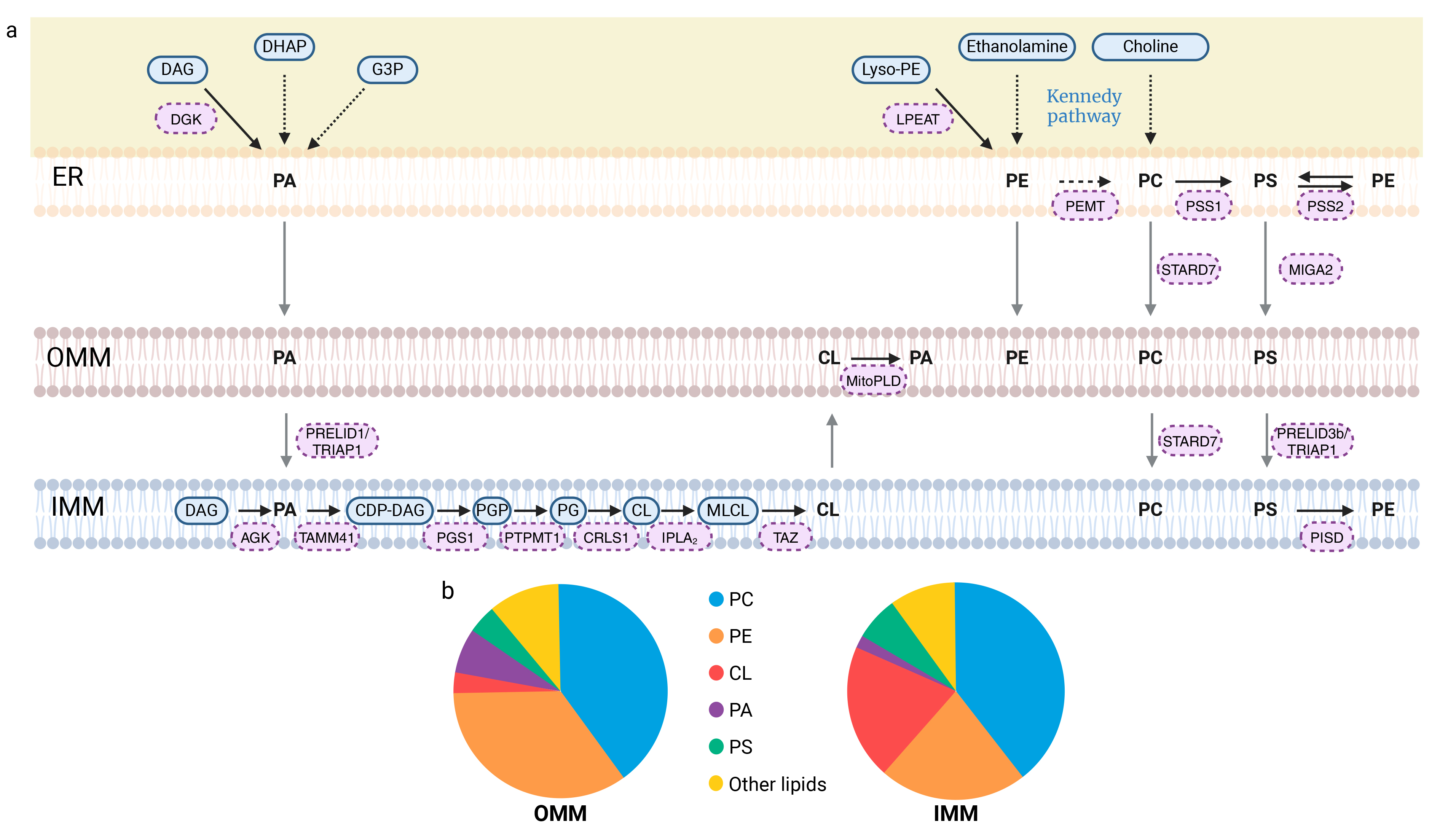
Phosphatidic acid (PA) can be generated through five different pathways (Fig. 2a). Two of these pathways involve de novo synthesis: the glycerol 3-phosphate (G3P) pathway and the dihydroxyacetone phosphate (DHAP) pathway [44]. PA can also be produced via the phospholipase D (PLD) pathway, in which PA is formed from phospholipids (e.g., phosphatidylcholine) through the action of phospholipase D. Another pathway for PA production is the mitochondrial cardiolipin hydrolase (MitoPLD) pathway, which converts cardiolipin into PA (Fig. 2a). Additionally, PA can be synthesized through the diacylglycerol kinase (DGK) pathway, where diacylglycerol (DAG) is phosphorylated by diacylglycerol kinase to form PA [44]. Another enzyme, acylglycerol kinase (AGK), catalyzes the phosphorylation of DAG, leading to PA formation [45]. Mitochondrial PA can be synthesized in the IMM or in the ER and subsequently transported to the IMM by a complex of two proteins: Protein of Relevant Evolutionary and Lymphoid Interest Domain 1 (PRELID1), associated with TP53-Regulated Inhibitor of Apoptosis 1 (TRIAP1) (Fig. 2a) [46, 47, 48].
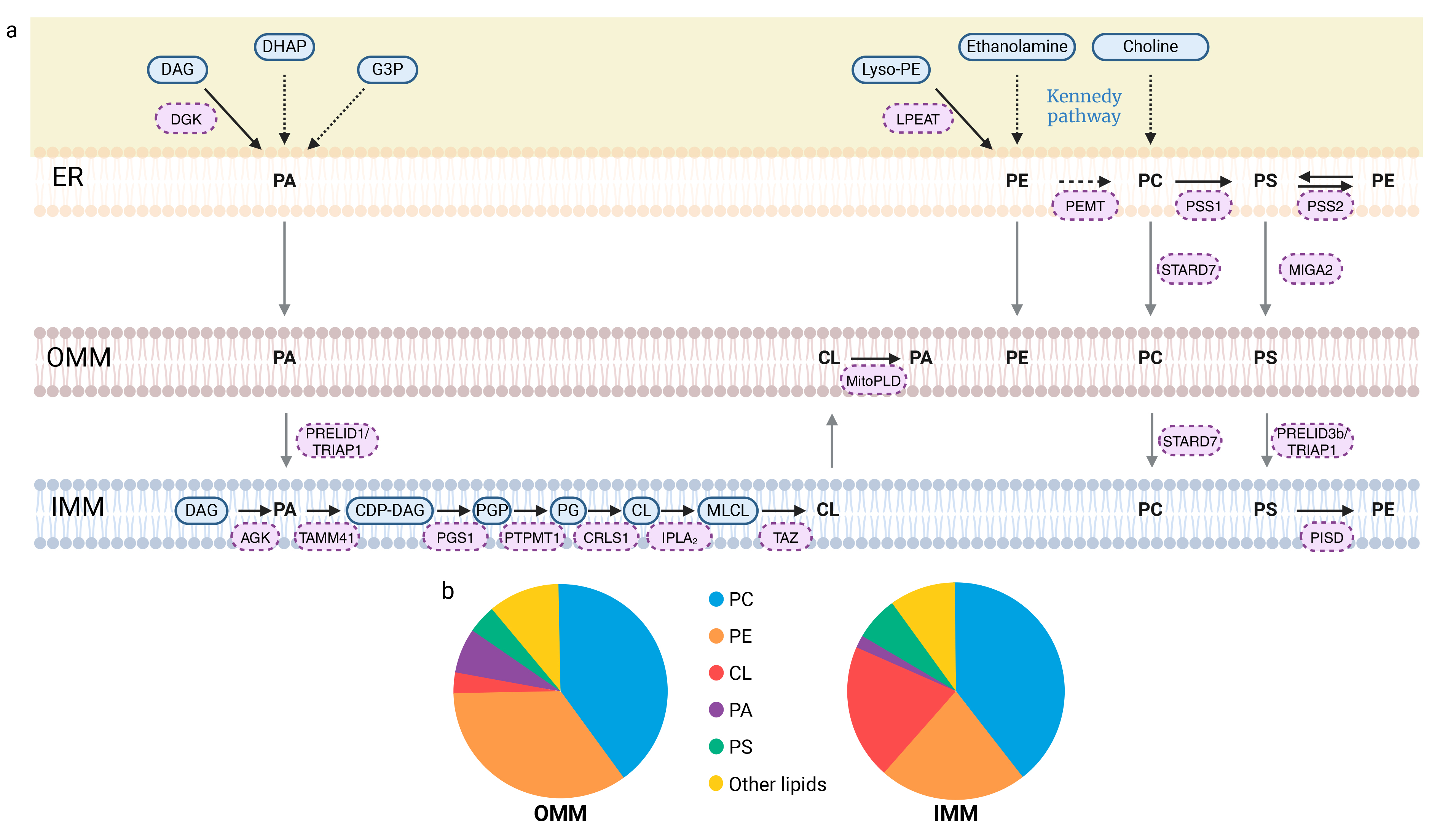 Fig. 2.
Fig. 2.
