


 , Yangxin Jiang 1,†, Jin Zeng 2, Dongchuan Zuo 1,3,*
, Yangxin Jiang 1,†, Jin Zeng 2, Dongchuan Zuo 1,3,*
1 Institute of Cardiovascular Research, Key Laboratory of Medical Electrophysiology, Ministry of Education, Southwest Medical University, 646000 Luzhou, Sichuan, China
2 Department of Orthodontics, The Affiliated Stomatology Hospital of Southwest Medical University, 646000 Luzhou, Sichuan, China
3 Department of Cardiology, The Affiliated Hospital of Southwest Medical University, 646000 Luzhou, Sichuan, China
†These authors contributed equally.
Abstract
Hypokalemia induces abnormal spontaneous pacemaker activities of cardiomyocytes, which is strongly associated with fatal cardiac arrhythmias caused by hypokalemia. However, the mechanism remains unclear.
For the study of the mechanisms associated with hypokalemia, optical mapping recordings were performed on isolated murine hearts perfused with hypokalemia solutions, which allows for the concurrent examination of membrane potential and calcium transient morphology and arrhythmogenesis. Human Kir2.1, Kir2.1-E224G mutant, or Kir4.1 channels were constructed with lentiviral vectors. Patch clamp recordings were performed to verify the corresponding currents of these constructed channels in the heterologous expression system chinese hamster ovary (CHO) cells, and to explore how Kir2.1 channels influence the resting membrane potentials of human iPSC-derived cardiomyocytes (hiPSC-CMs) when exposed to low [K+]e.
Isolated murine hearts perfused with hypokalemia solution (1 mmol/L) developed a high frequency of spontaneous ventricular tachycardia (VT), which was initiated as an after-depolarization triggered activity associated with Ca2+ overload. The VT was maintained by abnormal spontaneous pacemaker activities caused by membrane potential depolarization. In response to 1 mmol/L [K+]e, hiPSC-CMs overexpressing Kir2.1 channels exhibited membrane potential depolarization, leading to the induction of abnormal pacemaker activities. The cells overexpressing rectification-deficient Kir2.1-E224G mutant channels or weak rectification Kir4.1 channels exhibited membrane potential hyperpolarization without the occurrence of abnormal pacemaker activities.
Kir2.1 channel-mediated membrane potential depolarization contributes to hypokalemia-induced abnormal spontaneous pacemaker activities of cardiomyocytes. The inward rectification of Kir2.1 channels plays a critical role in this process.
Keywords
- Kir2.1 channel
- inward rectification
- resting membrane potential
- abnormal pacemaker activities
- cardiomyocyte
Hypokalemia (blood serum potassium levels lower than 3.5 mmol/L) is one of the most prevalent electrolyte disturbances, which can result in cardiac ectopic automaticity leading to life-threatening cardiac arrhythmias [1]. Hypokalemia-induced cardiac ectopic automaticity has been attributed to inducing after-depolarization triggered activities and abnormal spontaneous pacemaker activities in cardiomyocytes [2]. Hypokalemia increased the propensity for both early and delayed after-depolarizations of cardiomyocytes, due to the inhibitory effects of hypokalemia on potassium channels and the sodium-potassium pump, which causes prolonged action potential duration and Ca2+ overload [3, 4]. The hypokalemia-induced spontaneous pacemaker activities of cardiomyocytes have been thought to be a result of the abnormal depolarizing effects of hypokalemia on the membrane potential. The membrane potential of cardiomyocytes should be hyperpolarized in response to low [K+]e as predicted by the Nernst equation. Human cardiomyocytes show abnormal membrane depolarization instead of hyperpolarization in response to low [K+]e [5, 6, 7, 8, 9, 10]. Robinson et al. [11] reported that the membrane potential of murine cardiomyocytes also significantly depolarized, and suggested that the depolarized membrane potential induced by low [K+]e may be due to a combination of reduced conductance of the Ik1 channel. However, the depolarizing effects of hypokalemia in whole organ systems are still unclear.
The Kir2.1 channel is thought to be responsible for the membrane depolarization caused by hypokalemia. This channel displays classical inward rectification, manifested by a strong voltage-dependent decline of potassium conductance upon membrane depolarization, producing a distinctive region of negative slope conductance [12, 13]. We have previously reported that under hypokalemia conditions, Ik1 currents mediated by Kir2.1 nonlinear channels balanced with cation currents (such as TWIK-1 two-pore domain K+ channels mediated leak cation currents), cause membrane potential depolarization of human iPSC-derived cardiomyocytes (hiPSC-CMs) [14]. Based on these results, we hypothesize that Kir2.1 channel-mediated membrane potential depolarization contributes to hypokalemia-induced abnormal spontaneous pacemaker activities of human cardiomyocytes, and that the strong inward rectification of Kir2.1 channels plays a critical role in this process. In order to test this hypothesis, we utilized two more Kir channels (Kir4.1 channels and Kir2.1•E224G mutant channels) as controls. Kir4.1 channels display intermediate inward rectification [15], whereas the Kir2.1•E224G mutation abolishes the inward rectification of Kir2.1 channels [16, 17].
In this study, we first performed optical mapping recordings on isolated murine hearts perfused with a hypokalemia solution (1 mmol/L), which allows for the concurrent examination of action potentials and calcium transient morphology and arrhythmogenesis to study the mechanisms associated with hypokalemia. We next performed a patch clamp to monitor the membrane potential response to low [K+]e by using hiPSC-CMs with enhanced expression of Kir2.1 channels, rectification-deficient Kir2.1-E224G mutant channels, or weak rectification Kir4.1 channels. We demonstrated that hypokalemia depolarized cardiac membrane potential induces cardiac ectopic automaticity and that Kir2.1 channel-mediated membrane potential depolarization contributes to hypokalemia-induced cardiac ectopic automaticity. The inward rectification of Kir2.1 channels plays a critical role in this process.
Eight-week-old C57BL6/J mice weighing ~25 g were purchased from Chongqing Tengxin Biotechnology Co., Ltd., China, and housed in a temperature- and humidity-controlled animal facility. The procedures were approved by the Institutional Animal Ethics Committee at Southwest Medical University (Approval No. 20240903-003) in Luzhou, China, in accordance with the institution’s national guidelines that align with the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals.