Synthesis of glycerophospholipids and their transport to mitochondria and phospholipid membrane composition. (a) Mitochondria-associated membranes (MAMs) structure the relationship between the endoplasmic reticulum (ER) and mitochondria, promoting the import of lipids by mitochondria. Phosphatidic acid (PA) synthesis can occur via five distinct pathways. Three of these pathways occur within the ER: the G3P, DHAP and DAG pathways. PA can also be produced from DAG via the AGK protein. The last pathway for PA production is based on the modification of phospholipids, such as cardiolipins, which produce PA through MitoPLD. The transport of PA from the OMM to the IMM is facilitated by the PRELID1/TRIAP1 complex, where it undergoes a series of five enzymatic reactions, resulting in its conversion to CL. Additionally, the phospholipids phosphatidylethanolamine (PE) and phosphatidylcholine (PC) are produced in the ER via the Kennedy pathway. Phosphatidylcholine (PC) is synthesized exclusively in the ER. Indeed, the second synthesis pathway that occurs within the ER involves PE methylation by PEMT. Since PC is the most abundant PL in mitochondrial membranes, its transport to these membranes, mediated by STARD7, is essential. On the other hand, three additional synthetic pathways have been identified for PE: in the ER, from lyso-PE via the LPEAT enzyme or from phosphatidylserine (PS) via the PSS2 enzyme, and in the inner membrane from PS as well, but through the action of the PISD enzyme. With regard to the final PL, PS, two synthetic pathways have been identified, each corresponding to the modification of another PL. PC and PE produce PS via PSS1 and PSS2 in the ER. PS is then transported to the OMM by MIGA2, and from the OMM to the IMM via the PRELID3b/TRIAP1 complex. Thick black arrows indicate direct reactions, with enzymes labeled in pink boxes. Dotted black arrows correspond to a series of reactions, with the name of the pathway indicated in blue. Gray arrows represent phospholipid transport. (b) The pie charts illustrate the distribution of phospholipids within mitochondrial membranes: OMM on the left and IMM on the right. The “other lipids” category includes sphingolipids and additional phospholipids, such as phosphatidylinositol, not detailed in this review. Created with BioRender.com. AGK, Acylglycerol kinase; CDP-DAG, cytidine diphosphate-diacylglycerol; CL, Cardiolipin; CRLS1, Cardiolipin synthase; DAG, Diacylglycerol; DGK, Diacylglycerol kinase; DHAP, Dihydroxyacetone phosphate; ER, Endoplasmic reticulum; G3P, Glycerol 3-phosphate; IMM, Inner mitochondrial membrane; IPLA2, Calcium-independent phospholipase A2-gamma; LPEAT, Lyso-PE acyltransferase; Lyso-PE, Lyso-phosphatidylethanolamine; MIGA2, Mitoguardin-2; MitoPLD, Mitochondrial cardiolipin hydrolase; MLCL, Monolyso-cardiolipin; OMM, Outer mitochondrial membrane; PA, Phosphatidic acid; PC, Phosphatidylcholine; PE, Phosphatidylethanolamine; PEMT, Phosphatidylethanolamine N-methyltransferase; PG, Phosphatidylglycerol; PGP, Phosphatidyl-glycerol-phosphate; PGS1, phosphatidylglycerophosphate synthase 1; PISD, Phosphatidylserine decarboxylase; PRELID, Protein of Relevant Evolutionary and Lymphoid Interest Domain; PS, Phosphatidylserine; PSS1, Phosphatidylserine synthase-1; PSS2, Phosphatidylserine synthase-2; PTPMT1, Phosphatidylglycerophosphatase and protein-tyrosine phosphatase 1; STARD7, StAR-related lipid transfer protein 7; TAMM41, Phosphatidate cytidylyltransferase, mitochondrial; TAZ, Tafazzin; TRIAP1, TP53-regulated inhibitor of apoptosis 1.
Cardiolipins constitute 15–20% of all phospholipids and are the predominant component of the IMM (Fig. 2b) [49]. For CL synthesis, PA is first transformed into cytidine diphosphate-diacylglycerol (CDP-DAG) by the mitochondrial phosphatidate cytidylyltransferase (TAMM41). The subsequent reaction leads to the production of phosphatidyl-glycerol-phosphate (PGP) through the action of phosphatidylglycerophosphate synthase 1 (PGS1) [48]. PGP is then converted into phosphatidylglycerol (PG) by the phosphatidylglycerophosphatase and protein-tyrosine phosphatase 1 (PTPMT1) [50]. The final reaction, catalyzed by cardiolipin synthase (CRLS1), eventually lead to nascent cardiolipin (CL) from PG (Fig. 2a) [51].
Nascent CL can be further remodeled by varying the degree of unsaturation of its fatty acid chains. The first step in this remodeling process is the removal of an acyl chain from cardiolipin by calcium-independent phospholipase A2-gamma (IPLA2), resulting in the formation of monolyso-cardiolipin (MLCL) [52]. Subsequently, under the action of the protein tafazzin (TAZ), MLCL undergoes transacylation, leading to the addition of a new acyl chain and the production of mature cardiolipins (Fig. 2a) [40]. Of note, linoleate (C18:2) forms up to 80% of all acyl chains of CL in the mitochondria of human cardiac tissue [53].
The next representative phospholipid in mitochondrial membranes is phosphatidylethanolamine (PE), which accounts for approximately 30% of total PL (Fig. 2b) [54]. PE consists of a hydrophilic ethanolamine head group linked to a PA moiety. In mammalian cells, four pathways coexist for the generation of PE. The first is the Kennedy pathway, also known as the CDP-ethanolamine pathway, which consists in a sequence of enzymatic reactions starting with the phosphorylation of ethanolamine. Phosphoethanolamine is then converted into CDP-ethanolamine, and the final reaction, which occurs in the ER, leads to the production of PE [55]. The phosphatidylserine decarboxylase (PISD) pathway in the ER also contributes to the production of PE from phosphatidylserine (PS) (Fig. 2a). Interestingly, PISD has been identified in the inner mitochondrial membrane, emphasizing the role of mitochondria in PE synthesis [40]. In addition to these primary pathways for PE synthesis, the acylation of lyso-phosphatidylethanolamine (lyso-PE) by lyso-PE acyltransferase (LPEAT) and the transfer of the acyl chain from PS to ethanolamine by phosphatidylserine synthase-2 (PSS2) also drive the production of PE (Fig. 2a) [55].
The most abundant lipid in the mitochondrial membrane is phosphatidylcholine (PC), which consists of a choline head group and two acyl chains [54]. All the PC present in the mitochondrial membrane are imported from other organelles, as mitochondria cannot produce PC themselves [41, 56]. The first pathway for the synthesis of PC is the CDP-choline pathway, which corresponds to the Kennedy pathway, similar to the synthesis of PE, with the exception that the head group in this case is choline rather than ethanolamine. The final reaction in this pathway occurs in the ER, and the resulting PC is then transported into the mitochondria [57]. The second pathway involves the methylation of PE by phosphatidylethanolamine N-methyltransferase (PEMT), where three consecutive methylation reactions lead to the production of PC [58]. This process is also localized to the ER [54]. These roles of the ER in lipid biosynthesis highlight the dependence of mitochondrial phosphatidylcholine composition on MAM contacts, which consequently influences mitochondrial activity [40, 49]. It has been established that the process of PC synthesis takes place exclusively in the ER. Consequently, PC must be transported to the mitochondria, a process facilitated by StAR-related lipid transfer protein 7 (STARD7), a lipid transfer protein located in the cytosol and the OMM (Fig. 2a) [48, 56, 59, 60].
Less abundant but equally important, phosphatidylserine (PS) constitutes about 5% of all phospholipids in the mitochondrial membranes (Fig. 2b) [61]. The biosynthesis of PS, which contains a serine head group, begins with either PE or PC. The head group is then exchanged for serine through the action of PS synthase 1 (PSS1) for PC and PSS2 for PE (Fig. 2a). These enzymes are highly enriched in the MAM and PS is subsequently transported to the OMM [55]. Transport of PS from the ER to the OMM is mediated by a lipid transfer protein called mitoguardin-2 (MIGA2), while transport from the OMM to the IMM is facilitated by the PRELID3b/TRIAP1 complex (Fig. 2a) [47, 48, 62].
These large classes of phospholipids, defined by their head groups, are also subdivided into numerous subclasses, further increasing the diversity of lipids and their unique, lipid-specific properties. Indeed, the acyl chains are variable in terms of carbon number, position of unsaturation, and number of double bonds [63]. For example, as mentioned above, the linoleate moiety is almost the only fatty acyl residue found in CL from heart mitochondria, whereas four oleate (C18:1) residues are predominantly prevalent in CL from mouse cerebellar mitochondria [64]. In addition, unsaturation can occur in the form of cis or trans isomers. A fatty acid is considered as a cis isomer when the hydrogen atoms attached to the carbon atoms involved in the double bond are on the same side of the double bond. This configuration results in a curvature of the fatty acid chain that prevents the molecules from packing closely together. Conversely, trans isomers have hydrogen atoms on opposite sides of the double bond, resulting in a straighter structure, similar to saturated fatty acids [65]. All of these differences modulate the shape of lipids, their ability to interact, and their biophysical and biochemical properties, adding even more complexity to lipid biology [63, 66]. And indeed, although technological advances in metabolomics and lipidomics have significantly improved our understanding of lipid diversity, they have also highlighted the remarkable complexity of lipid metabolism [67]. In addition to phospholipids, sphingolipids are also present in mitochondrial membranes [68]. Sphingolipids are composed of an acyl chain, a sphingoid base backbone, and a head group, which can include phosphate, glucose, galactose, or choline [63]. Ceramides, a type of sphingolipid with a hydroxyl head group, are present in mitochondrial membranes and have been shown to play a major role in membrane fluidity.
These lipids are crucial not only for membrane structure, but also to modulate their fluidity and ensure their cohesion, impermeability and mechanical protection [66]. The term membrane fluidity is used to describe the viscosity of the lipid bilayer in a cell membrane, affecting its ability to undergo movements, bending, and shape changes. It characterizes the capacity of lipid molecules, in conjunction with proteins and other membrane-embedded components, to move laterally within the bilayer with minimal resistance [69]. This fluidity is critical for various cellular processes, in particular the movement and clustering of molecules across the membrane, such as receptors, and the adaptation to temperature changes [70]. Several factors influence membrane fluidity, including the composition of fatty acids, the amount of cholesterol and the temperature [71, 72]. Ether phospholipids are lipids with ether bonds instead of ester bonds, connecting alkyl/alkenyl chains with the glycerol backbone and also supporting membrane fluidity [63, 73]. Moreover, plasmalogens are a unique class of phospholipids characterized by a vinyl ether bond at the sn-1 position of the glycerol backbone. They contribute significantly to the structural and functional integrity of lipid rafts, as they promote the clustering of cholesterol and other lipids [74, 75, 76]. Maintaining optimal membrane fluidity is mandatory to ensure proper cellular function. While assessing membrane fluidity at the mitochondrial level is challenging, it can be studied indirectly by measuring the diffusion of fluorescent Green Fluorescent Protein (GFP) or HaloTag-tagged proteins using photoactivation or tracking techniques (Fig. 1e and Motion 1). A significant amount of active research is focused on this area [77, 78, 79, 80]. Numerous fluorescent probes have been developed to study the highly dynamic nature of mitochondrial membranes [81]. Time-lapse microscopy has revealed the remarkable mobility of these structures, which are able to cross cell surface and migrate along cell extensions via a tubular conformation [82, 83]. Mitochondria can undergo shape changes by creating branches or thinner tubes, folding, and even forming donut-shaped structures [84, 85, 86]. This constant remodeling is closely linked to fission and fusion events, with lipids playing an active role in these processes, as further discussed below.
Many lipid metabolism pathways take place at least partially in the mitochondria including fatty acid oxidation, oxidative phosphorylation or mitochondrial hormone biosynthetic pathways. As excellent review articles exhaustively address these subjects, they will not be covered in depth in this review [87, 88, 89, 90]. However, our discussion of mitochondrial membrane dynamics must indirectly consider these biochemical pathways, since mitochondrial import of lipids across membranes cannot take place without interaction with the surrounding membrane lipids. Furthermore, the presence of specific lipid microenvironments can facilitate or hinder the efficient import of these lipids, underscoring the importance of lipid dynamics in mitochondrial metabolism and overall cellular energy homeostasis.
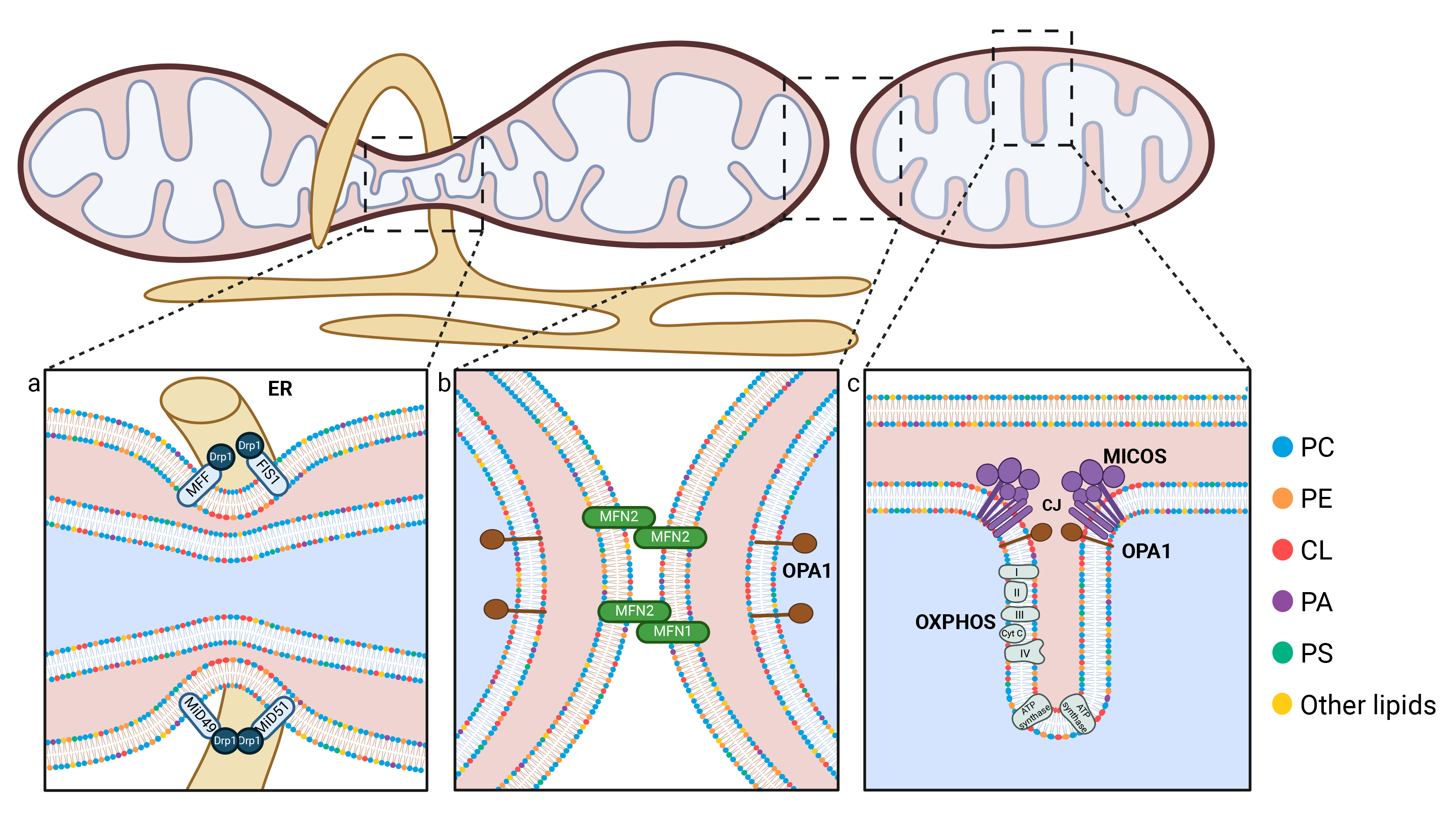
Fission begins when the ER envelops the mitochondrion, facilitating its constriction [91]. This interaction serves as a signal for the recruitment of Dynamin-related protein 1 (Drp1), a cytosolic protein essential for mitochondrial fission. Drp1 recruitment is facilitated by several other protein factors, including Mitochondrial Fission 1 (FIS1), Mitochondrial dynamics protein of 49 kDa (MiD49), Mitochondrial dynamics protein of 51 kDa (MiD51), and Mitochondrial Fission Factor (MFF) (Fig. 3a) [92, 93, 94, 95]. Once recruited to the OMM, Drp1 forms helical rings around the mitochondrion and further tightens the constriction [96, 97]. Additionally, actin is believed to play a role in this fission process [98, 99]. As constriction progresses, the mitochondrion eventually splits into two distinct mitochondria. Increased mitochondrial mass (mitochondrial biogenesis) is accompanied by enhanced mitochondrial fission, which separates mitochondria and facilitates their migration. In response to mitochondrial stress or as part of the renewal process, fission is triggered to eliminate damaged mitochondria. This occurs at the periphery of the mitochondrial network, and specifically targets mitochondria that are enriched in ROS for degradation by mitophagy [24, 100, 101]. The availability of energetic substrates plays a role in the regulation of fission key players, such as the phosphorylation of MFF at specific residues [102, 103]. Consequently, mitochondrial fission is a tightly regulated process that adapts to energy consumption and production within the cell, as well as a response to apoptotic stress [32].