Mice were humanely anesthetized by the administration of ketamine (80 mg/kg) and
xylazine (5 mg/kg). Anesthesia was confirmed by no obvious reflex response to
hind feet pinching. After deep anesthesia, animals were euthanized by cervical
dislocation. Hearts were immediately excised and loaded onto a Langendorff
perfusion system. Electrophysiological abnormalities and arrhythmias of the
intact hearts perfused with hypokalemia solution (1 mmol/L) were assessed by an
optical mapping system, as previously described [18]. Briefly, the murine hearts
were quickly removed and loaded onto a Langendorff perfusion system, and were
perfused with an oxygen-containing physiological salt solution at a constant rate
of 3 mL/min at 37 °C for 10 min. The hearts were then incubated with
Rhod-2 AM (a Ca2+ dye), 10% Pluronic F127, and RH237 (a voltage-sensitive
dye) for 10–20 min. Rhod-2 AM and RH237 were stimulated using four
light-emitting diode macro LED lights (530 nm, Kane Research). The fluorescence
signal was captured by an optical mapping system and fitted with a
custom-designed EMCCD camera (Evolve 512 Photometrics, Tucson, AZ, USA). The
membrane potential and Ca2+ were measured at maximum resolution (512
Lentiviral vectors encoding human Kir2.1, Kir2.1-E224G mutant, and Kir4.1
channels were engineered and packaged into corresponding lentiviruses (titer 1
hiPSC-CMs were purchased from Help Therapeutics (Nanjing, China) and cultured
according to the instructions [14]. All cell lines were validated by STR (short
tandem repeat) profiling and tested negative for mycoplasma. Cells were
transduced with lentiviral vectors encoding GFP (green fluorescent protein)-Kir2.1, GFP-Kir2.1-E224G, or GFP-Kir4.1 at concentrations between 1.8 and 3
hiPSC-CMs plated on slides were treated as follows: they were fixed in 4% paraformaldehyde for 10 min, permeabilized using 0.5% Triton X-100 for 15 min, and then blocked with 1% bovine serum albumin in phosphate buffered saline (PBS) for 30 min. Following the blocking step with 10% normal goat serum, the cells were subsequently incubated with primary antibodies targeting cTnT (Cardiac troponin T, Abcam, Cambridge, UK, ab8295, 1:100) overnight at 4 °C. The following day, the cells were incubated with the secondary antibody for 1 h. The nuclei were then stained with 4′,6-Diamidino-2-phenylindole, Dihydrochloride (DAPI, C1005, Beyotime Biotechnology, Shanghai, China). A fluorescence microscope (Leica DM2500, Leica Optical, Wetzlar, Germany) was used to capture the images.
A HEKA patch-clamp system (EPC10, HEKA Elektronik, Reutlingen, Germany) was used