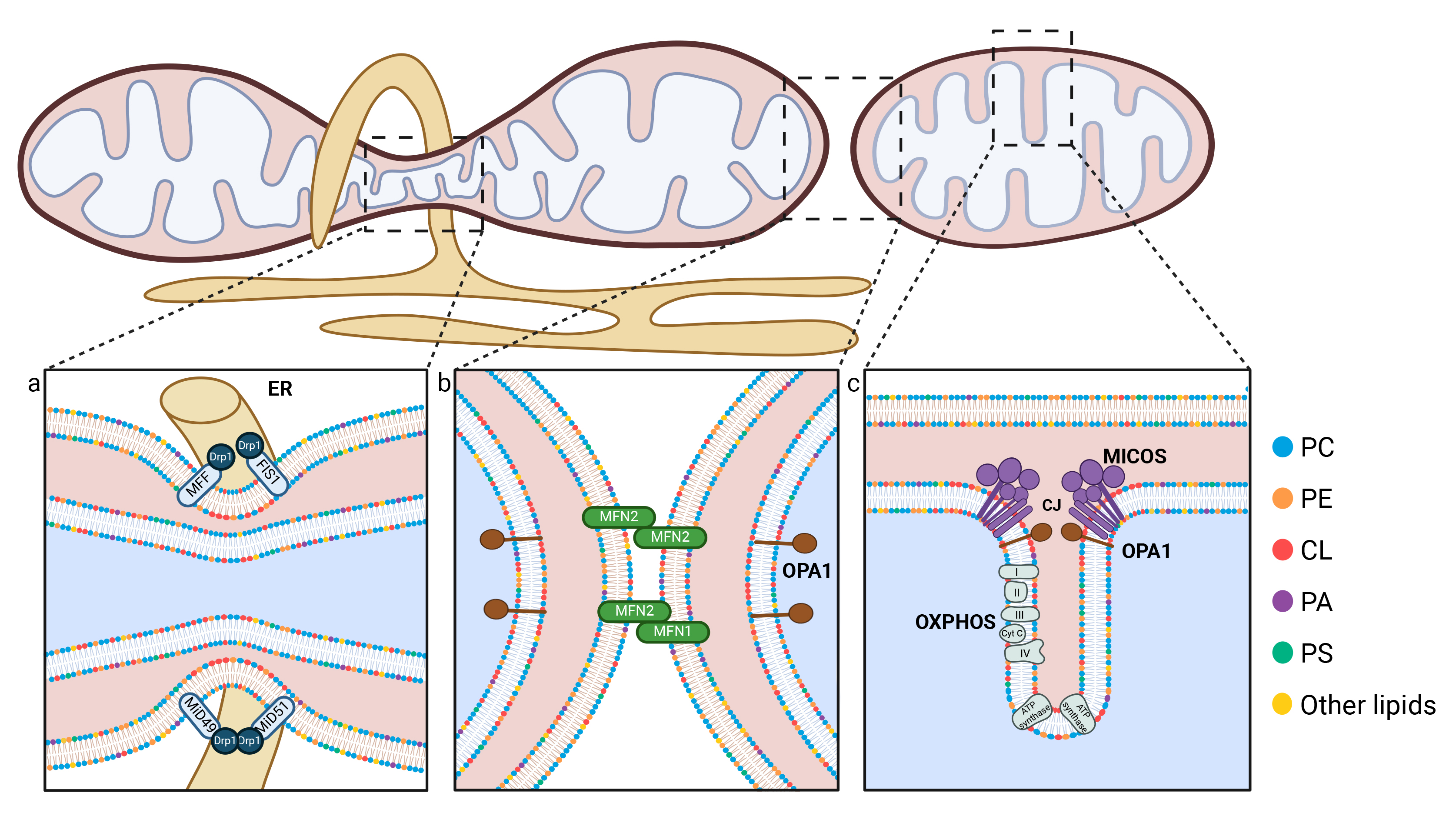 Fig. 3.
Fig. 3.
Schematic representation of the key mechanisms involved in mitochondrial dynamics. (a) During fission events, the Drp1 protein is recruited to the OMM as a result of the presence of Drp1 receptors, including MiD49, MiD51, FIS1 and MFF. The endoplasmic reticulum (ER, shown in yellow) participates in mitochondrial constriction by partially encircling the mitochondria. (b) Mitochondrial fusion occurs in two distinct stages: the fusion of the outer mitochondrial membrane, mediated by MFN1 and MFN2, and the fusion of the inner mitochondrial membrane, regulated by OPA1. Phospholipids play a crucial role in both fission and fusion processes, and interact with mitofusins through their HR1 domain, and with Drp1. Furthermore, they facilitate the generation of the negative membrane curvature that is necessary for these processes to occur. (c) The IMM forms invaginations known as cristae, whose morphology is regulated by the MICOS complex and OPA1. The oxidative phosphorylation (OXPHOS) chain and ATP synthase are located within the cristae. Phospholipids, particularly cardiolipin (shown with red polar heads) and phosphatidylethanolamine (PE, shown in orange), are essential for maintaining the cristae structure. Created with BioRender.com. CJ, Cristae junction; CL, Cardiolipin; Cyt c, Cytochrome c; Drp1, dynamin-related protein 1; ER, Endoplasmic reticulum; FIS1, Mitochondrial fission 1 protein; MFF, Mitochondrial fission factor; MICOS, Mitochondrial contact site and Cristae Organizing System; MiD49, Mitochondrial dynamics protein of 49 kDa; MiD51, Mitochondrial dynamics protein of 51 kDa; MFN1, Mitofusin 1; MFN2, Mitofusin 2; OPA1, Optic atrophy protein 1; OXPHOS, Oxidative phosphorylation; PA, Phosphatidic acid; PC, Phosphatidylcholine; PE, Phosphatidylethanolamine; PS, Phosphatidylserine.
The action of actin polymers and the ER on the OMM, as well as the constriction
of the IMM compartments, which involves an influx of Ca2+ into the
mitochondria and a subsequent influx of K+, have been shown to contribute to
pre-constriction and fission process [104]. This implies that mitochondrial
fission requires the pre-constriction of mitochondrial tubules, as their diameter
frequently exceeds the capacity of Drp1 polymers to coil them. Certain
lipid-mediated pathways have been shown to regulate the expression and activity
of Drp1. Ceramides, a class of sphingolipids found in mitochondrial membranes,
play a role in mitochondrial fission [105]. For instance, treatment with
N-acetylsphingosine, a cell-permeable ceramide analog, has been shown to increase
mitochondrial fission in cardiomyocytes by increasing Drp1 and FIS1 expression
levels [105]. This finding prompts further investigation into the regulatory
mechanisms involving lipids in the control of Drp1 content and distribution.
Ceramides are also involved in the activation of mitophagy. For instance,
C18-pyridinium ceramide treatment or the endogenous generation of C18-ceramide by
ceramide synthase 1 (CerS1) can trigger mitophagy [106]. CerS1 has been
demonstrated to promote the lipidation of microtubule-associated protein 1 light
chain 3
The fission process also includes the reorganization of numerous lipid components. These lipids influence the shape and flexibility of the mitochondrial membranes, which is essential for membrane dynamics and mitochondrial fission. Phospholipids, the primary lipids in the mitochondrial membranes, play a crucial role in this process.
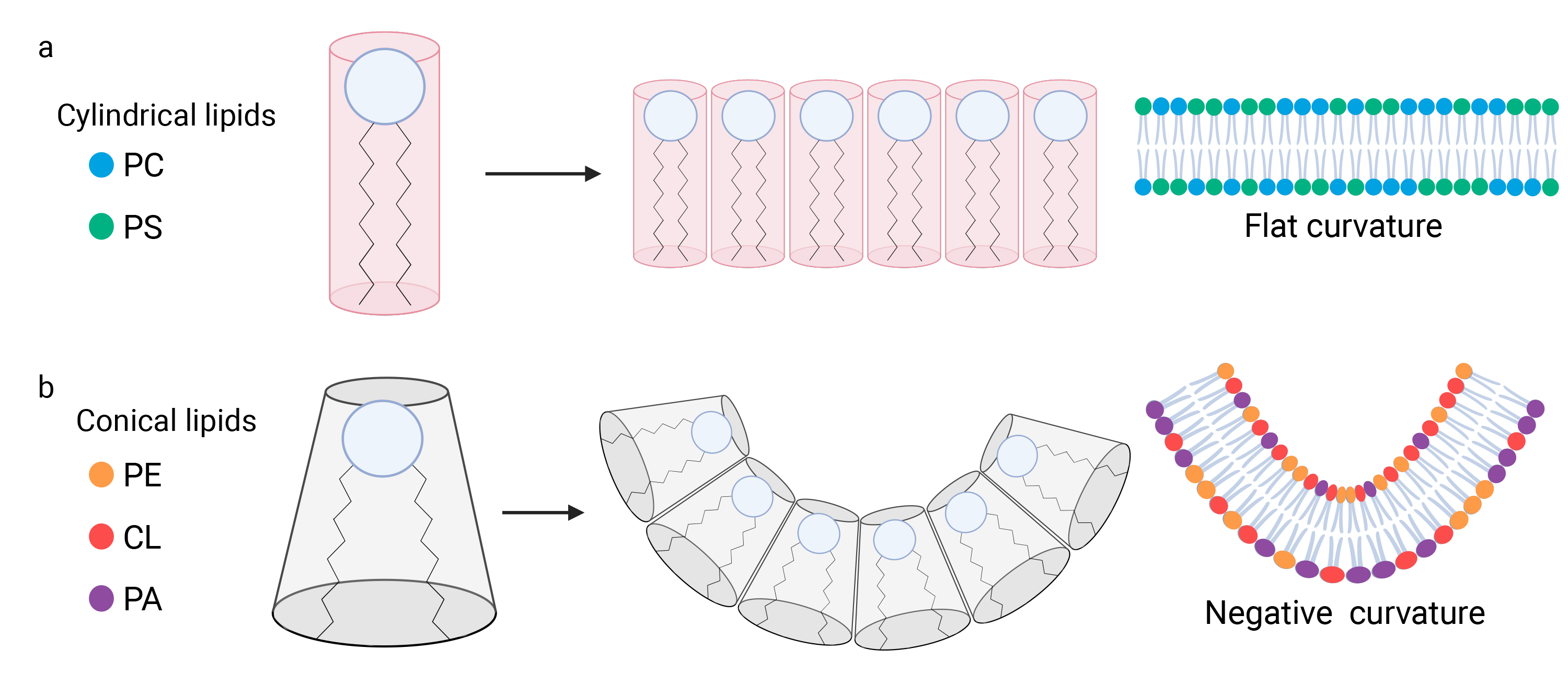
For instance, it has been shown that a reduction in PC levels, driven by chemical inhibition of its synthesis, leads to fragmentation of the mitochondrial network. This indicates that PC, a PL of cylindrical shape, acts as a negative regulator of mitochondrial fission (Fig. 4a) [107].
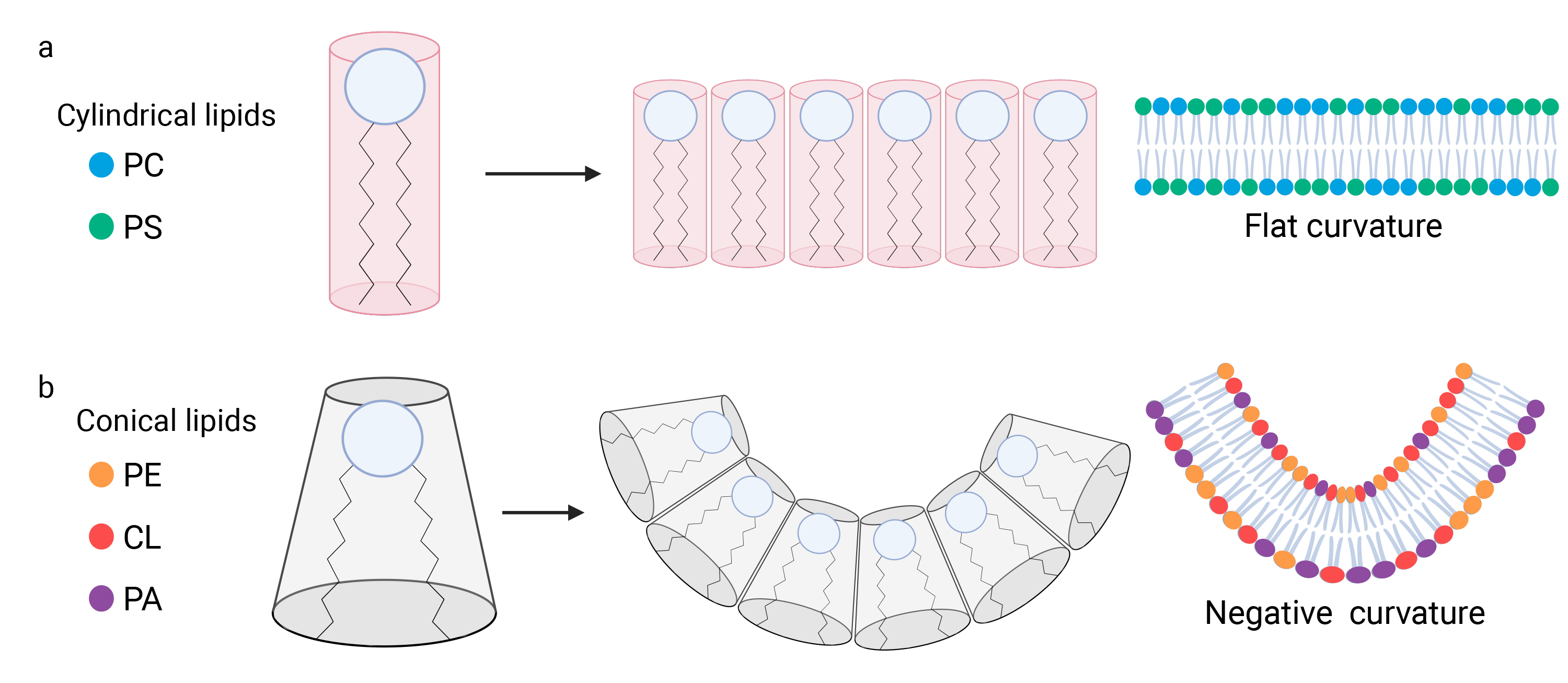 Fig. 4.
Fig. 4.
Phospholipids influence the curvature of mitochondrial membranes. (a) Phospholipids (PL) are composed of a polar head and hydrophobic acyl chains. The hydrophilic nature of the polar heads facilitates interactions among them. The structural diversity of phospholipids stems from their composition. Specific lipids such as phosphatidylcholine (PC) and phosphatidylserine (PS), display a cylindrical configuration, with the acyl chains exhibiting a width that is approximately equivalent to that of the polar head. Consequently, when the polar heads of cylindrical lipids interact, they form flat membranes devoid of any curvature. (b) Other lipids such as cardiolipin (CL), phosphatidic acid (PA), and phosphatidylethanolamine (PE), exhibit a conical shape, wherein width of the polar head is narrower than the width between the acyl chains. Consequently, when the polar heads of these lipids come together and interact, the membrane displays a negative curvature. Created with BioRender.com. CL, Cardiolipin; PA, Phosphatidic acid; PC, Phosphatidylcholine; PE, Phosphatidylethanolamine; PS, Phosphatidylserine.
On the other hand, non-bilayer PL of conical shape, such as CL and PE, may promote mitochondrial fission by inducing membrane curvature when they cluster together, rather than maintaining a flat structure (Fig. 4b) [108, 109]. This curvature is critical for mitochondrial fission, as it contributes to the constriction required to split the mitochondria. In summary, CL and PE generate a negative curvature, which is essential for the OMM at the fission site [110]. This curvature is equally important in the IMM region adjacent to the fission site, facilitating the separation of the two mitochondrial compartments [108]. The role of cardiolipin in this process extends beyond its contribution to negative curvature and mitochondrial fission, as it also interacts with Drp1. CL has been shown to cooperate with Drp1 by stimulating its self-assembly into helical polymers and by enhancing its GTPase activity, which is crucial for membrane constriction [111].
A recent study revealed that a protein involved in cardiolipin synthesis also influences fission regulation. Gene screening linked to mitochondrial function demonstrated that inhibiting the PGS1 gene causes PA buildup in the IMM and results in a hyperfused mitochondrial network. Inhibition of both PGS1 and PRELID1 disrupts PA transport from the ER to the IMM, blocking CL production and preventing PA accumulation in the IMM, which leads to mitochondrial fragmentation. Interestingly, the study concluded that PA accumulation inhibits Drp1-dependent mitochondrial fragmentation [112]. Additionally, another study showed that PA interacts with Drp1 and inhibits its activity [113].
Mitochondrial fission is an essential process for two key cellular events: mitophagy and apoptosis. Since these two events are interconnected, altering mitophagy can lead to the accumulation of nonfunctional mitochondria, causing cellular damage that may ultimately trigger apoptosis. Conversely, certain apoptotic pathways can compromise mitochondrial integrity, thereby promoting mitochondrial fission. Lipid metabolism plays a crucial role in these processes by facilitating membrane remodeling. On the other hand, lipid abnormalities can trigger premature apoptosis or inhibit mitophagy, which is essential for the clearance of damaged mitochondria [114, 115]. A key step in apoptosis is the permeabilization of the mitochondrial outer membrane (MOMP). Pro-apoptotic proteins such as Bcl-2 associated X protein (Bax) and Bcl-2 homologous antagonist/killer (Bak), can form pores in the OMM. Bax typically resides in the cytoplasm. However, when exposed to apoptotic stimuli, it is recruited to the OMM through interactions involving its transmembrane domain. The recruitment and activation of Bax and Bak are known to be mediated by specific lipids, including cardiolipins and sphingolipids, specifically sphingosine-1-phosphate. These lipids facilitate Bax translocation to the OMM and support pore formation, thereby promoting the apoptotic process [116, 117]. A recent study shows that pore formation is preceded by a significant enrichment of PC and PE with unsaturated fatty acyl chains in the vicinity of Bak and Bax in apoptotic conditions [118]. The oligomerization of these proteins leads to the formation of proteolipid pores in the OMM, thus creating discontinuities in the lipid bilayer. This allows the release of cytochrome c into the cytosol, triggering the caspase cascade that leads to apoptosis [119, 120, 121, 122, 123]. Consequently, the IMM is exposed to the cytosol due to this rupture of the OMM. Moreover, this permeabilization promotes the release of other pro-apoptotic factor as well as mitochondrial DNA into the cytosol [124, 125, 126, 127]. In summary, the apoptosis process alters mitochondrial membrane fluidity with the insertion of Bax and the formation of proteolipid pore. Apoptosis also impacts mitochondrial dynamics in other ways. Indeed, Drp1 may be recruited to lipid raft domains on the mitochondrial membrane. Supporting this, a study demonstrated that Drp1 and FIS1 are localized at lipid rafts following treatment with CD95/Fas, which induced apoptosis. Moreover, lipid raft dispersion, through the use of a ceramide synthase inhibitor or a glucosyltransferase inhibitor, was found to reduce mitochondrial fission and decreased Drp1 recruitment [128]. When cells are exposed to an apoptotic environment, mitochondria undergo fragmentation and remodeling of the cristae [129, 130, 131]. Karbowski et al. [132] originally demonstrated the colocalization of MFN2 and Drp1 with Bax at the OMM in discrete foci during the initial stages of apoptosis. Recent results suggest that Drp1 can only promote Bax pore activity when already bound to membranes, consistent with Drp-1-Bax interaction in an exclusively lipidic environment [133, 134, 135, 136].