to record the resting membrane potentials and the whole-cell currents of the
cells. A standard voltage ramp protocol lasting 2.2 seconds, ranging from –120
mV to +20 mV, was applied. analysis of the patch clamp data was performed with a
PatchMaster version 2
Internal solutions for CHO cells contained (mmol/L): 140 KCl, 1 MgCl2, 10 EGTA, 1 K2-ATP, and 5 Hepes (pH 7.4); external solutions contained (mmol/L): 135 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 15 glucose, and 10 Hepes (pH 7.4). Internal solutions for hiPSC-CMs contained (mmol/L): 20 KCl, 120 K aspartate, 1 MgCl2, 5 Na2-ATP, 0.5 Na2-GTP, 10 EGTA, and 5 Hepes (pH 7.4); external solutions contained (mmol/L): 140 NaCl, 5.4 KCl, 1.8 CaCl2, 1 MgCl2, 10 glucose, and 10 Hepes (pH 7.4). Bath solutions containing low [K+]e were prepared by decreasing the [K+]e and supplementing it with equimolar Na+. Voltage-gated Ca2⁺ and Na⁺ currents were blocked by adding 2 mM CoCl2 and 3 µM TTX (tetrodotoxin) to the bath solutions [14].
Statistical analysis was performed using GraphPad Prism software version 9
(GraphPad Software, San Diego, CA, USA). Data are presented as mean
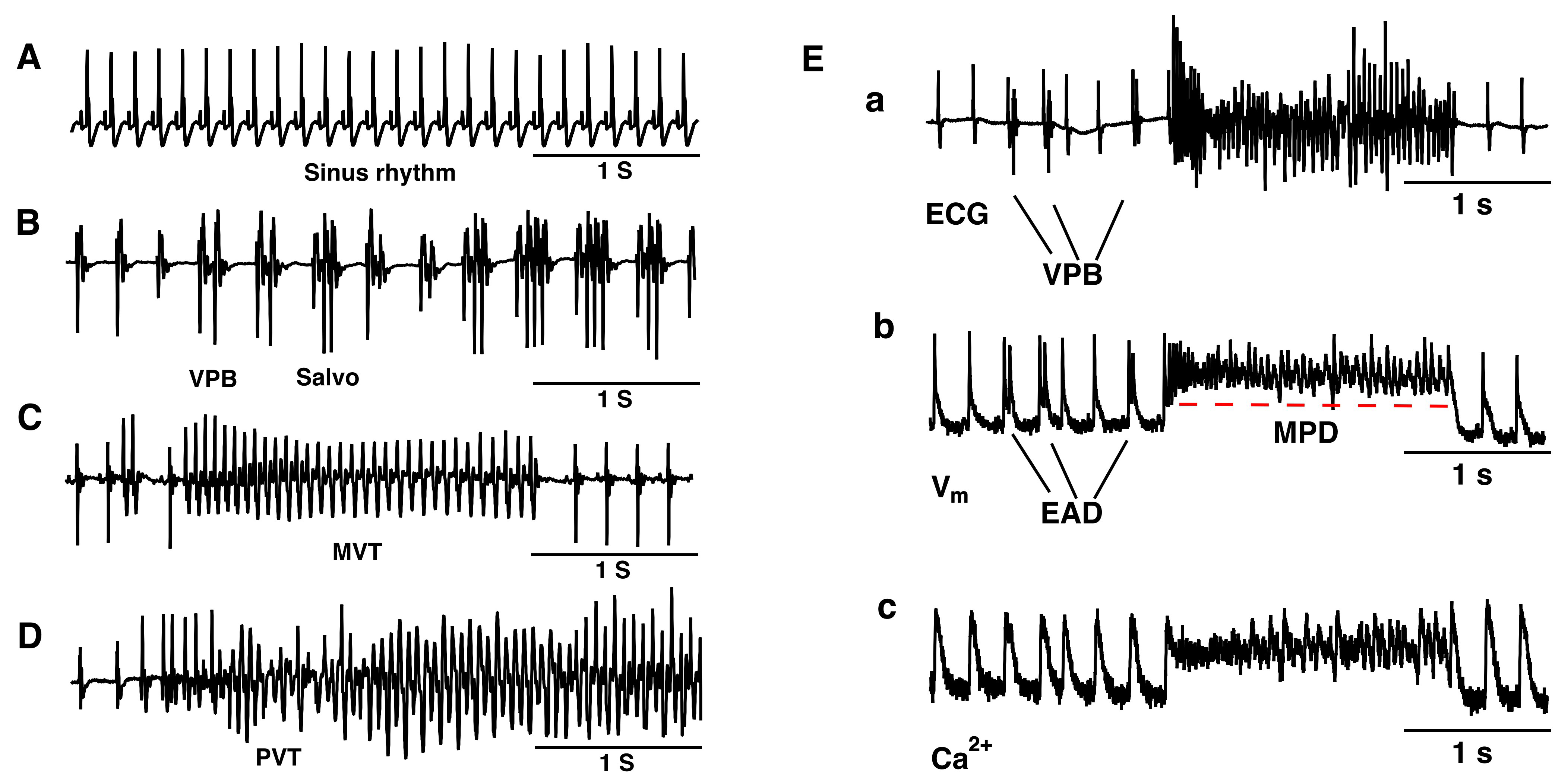
We performed pseudo-ECG recordings to monitor the extracellular potentials of isolated hearts of the mice. Under 5.4 mM [K+]e conditions, all isolated hearts maintained sinus rhythm (n = 6) (Fig. 1A). Within 30 minutes of perfusion with 1 mM [K+]e, all hearts consistently exhibited ventricular premature beats (VPBs, defined as 3 or more sinus beats between two consecutive ectopic beats), salvo (difined as 2 or 3 consecutive ectopic beats), monomorphic ventricular tachycardia (MVT, VT was defined as 4 or more consecutive ectopic beats), and polymorphic ventricular tachycardia (PVT) (Fig. 1B–D) (n = 6).
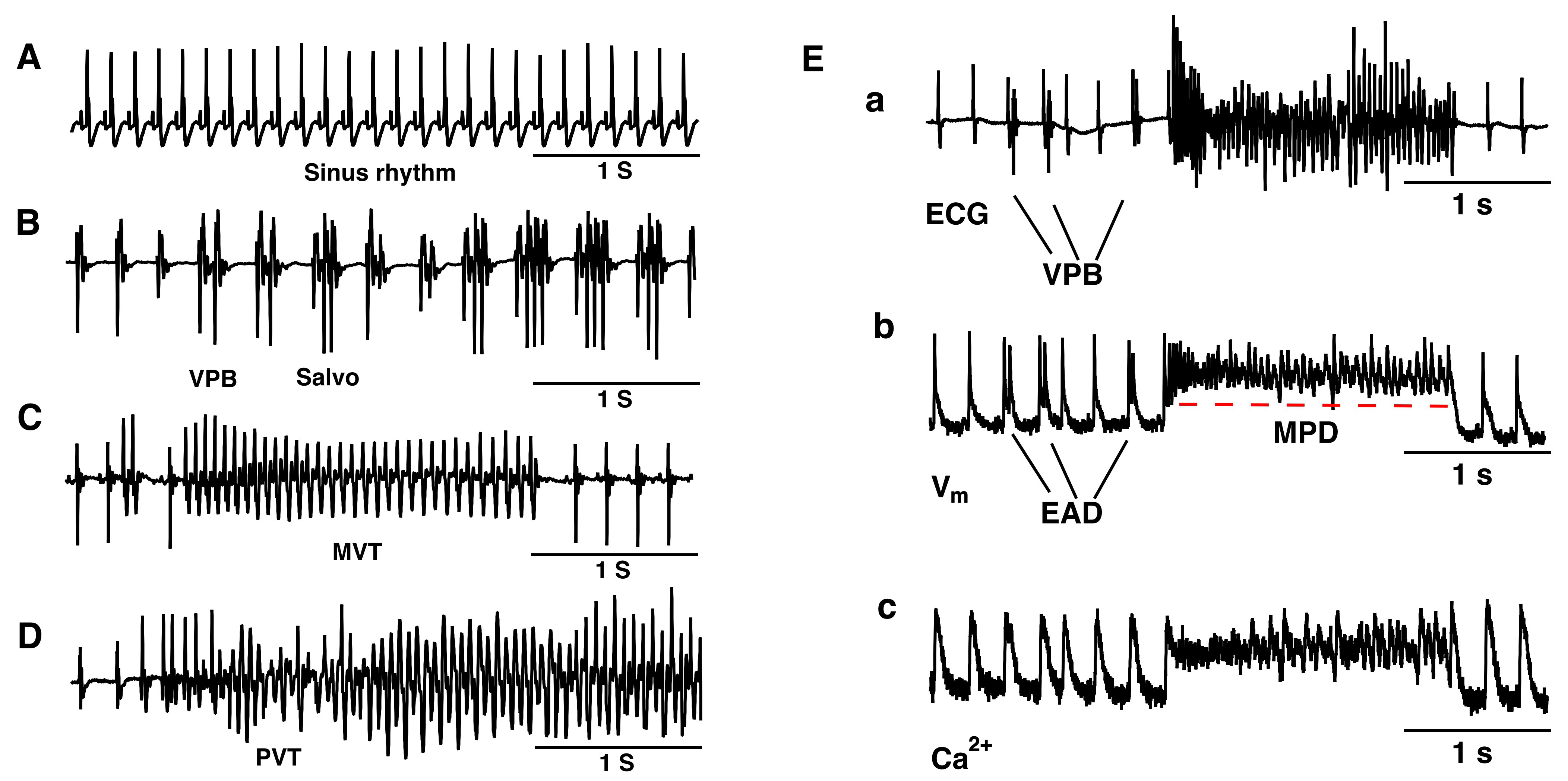 Fig. 1.
Fig. 1.
Hypokalemia-induced various types of ventricular ectopic beats in a murine whole heart. Langendorff preparation. (A) Representative pseudo-ECG of sinus rhythm under 5.4 mM [K+]e conditions. (B–D) Representative pseudo-ECG of isolated VPB, Salvo (B), MVT (C), and PVT (D). (E) Typical pseudo-ECG (a), membrane potential (b), and Ca2+ transient trace (c) simultaneously recorded from a spontaneously beating isolated heart during VT onset after exposure to a hypokalemia solution. n = 6. EAD, early afterdepolarization; MPD, membrane potential depolarization; ECG, electrocardiograms; VPB, ventricular premature beat; MVT, monomorphic ventricular tachycardia; PVT, polymorphic ventricular tachycardia.
The mechanisms of the initiation and maintenance of VT in the isolated murine hearts under hypokalemic conditions were analyzed. Fig. 1E shows a typical episode of VT, which was initiated by after-depolarization triggered activity associated with Ca2+ overload, and was maintained by abnormal spontaneous pacemaker activities caused by cardiac membrane potential depolarization.
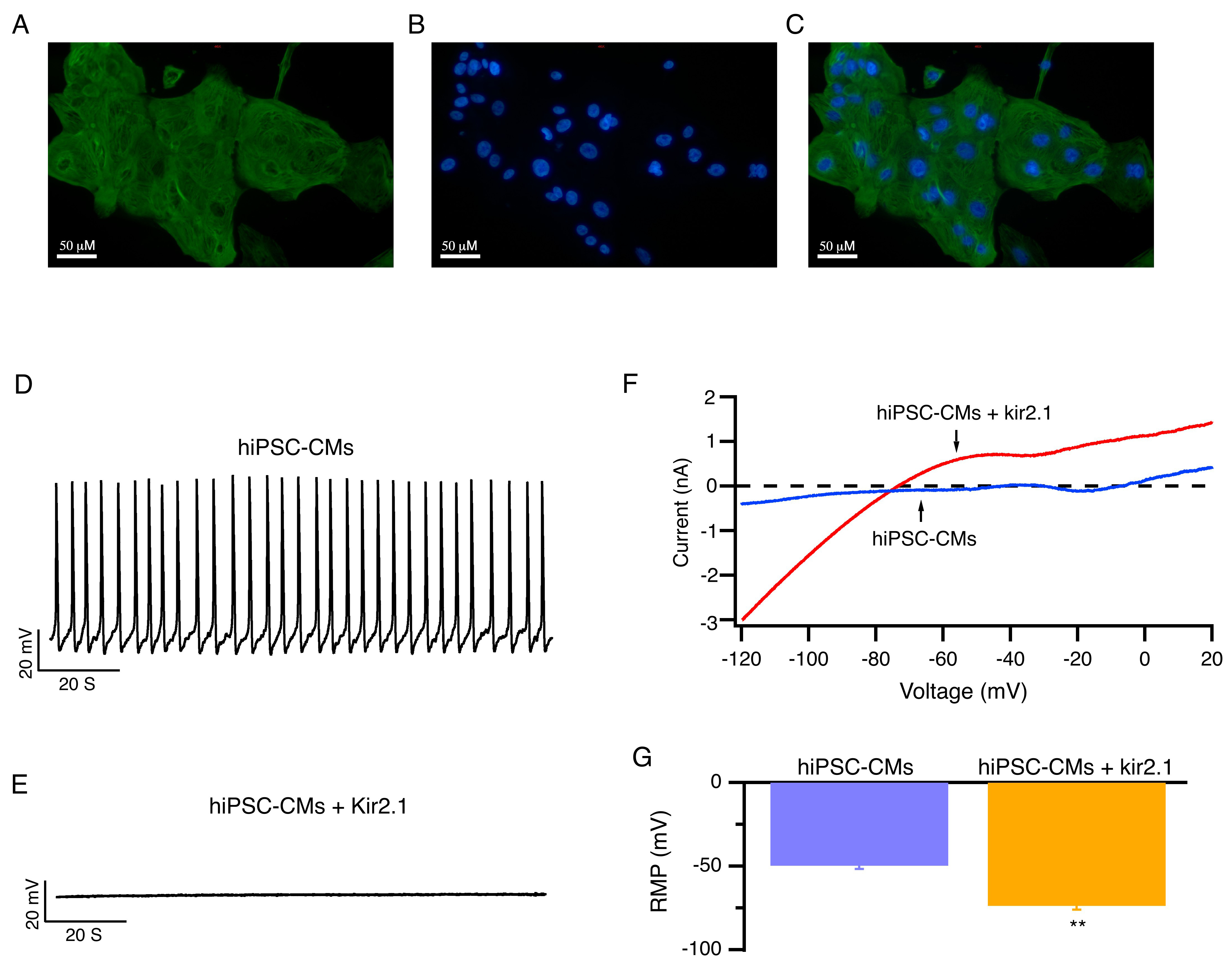
The hiPSC-CMs were verified by immunofluorescence staining with
cardiomyocyte-specific markers cTnT (Fig. 2A–C). hiPSC-CMs exhibit
electrophysiological properties that are similar to those of human native
cardiomyocytes. However, due to the lack of expression of the Kir2.1 channels,
hiPSC-CMs have lower resting membrane potential levels (–49.7
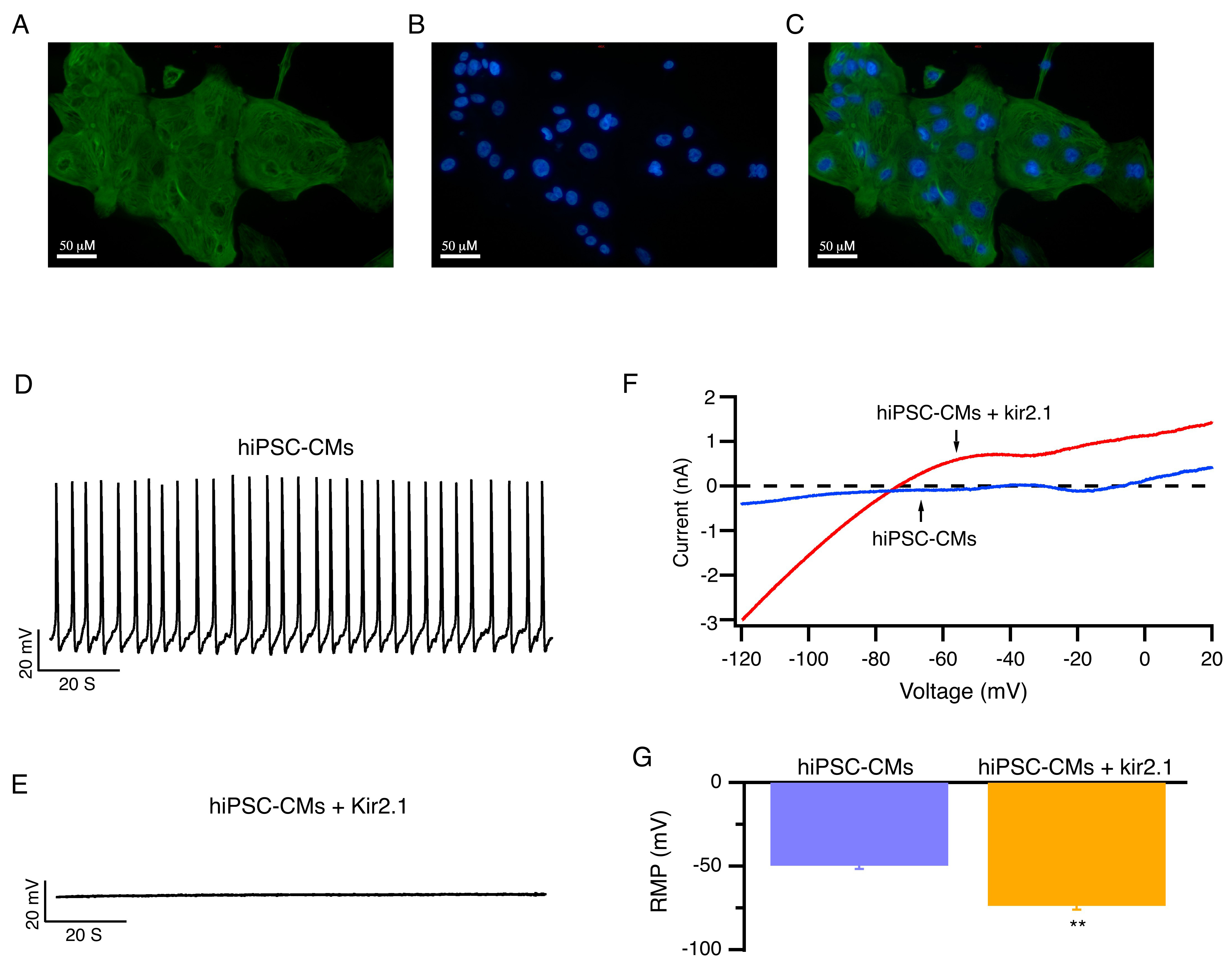 Fig. 2.
Fig. 2.