Fusion is a process that involves the merging of two mitochondria into a single mitochondrion. Mitochondria move along the cytoskeleton primarily through motor proteins such as dynein and kinesin, which travel along microtubules [137]. Other cytoskeletal elements, including actin microfilaments and some intermediate filaments, facilitate the contact of adjacent mitochondria, enabling fusion or self-fusion to form donut-shaped structures and other rearrangements within the mitochondrial network. Fusion is a crucial process for mitochondrial functionality, as it facilitates the mixing of mitochondrial matrix contents, including metabolites, ions, lipids, and mitochondrial DNA, which is organized into nucleoprotein complexes called nucleoids. This mixing ensures an even distribution of critical mitochondrial components, such as proteins and lipids, throughout the network, promoting metabolic flexibility and cellular adaptability. In particular it allows for mtDNA complementation, i.e., wild-type mtDNA molecules from one mitochondrion can compensate for mutated mtDNA molecules in another mitochondrion [138].
As demonstrated by time-lapse imaging of mitochondrial fusion and the tracking of fluorescent signal propagation, the distribution and homogenization of lipid contents occur rapidly, taking only seconds to minutes to reach the entire mitochondrial network. This process consists of two sequential steps, OMM fusion and IMM fusion, which involve different players. OMM fusion is mediated by mitofusins, which are GTPase proteins localized in this membrane (Fig. 3b). There are two types of mitofusins, MFN1 and MFN2, which share 80% of identity and have redundant roles [139]. Inhibition of both MFN1 and MFN2 results in fragmentation of the mitochondrial network due to impaired fusion activity [140]. Although MFN1 and MFN2 share a similar domain structure, MFN2 and some of its shorter isoforms are also localized to the ER membrane, promoting the formation of MAMs, which is crucial for mitochondrial lipid synthesis and import [141]. IMM fusion is mediated by another GTPase protein, OPA1 (Fig. 3b). This protein exists in different forms; specifically, cleavage of OPA1 by the proteases Yme1l, or OMA1 under stress conditions, is necessary for its activity [142]. Eight OPA1 isoforms coexist, which can be classified into two categories: short isoforms (s-OPA1) and long isoforms (l-OPA1). The short isoforms are soluble and localized in the intermembrane space, while the long isoform is a transmembrane protein localized in the IMM [142, 143]. The balance between the rates of s-OPA1 and l-OPA1 is crucial for regulating mitochondrial morphology [142]. A decrease in the levels of long forms leads to the inhibition of fusion thereby promoting fission and leading to the fragmentation of the mitochondrial network. The study conducted by Ciarlo and colleagues suggested that proteins facilitating the fusion of mitochondrial membranes could be recruited to plasmalogen-rich lipid rafts, and that this recruitment is a prerequisite for the fusion of two mitochondria. Indeed, in their study, Drp1 inhibition with mitochondrial division inhibitor 1 (Mdivi-1) led to the co-localization of MFN2 with ganglioside GD3, a protein localized in lipid rafts. Furthermore, they demonstrated that OPA1 is also co-localized with ganglioside GD3 and concluded that the localization of OPA1 and MFN2 in lipid rafts may be a crucial step in the fusion of mitochondria [144]. Phospholipids with non-bilayer shapes play a crucial role in both mitochondrial fusion and fission. It has been shown that mitochondria become fragmented when both CL and PE are absent. However, the presence of at least one of these lipids allows the mitochondria to remain connected, suggesting redundancy in their roles [145]. Given the conical structure of CL and PE, it is hypothesized that their ability to induce negative membrane curvature is crucial to facilitate mitochondrial fusion (Fig. 4b).
Recent studies have demonstrated that various phospholipids interact with the heptad repeat domains (HR1) of mitofusins, regulating mitochondrial fusion [146]. Phosphatidylethanolamine promotes fusion by inducing strong local curvature, an effect mediated by the HR1 domain. Furthermore, in the presence of calcium ions, PA enhances HR1 domain-mediated fusion. Calcium facilitates the formation of PA-rich domains by attracting the phosphatidic acid head groups. This interaction exposes the hydrophobic chains of PA, enabling the HR1 helix to interact with the membrane by electrostatic attraction. Conversely, in the absence of cations, PA and CL do not activate HR1-mediated fusion. Indeed, in this context, HR1 is unable to bind to hydrophobic regions of the membrane due to the negative charges of PA and CL, resulting in electrostatic repulsion [146].
Mitochondrial cardiolipin hydrolase, known as MitoPLD, is a phospholipase that catalyzes the hydrolysis of CL to produce two PA molecules. MitoPLD has been shown to influence mitochondrial fusion. Specifically, depletion of MitoPLD reduces the rate of mitochondrial fusion, while its overexpression has opposite effects [147]. This suggests that the conversion of CL to PA by MitoPLD plays a key role in regulating the mitochondrial fusion dynamics. These findings also highlight an important role for PE and PA in mitochondrial fusion. While cardiolipin appears to play a modest role in the OMM fusion influenced by MitoPLD, CL is still crucial for IMM fusion. In fact, it has been shown that high concentrations of CL are necessary for OPA1-mediated IMM fusion in vitro, particularly in membrane fusion assays [148]. This suggests that CL is more critical for the fusion of the IMM, while PE and PA may be more involved in OMM fusion events. This finding is consistent with the distribution pattern of phospholipids in the OMM and IMM.
Another protein involved in modulating mitochondrial fusion is mitochondrial transporter homolog 2 (MTCH2), which is localized in the OMM. It has been predicted that MTCH2 functions as a scramblase, i.e., it is responsible for translocating phospholipids between membrane layers without requiring metabolic energy [149, 150, 151]. This activity could play a role in facilitating membrane curvature and lipid distribution, both of which are crucial for the fusion process. MTCH2 also appears to play a role in the regulation of mitochondrial fusion. Interestingly, inhibition of MFN2 or MTCH2 alone does not significantly affect mitochondrial network morphology. However, simultaneous inhibition of both proteins causes mitochondrial fragmentation [152]. This suggests that MTCH2 may function in a pathway that complements MFN2 activity, and that the combined actions of these proteins contribute to the maintenance of mitochondrial fusion and network integrity. Goldman and colleagues have suggested that mitochondrial fusion may be regulated by two distinct pathways. One pathway depends on MTCH2 and MFN1, while the other relies on MFN2. This dichotomy underscores the potential for distinct regulatory mechanisms to control mitochondrial fusion, to ensure a fine modulation of mitochondrial dynamics according to the cellular context or specific metabolic requirements [152].
The MFN2-dependent pathway appears to require lysophosphatidic acid (LPA), which is produced either through the addition of an acyl-CoA to glycerol-3-phosphate or via the modification of phosphatidic acid by phospholipase A1, to maintain mitochondrial fusion [153]. Inhibition of both MTCH2 expression and LPA synthesis results in the loss of the ability of mitochondria to fuse, suggesting a crucial role for LPA in this process. MTCH2 is thought to modulate LPA levels, as proposed by Labbé and colleagues [154]. Collectively, their findings suggest that mitochondrial fusion is regulated by the presence of LPA and the translocation of phospholipids facilitated by MTCH2.
A study published in 2016 showed that the solute carrier family 25 member 46 (SLC25A46), a mitochondrial metabolite carrier protein interacting with MFN2, MFN1, OPA1, and MICOS, facilitates lipid transfer between the ER and mitochondria [155]. Loss of function of SLC25A46 resulted in a hyperfused mitochondrial network, abnormal cristae, and altered mitochondrial membrane lipid composition. More recently, the same research group confirmed the altered mitochondrial lipid composition but observed a fragmented network when SLC25A46 was knocked out [156]. Similarly, two other studies reported mitochondrial fragmentation in the absence of SLC25A46, suggesting its regulatory role in MFN1 and MFN2 oligomerization [156, 157, 158]. More research is needed to uncover how SLC25A46 facilitates lipid transfer, but similar to MTCH2, this further highlights the role of proteins interacting with MFN2/MFN1 in shaping the lipid environment.
Recent studies have also revealed that the absence of OPA1 or mitofusins leads to alterations in the phospholipid profile of mitochondria [159]. These changes are likely due to altered phospholipid transport, suggesting that mitofusins and/or OPA1 may play a role in the regulation of this process. This suggests a link between mitochondrial dynamics, mediated by fusion proteins, and the regulation of mitochondrial membrane composition, which could have a more general impact on mitochondrial function.
The IMM can be subdivided into two distinct regions: the peripheral inner membrane, which faces the OMM, and the cristae, which are invaginations of the IMM extending into the matrix. These cristae pockets are connected by nanotubes to the peripheral inner membrane forming cristae junctions (Fig. 3c). Each cristae junction serves as a gateway to one or more cristae pockets. With an internal diameter of around 10 to 15 nm, these junctions are indeed similar to collets with adjustable diameters, and they are regulated by the Mitochondrial Contact Site and Cristae Organizing System (MICOS) complex (Fig. 3c) [160, 161]. The cristae are thought to host the majority of OXPHOS complexes. Their pocket-like structure provides an optimal environment for the accumulation of protons (H⁺), facilitating the creation of a gradient. This proton gradient is essential for ATP synthase to produce ATP, thus stimulating cellular energy production. Transmission electron microscopy (TEM) and electron tomography data revealed a great diversity in cristae shape and size, depending on mitochondrial diameters and in which tissues they are observed [162]. Recently, super-resolution Stimulated emission depletion (STED) microscopy has provided remarkable insights into the dynamic nature of cristae within mitochondria, revealing their constant remodeling and movements [83, 163]. These structures appear to be far more dynamic than previously thought, as they continuously adjust their structure in response to specific metabolic needs. The regulation of cristae shape is not restricted to MICOS complexes or the OPA1 fusion protein; ATP synthase dimerization also plays a significant role, contributing to the curvature of the IMM (Fig. 3c) [164]. The MICOS complex, localized at cristae junctions, consists of at least seven subunits. In the absence of key proteins regulating cristae structure, cristae lose their tubular shape, become disorganized and eventually shorten [161]. This disorganization impairs the optimal function of OXPHOS leading to inefficient ATP production and reduced cellular energy output [165, 166, 167]. The IMM is densely packed with membrane-bound and transmembrane proteins. Specific structural motifs within these proteins create mechanical constraints, either within the proteins themselves or between protein homodimers, imposing curvature of the membrane. Additionally, the high concentration of these membrane proteins amplifies their impact on its structure, further contributing to its dynamic shape and function [168, 169].
Lipids, particularly PL, play a pivotal role in regulating both the shape and function of mitochondrial cristae. As previously mentioned, conical lipids, such as CL and PE, due to their non-bilayer properties, contribute to membrane curvature (Fig. 4b) [110]. This curvature is critical for the formation and maintenance of cristae architecture, which is essential for optimizing mitochondrial function. CL not only helps to shape the IMM but also stabilizes the protein complexes involved in OXPHOS [170].
A specific mixture of lipids, known as the IMM lipidome, is essential for shaping cristae. This lipidome predominantly consists of cardiolipins, which are crucial for establishing the curvature of the cristae. Additionally, other phospholipids are involved, as demonstrated by Kojima and colleagues, which showed that the import of specific phospholipids is necessary for the formation of tubular cristae [171]. Another important factor in mitochondrial cristae formation is not only the presence of specific phospholipids, but also their degree of unsaturation. Saturated phospholipids inhibit ATP synthase oligomerization, resulting in flattening of the IMM. On the other hand, unsaturated phospholipids promote the curvature necessary for cristae formation [172]. The level of phospholipid saturation is modulated by the oxygenation status of the cell. In low-oxygen environments, phospholipids tend to be more saturated, which can disrupt cristae structure and impair mitochondrial function. Cristae constantly changes shape in relation to CL structure. Local pH differences between the mitochondrial matrix and the intermembrane space promotes CL synthesis, thus resulting in the negative curvature necessary for optimal cristae architecture [172]. However, this dependence on CL remodeling is only observed in lipid environments that are rich in saturated fats. When the lipid environment is unsaturated, CL remodeling becomes less critical for IMM structure and function [173]. The saturation level of the CL is modulated by the surrounding fatty acid environment. Depending on the fatty acids present, the chain length and degree of unsaturation of the CL can vary. For instance, in the presence of fourteen-carbon saturated myristic acid, CL chains become shorter and more saturated, whereas polyunsaturated eighteen-carbon linoleic acid leads to longer, less saturated CL chains [64]. CL remodeling is also regulated by OXPHOS [174]. The study by Xu and colleagues demonstrated that deletion of specific subunits of the OXPHOS system can modify CL composition. Furthermore, they showed that CL remodeling is influenced by the assembly state of OXPHOS complexes, indicating the importance of the structural organization of these proteins in maintaining a proper CL profile. This remodeling is essential not only for CL stability, but also for the overall structural integrity of cristae, which are essential for proper mitochondrial function [174].
Diseases involving mitochondrial dynamics proteins represent a distinct category of pathologies linked to defects in mitochondrial structure [175]. These diseases are associated with mutations in genes of the MICOS complex, which impair mitochondrial organization, as well as mutations in genes regulating fusion, such as OPA1 in dominant optic atrophy, and fission, such as Drp1 (DNM1l gene) in neurological disorders. These mitochondrial defects result in muscle deficits, neuronal damage and, in particular, optic nerve damage, often leading to partial or total loss of vision [176]. A metabolomics study using mouse fibroblasts with human OPA1 missense variants suggested a link between lipid composition and disease severity. Indeed, as neurological impairments severity increased, the proportion of PC with unsaturated fatty acyl chains decreased [177].
Conversely, changes in the mitochondrial lipidome, such as altered lipid membrane composition or disrupted lipid transport, can worsen and accelerate the progression of mitochondrial diseases. Given the critical role of phospholipids in mitochondrial structure, alterations in the mitochondrial lipidome are linked to rare genetic diseases. Additionally, mutations in genes coding for lipid metabolism can cause significant mitochondrial dysfunction and lead to serious mitochondrial disorders [52, 177].
Table 1 (Ref. [50, 58, 59, 60, 62, 112, 155, 156, 178, 179, 180, 181, 182, 183, 184, 185, 186, 187, 188, 189, 190, 191, 192, 193, 194, 195]) presents the current knowledge on mitochondrial proteins involved in phospholipid synthesis and import, as well as their associated human pathologies. For instance, mutations in the enzyme acylglycerol kinase (AGK) disrupt both phospholipid metabolism and mitochondrial protein biogenesis, thereby contributing to the pathogenesis of Sengers syndrome [178]. The disease is characterized by congenital cataracts, hypertrophic cardiomyopathy, muscle weakness and lactic acidosis, reflecting a general disturbance in mitochondrial energy production. AGK, a multi-substrate kinase, catalyzes the conversion of mono- and diacylglycerol into PA and lyso-PA. The catalytic activity of AGK is essential for preserving mitochondrial structure (Table 1) [179].
| Lipid | Gene/pathways | Role | Consequence on mitochondrial dynamics | Human disease OMIM reference | Clinical feature | Ref |
| Phosphatidic acid | AGK | Converts DAG into PA | AGK is required to preserve the tubulated morphology and ultrastructure of mitochondria | Sengers syndrome #212350 | Cataracts, hypertrophic cardiomyopathy, skeletal myopathy | [178, 179] |
| Cardiolipin | TAMM41 | CL Synthesis pathway | Not yet described | Neonatal mitochondrial disease #620139 | Myopathy, chronic progressive external ophthalmoplegia | [180] |
| PGS1 | KO PGS1: Hyperfusion | Not yet described | [112] | |||
| PTPMT1 | KO PTPMT1: fragmentation, rupture of the OMM, abnormal cristae | New autosomal recessive primary mitochondrial disease (PMD) | Neurologic disorder: developmental delay, microcephaly, epilepsy, spasticity | [50, 181] | ||
| CRLS1 | Fragmentation of mitochondria | Autosomal recessive mitochondrial disease #620167 | Severe encephalopathy with multi-systemic involvement myopathy | [182, 183] | ||
| IPLA2 (gene PNPLA8) | Abnormal cristae structure and subsarcolemmal aggregated of mitochondria | Mitochondrial myopathy #251950 | Central hypotonia, dystonia, seizure, cerebellar atrophy | [184, 185, 186] | ||
| TAZ | Knockdown of tafazzin: Cristae morphology disruption | Barth syndrome #302060 | Cardiomyopathies | [187, 188] | ||
| Phosphatidylethanolamine | PISD | Synthesis of PE from PS | Fragmentation of the mitochondrial network | Liberfarb syndrome (LIBF) #618889 | An autosomal recessive multisystem disorder affecting the eye, ear, bone, brain development | [189, 190] |
| Phosphatidylcholine | PEMT | Synthesis of PC from PE | Mice PEMT-/-: Smaller mitochondria and elongated | Pathogenic link with obesity, liver and cardiovascular diseases | [58, 191] | |
| STARD7 | Transport of PC from ER to mitochondria | KO STARD7: Fragmentation | Myoclonic epilepsy-2 FAME2 #607876 | Neurologic disorder | [59, 60, 192] | |
| Phosphatidylserine | PSS1 (gene PTDSS1) | Synthesis of PS from PC | Not yet described | Lenz-Majewski hyperostotic dwarfism (LMHD) #151050 | Intellectual disability, sclerosing bone dysplasia | [193] |
| MIGA2 | Transport of PS from ER to mitochondria | KO MIGA2: Mitochondrial fragmentation | Not yet described | [62, 194, 195] | ||
| Other lipids | SLC25A46 | Fatty acids transport across the IMM | Pathogenic variants leas to hyperfused mitochondrial network | Neurodegenerative disease #616505 | Early-onset optic atrophy | [155, 156] |
AGK, Acylglycerol kinase; CL, Cardiolipin; CRLS1, Cardiolipin synthase; DAG, diacylglycerol; ER, Endoplasmic reticulum; IMM, Inner mitochondrial membrane; IPLA2, Calcium-independent phospholipase A2-gamma; KO, Knock-out; LIBF, Liberfarb syndrome; LMHD, Lenz-Majewski hyperostotic dwarfism; MIGA2, Mitoguardin-2; PA, Phosphatidic acid; PC, Phosphatidylcholine; PE, Phosphatidylethanolamine; PEMT, Phosphatidylethanolamine N-methyltransferase; PGS1, phosphatidylglycerophosphate synthase 1; PMD, Primary mitochondrial disease; PISD, Phosphatidylserine decarboxylase; PS, Phosphatidylserine; PSS1, Phosphatidylserine synthase-1; PTPMT1, Phosphatidylglycerophosphatase and protein-tyrosine phosphatase 1; SLC25A46, Solute carrier family 25 member 46; STARD7, StAR-related lipid transfer protein 7; TAMM41, Phosphatidate cytidylyltransferase; TAZ, Tafazzin.