hiPSC-CMs with enhanced expression of Kir2.1 channels exhibited
hyperpolarized resting membrane potentials and became electrically quiescent.
(A–C) Representative images of fluorescent immunohistochemistry staining for
cTnT and DAPI with hiPSC-CMs. Scale bar: 50 µM. (D,E) Representative
RMP of hiPSC-CMs (D) and hiPSC-CMs that overexpress Kir2.1 channels (E) at 5.4
mmol/L [K+]e are shown. (F) Representative whole-cell ramp currents of
hiPSC-CMs (blue) and hiPSC-CMs overexpressing Kir2.1 channels (red). (G) Average
RMP of hiPSC-CMs and hiPSC-CMs that overexpress Kir2.1 channels at 5.4 mmol/L
[K+]e. Mean
When [K+]e was decreased from 5.4 mmol/L to 1 mmol/L, current clamp
results showed that the membrane potentials of hiPSC-CMs overexpressing Kir2.1
channels initially hyperpolarized from –73.7
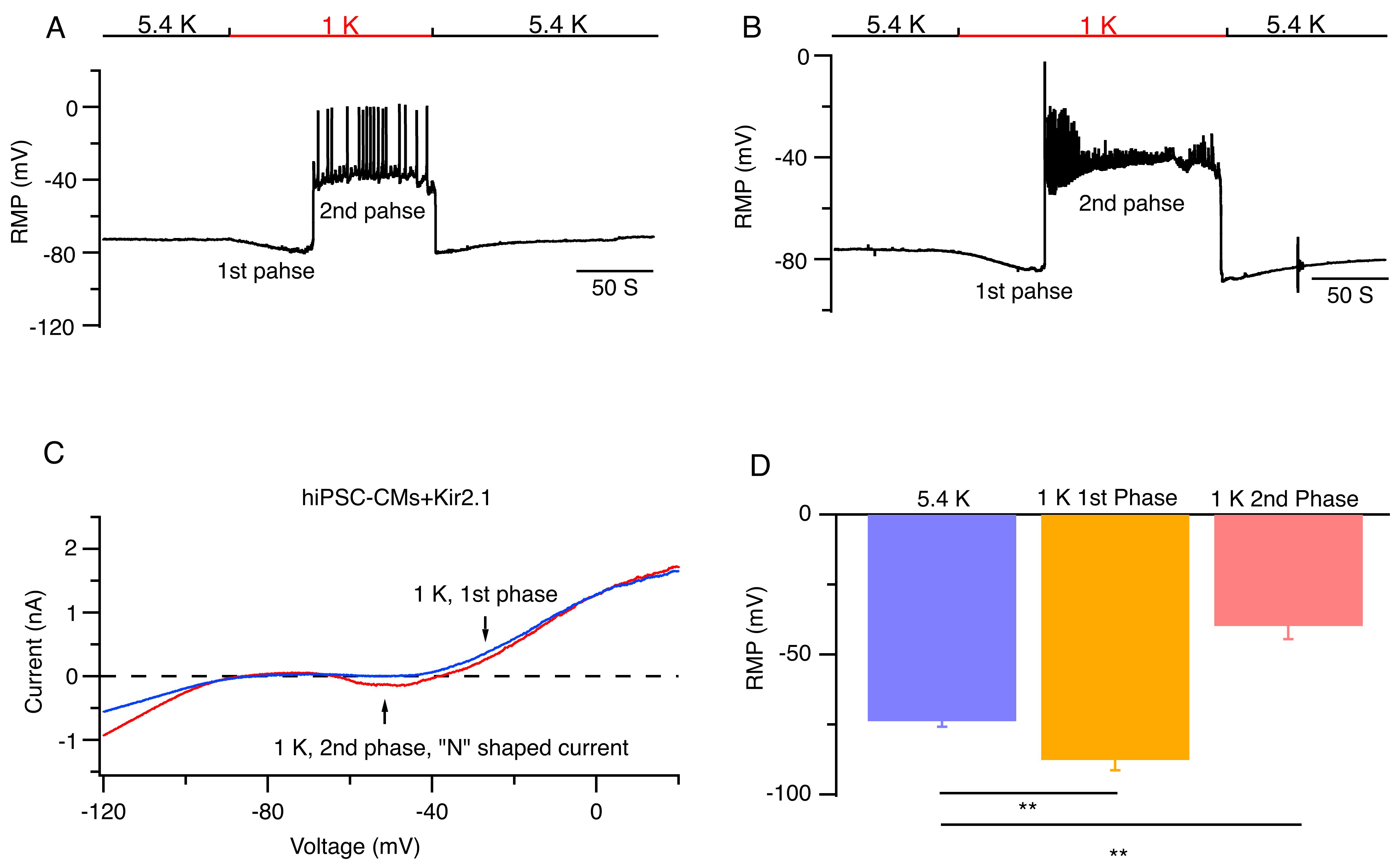 Fig. 3.
Fig. 3.
Low [K+]𝐞 induced abnormal pacemaker activities in
hiPSC-CMs overexpressing Kir2.1 channels. (A,B) Representative RMPs of hiPSC-CMs
overexpressing Kir2.1 channels are shown when bath solutions were reversibly
changed from 5.4 (black bar) to 1 mmol/L [K+]e (red bar). (C)
Representative current traces recorded at 1 mmol/L [K+]e corresponding
to the first phase (blue) and the second phase (red) in hiPSC-CMs overexpressing
Kir2.1 channels. (D) Average RMP of hiPSC-CMs overexpressing Kir2.1 channels at
5.4 and 1 mmol/L [K+]e. Mean
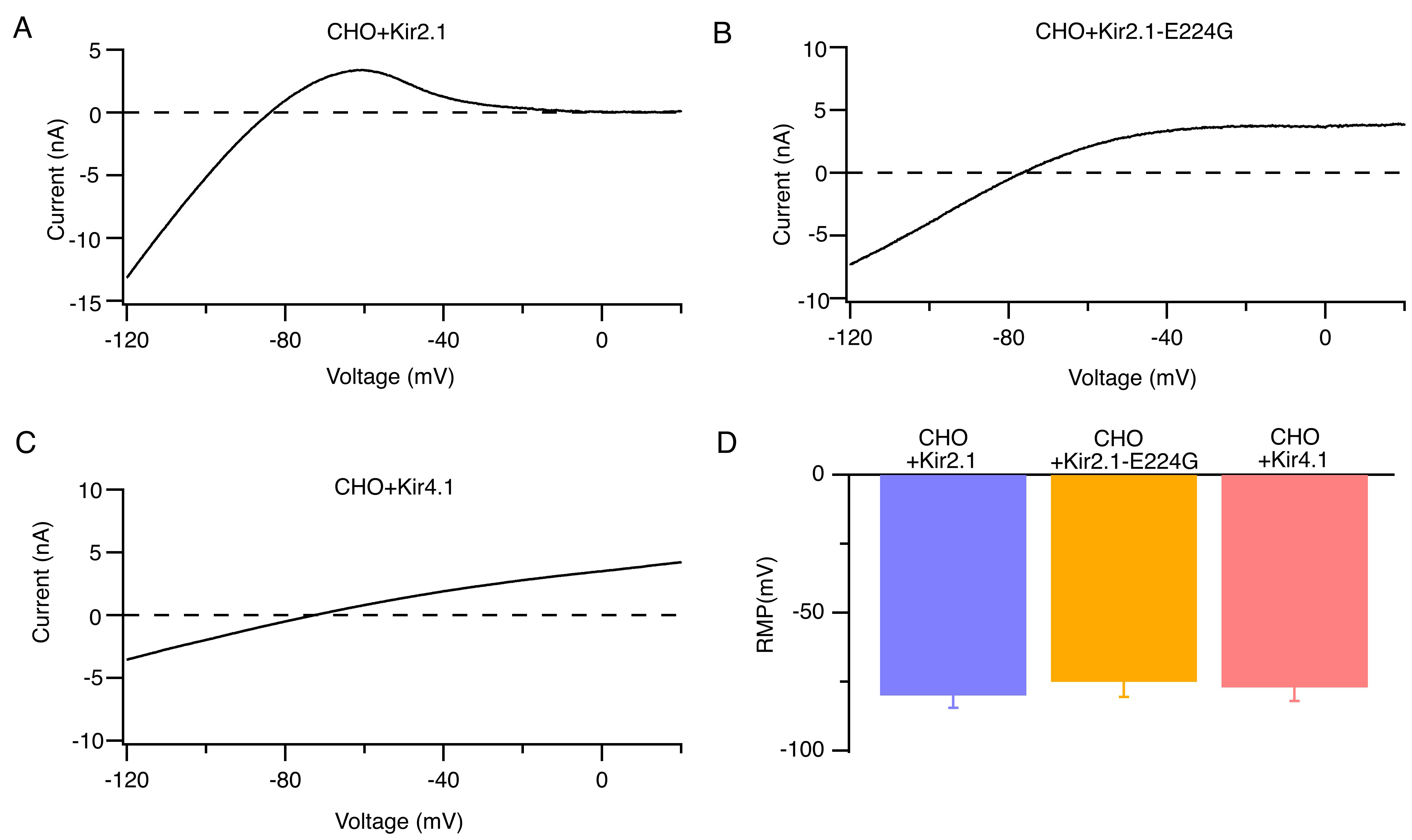
We constructed human Kir2.1, Kir2.1-E224G mutant, and Kir4.1 channels with lentiviral vectors and verified the corresponding currents in a heterologous expression system of CHO cells. Fig. 4A–C show the typical whole-cell currents of Kir2.1, Kir2.1-E224G mutant, and Kir4.1 channels. CHO cells expressing Kir2.1, Kir4.1, or the Kir2.1-E224G mutant channels exhibited similar resting membrane potentials at 5.4 mmol/L [K+]e (Fig. 4D).