In this classification, the most studied gene, the best established diagnosis with recent data and the most advanced therapy concern Barth syndrome. This rare genetic disorder is caused by mutations in the TAZ gene, which encodes the mitochondrial tafazzin enzyme responsible for CL remodeling [196]. Patients diagnosed with Barth syndrome suffer from cardiomyopathy, skeletal muscle weakness, neutropenia and stunted growth, illustrating how an imbalance in mitochondrial lipid can disrupt organ function [187, 188, 197]. More specifically, this phospholipid-lysophospholipid transacylase, which enables the exchange of fatty acids between phospholipids such as PE or PC and lysophospholipids such as MLCL, causes a decrease in CL and plasmalogen levels, and an accumulation of MLCL when mutated [198, 199]. Furthermore, in the tissues of patients suffering from Barth’s syndrome, the CL species are modified, as shown by the form tetralinoleoyl CL (18:2)4, the predominant CL in the cardiac or skeletal muscle, which are no longer detected when the tafazzin is mutated [200, 201]. Such abnormal CL impairs the integrity and function of the IMM, affecting the assembly of the mitochondrial respiratory chain supercomplexes, the OXPHOS efficiency and the nicotinamide adenine dinucleotide (NAD) redox metabolism as well as mitochondrial dynamics (Table 1) [202, 203, 204, 205]. Recently, Kagan et al. [206] have shown a new impact of MLCL accumulation that leads to the formation of a complex with cytochrome c peroxidase and causes an increase in the peroxidation of polyunsaturated fatty acid phospholipids.
Tafazzin is not the only protein linked to CL production and related diseases; proteins involved in CL biosynthesis from PA are also linked to human disorders. For example, a recent study identified a biallelic TAMM41 variant in three unrelated patients with neonatal mitochondrial disease, marked by lethargy, hypotonia, developmental delay, myopathy, and ptosis. This mutation altered OXPHOS complex assembly and reduced CL levels in muscle tissue, though its impact on mitochondrial dynamics remains unclear (Table 1) [180]. To date, no disease has been linked to PGS1; but a study by Cretin et al. (2021) [112] showed that PGS1 inhibition via siRNAs in MEF WT cells caused mitochondrial network hyperfusion. The PTPMT1 example, however, encourages further clinical exploration: in 2011, mutations of PTPMT1 introduced in mice were shown to be lethal during embryonic development [50]. Several years later, biallelic variants of PTPMT1 were identified in six patients from three unrelated families, who exhibited neurological impairments such as developmental delay, microcephaly, facial dysmorphia, and epilepsy. These mutations caused mitochondrial fragmentation in patient-derived fibroblasts and reduced CL levels in skeletal muscle (Table 1) [181].
Similarly, biallelic variants in CRLS1 were identified in four patients from three unrelated families, resulting in an autosomal recessive mitochondrial disease characterized by encephalopathy and multi-system involvement. Mitochondria in patient-derived fibroblasts exhibited fragmentation [182]. An in vivo study also linked CRLS1 to age-related muscle deterioration, revealing downregulation of cardiolipin and CRLS1 in aged skeletal muscle (Table 1) [183].
IPLA2, encoded by the PNPLA8 gene, is responsible for remodeling nascent CL into MLCL. Several biallelic variants of PNPLA8 are associated with a neurodegenerative mitochondrial disease characterized by central hypotonia, dystonia, seizures, and cerebellar atrophy [184, 185, 186]. In patient-derived cells, the mitochondria have been described with abnormal cristae architecture, and mitochondria formed subsarcolemmal aggregates in muscle biopsies (Table 1) [184].
Fragmented mitochondrial morphology was also seen in fibroblasts from patients with various biallelic variants of the PISD gene coding for phosphatidylserine decarboxylase, an enzyme crucial for PE synthesis from PS and mitochondrial fusion (Table 1) [189, 190]. Further systematic exploration of mitochondrial structure and migration in patient cells and preclinical models should unravel new connections between lipid metabolism genes and mitochondrial dynamics.
STARD7 is a protein that facilitates the transfer of PC from the ER to the IMM [56]. Despite the absence of pathogenic variants, modulations in STARD7 expression are observed in several diseases, in particular cancer [207, 208]. Interestingly, it has been shown that inhibiting STARD7 causes mitochondrial network fragmentation, abnormal cristae shape and compromised mitochondrial bioenergetic functions. This fragmentation depends on Drp1 activity (Table 1) [59, 60, 192].
Although PEMT is no currently linked to mitochondrial disease, it is believed to contribute to the development of liver and cardiovascular diseases [58]. In mice, PEMT-/- mitochondria were smaller and more elongated (Table 1) [191]. Mitoguardin-2 (MIGA2), responsible for transferring PS from the ER to the mitochondria, has not been yet associated with pathogenic variants, but its inhibition induces mitochondrial fragmentation, which is rescued only by expressing MIGA2 with lipid transfer activity (Table 1) [194, 195].
In 2014, variants of PTDSS1, the gene coding for PSS1, were identified as the cause of Lenz-Majewski syndrome, characterized by sclerosing bone dysplasia, intellectual disability, and distinct craniofacial anomalies. The impact of PSS1 mutations on mitochondrial dynamics remains to be elucidated (Table 1) [193].
Last, mutations in SLC25A46, which regulates mitofusins oligomerization and lipid environment, are linked to multiple neurological diseases, including Charcot-Marie-Tooth disease, Leigh syndrome, and Parkinson’s disease [155, 209, 210]. Recently, mitochondrial hyperfusion has been documented in cases of SLC25A46 mutations (Table 1) [156].
Altered mitochondrial dynamics, frequently accompanied by changes in lipid composition, are not limited to mitochondrial disorders or genetic conditions related to lipid metabolism. Indeed, several common diseases, including Alzheimer’s disease, cancer and diabetes, also exhibit disturbances in mitochondrial dynamics and lipid homeostasis. In Alzheimer’s disease, for instance, mitochondrial dysfunction and altered lipid metabolism contribute to neuronal degeneration and cognitive decline [211, 212]. Similarly, in cancer, tumor cells often display altered mitochondrial morphology and lipid composition, favoring rapid growth and survival [213]. In diabetes, changes in mitochondrial function and lipid accumulation in tissues such as muscle and liver are linked to insulin resistance and metabolic dysfunction [214, 215]. These findings suggest that mitochondrial dynamics and lipid metabolism are interconnected processes that play a crucial role in the pathogenesis of various diseases, underscoring the potential of therapeutic strategies targeting both pathways.
TAZ gene replacement therapy has been developed in recent years and has been shown to improve cardiac and muscular function when administered to mice with the TAZ gene knock-out [216, 217]. But the use of lipid-based therapeutic strategies has recently emerged as a new approach for the management of certain diseases, including Barth syndrome. These strategies involve lipid replacement therapy, in which a specific lipid is administered. For example, CL nanodisks demonstrated efficacy in 2015 when tested on cells. However, subsequent in vivo studies in 2018 revealed proved less successful, with no elevation of CL levels [218, 219]. Another approach is plasmalogen replacement therapy, which has been shown to raise CL levels in cells derived from Barth syndrome patients [220]. Interestingly, and especially following on from the above review, recent research has also demonstrated the efficacy of ether-glycerophospholipid precursors in restoring mitochondrial morphology in cells with defects in ER biogenesis [73]. Another therapeutic strategy for Barth syndrome is the administration of elamipretide (SS-31), a peptide that binds to CL. It is noteworthy that this treatment facilitates the restoration of cristae architecture. Elamipretide, evaluated in a Phase 2/3 trial for Barth syndrome, demonstrated sustained long-term tolerability, efficacy, and improvements in functional and cardiac assessments [221, 222, 223].
Mitochondrial membranes are highly dynamic structures that depend on extensive lipid exchange and lipid composition. Their relationship with the endoplasmic reticulum is essential for lipid import, synthesis, and maturation and our understanding of these exchanges has progressed significantly in recent years. The control of the mitochondrial lipidome is crucial for maintaining mitochondrial structure and function. The diffusion and asymmetric distribution of lipids across membrane bilayers, as well as the non-cylindrical structures of certain lipids, in particular cardiolipins, play an essential role in inducing negative membrane curvature, which is critical for fission and fusion processes as well as for proper cristae structure. Disruptions in these lipid dynamics can result in severe mitochondrial dysfunction, with considerable effects on cellular energy production and metabolic homeostasis. Rare genetic pathologies that disrupt lipid metabolism or synthesis pathways underline the critical role that lipids play in human health. Thus, a deeper understanding of mitochondrial lipids composition, of their exchanges within organelles, and their biosynthesis pathways will allow us to explore their potential as therapeutic tools or targets.
SP, JCDLB and AC worked on the bibliographic synthesis, data formatting and production of the original images of the Figures. SP and AC drafted the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Primary fibroblasts were obtained from skin biopsy from healthy controls. This was carried out in accordance with the guidelines of the Declaration of Helsinki. Written informed consent was obtained from all participants (Ethics Committee from the Angers University Hospital approval: CPP Ouest II – Angers, France; number: CPP CB 2014/02).
We thank Florence Pascaretti, Anaïs Girardeau for image acquisition and Florence Manero from the SCIAM microscopy core facility (Angers University SFR ICAT) and Christian Jüngst from CECAD Imaging (University of Cologne) and Dr. Olivier R Baris for proofreading.
This research received no external funding.
The authors declare no conflict of interest.
We used ChatGPT and Deepl software to improve our English. The authors checked the article, made appropriate revisions, and are responsible for the article.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.