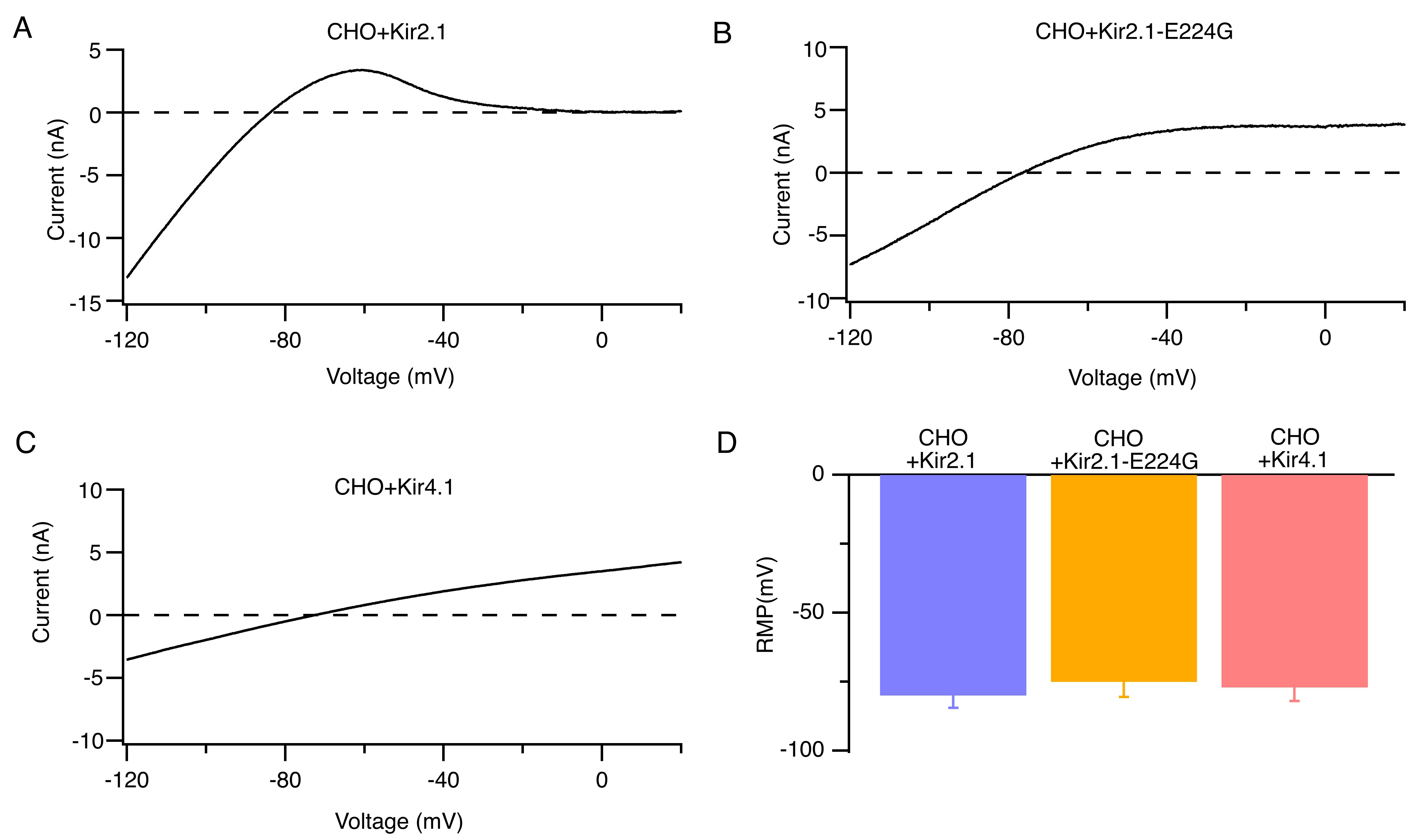 Fig. 4.
Fig. 4.
Identification of Kir2.1, Kir2.1-E224G, and Kir4.1 currents in CHO heterologous expression systems. (A–C) Representative whole-cell ramp currents of Kir2.1 (A) Kir2.1-E224G (B), and Kir4.1 channels (C) at 5.4 mmol/L [K+]e. (D) Average RMP of CHO cells that overexpress Kir2.1, Kir2.1-E224G, or Kir4.1 channels at 5.4 mmol/L [K+]e. n = 9. CHO, chinese hamster ovary.
To assess the contribution of the Kir2.1 inward rectification to the induction
of abnormal spontaneous pacemaker activities in human cardiomyocytes under
hypokalemic conditions, we observed the effects of low [K+]e on
hiPSC-CMs that overexpress Kir2.1, Kir4.1, or Kir2.1-E224G mutant channels. Under
5.4 mmol/L [K+]e conditions, the resting membrane potentials of
hiPSC-CMs that overexpress Kir2.1, Kir4.1, or Kir2.1-E224G mutant channels have
similar resting membrane potential levels. When [K+]e was reduced from
5.4 mmol/L to 1 mmol/L, the resting membrane potentials of hiPSC-CMs that
overexpress Kir4.1 or Kir2.1-E224G mutant potassium channels were hyperpolarized
(Fig. 5F), and the reversal potentials of the whole-cell currents were maintained
at –101.3
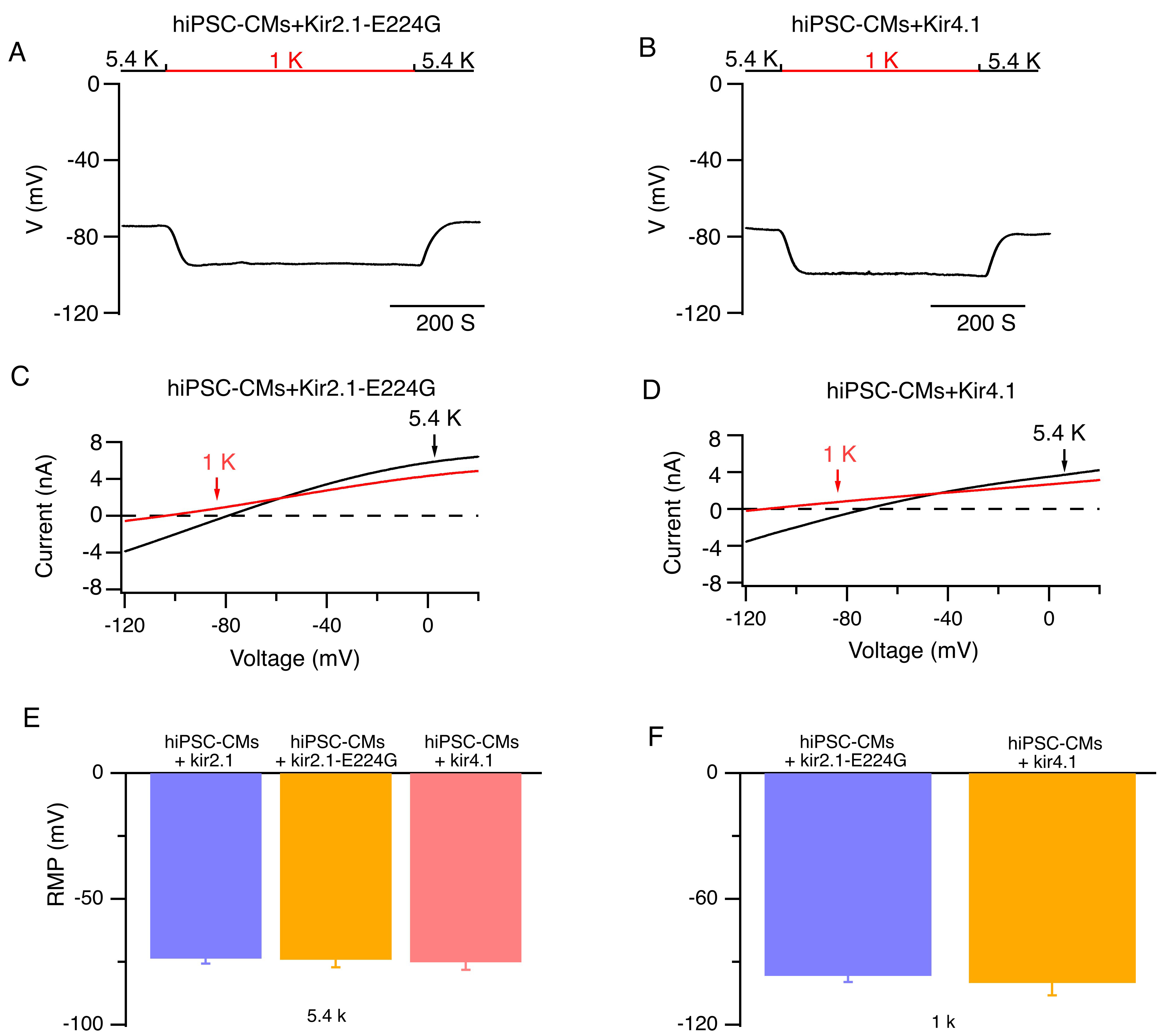 Fig. 5.
Fig. 5.
Impacts of low [K+]e on the RMP and whole-cell
currents of hiPSC-CMs that overexpress Kir2.1-E224G or Kir4.1 channels. (A,B)
Representative RMPs of hiPSC-CMs that overexpress Kir2.1-E224G or Kir4.1 channels
are shown when external solutions were reversibly switched from 5.4 (black bar)
to 1 mmol/L [K+]e (red bar). (C,D) Representative whole-cell currents
in hiPSC-CMs that overexpress Kir2.1-E224G or Kir4.1 channels at 5.4 mM
[K+]e (black lines) and 1 mM [K+]e (red lines), respectively.
(E) Average RMP of hiPSC-CMs that overexpress Kir2.1, Kir2.1-E224G, or Kir4.1
channels at 5.4 mmol/L [K+]e. (F) RMP of hiPSC-CMs that overexpress
Kir2.1-E224G or Kir4.1 channel at 1 mmol/L [K+]e. Mean
In this study, we first showed that Langendorff-perfused murine hearts replicate hypokalemia-induced arrhythmias and demonstrated that spontaneous VT developed in the intact murine hearts when hypokalemia was mediated by cardiac membrane potential depolarization. To the best of our knowledge, this is the first report on isolated heart models to demonstrate that hypokalemia causes cardiac membrane potential depolarization, thereby inducing cardiac ectopic automaticity. We then showed that hiPSC-CMs overexpressing Kir2.1 channels recapitulate the abnormal pacemaker activity response to low [K+]e, and demonstrated that inwardly rectifying Kir2.1 channels mediated membrane potential depolarization, contributing to hypokalemia-induced abnormal pacemaker activities of human cardiomyocytes.
According to the Nernst equation, cardiomyocytes should exhibit hyperpolarized membrane potentials in response to low [K+]e. However, previous studies demonstrated that human cardiomyocytes, including ventricular, atrial myocytes, and Purkinje fibers, exhibited depolarized resting membrane potentials at low [K+]e [7, 8, 9]. Such an abnormal membrane potential depolarization would increase the propensity of cations (the majority of which are Ca2+) influx, leading to the generation of abnormal pacemaker activities. However, these arrhythmogenic effects of hypokalemia were previously studied in single cardiomyocytes. While the approach was valid, it did not allow for the study of arrhythmogenic mechanisms in the more complex cardiac environment. In this study, by using optical mapping techniques that enable simultaneous study of membrane potential and Ca2+ transient and arrhythmogenesis at the whole heart level, we confirmed that the spontaneous VT developed in the intact murine hearts with hypokalemia was mediated by membrane potential depolarization. The results clearly showed that the hypokalemia-induced VT is a consequence of spontaneous pacemaker activities associated with Ca2+ overload, which is caused by membrane potential depolarization.
In response to low [K+]e, the decreased K+ conduction caused by the reduced K+ gradient leads to two changes in the cardiomyocytes. First, it prolongs the action potential duration, subsequently inducing both early and delayed after-depolarizations. Second, it may depolarize the resting membrane potential, which promotes a transition from quiescent to spontaneous electrical activity [2]. Both of these factors account for hypokalemia-induced cardiac ectopic automaticity. The membrane potential of cardiomyocytes is determined by the balance of outward and inward ionic currents across the cell membrane. In response to low [K+]e, Kir2.1-overexpressing hiPSC-CMs initially rapidly hyperpolarized (phase 1) due to the function of Kir2 channels. As the perfusion time of the low [K+]e solution was prolonged, the conductance of the Ik1 was further reduced. When the reduced Ik1 currents mediated by nonlinear Kir2.1 channels are balanced with cation currents (such as TWIK-1 two-pore domain K+ channels mediated leak cation currents), “N” shaped I–V relationships are generated and lead to abnormal membrane potential depolarization of hiPSC-CMs (phase 2) [13]. However, the mechanism by which Kir2 channels contribute to abnormal pacemaker activity associated with membrane potential depolarization induced by low [K+]e is unknown. We now present evidence supporting the hypothesis that the inward rectification of Kir2.1 channels is required for hypokalemia-induced abnormal spontaneous pacemaker activities of human cardiomyocytes. First, the hiPSC-CMs over-expressing Kir2.1 channels exhibited membrane potential depolarization resulting in spontaneous abnormal pacemaker activities, as observed in human native cardiomyocytes. Second, the hiPSC-CMs over-expressing rectification-deficient Kir2.1-E224G mutant channels or weak rectification Kir4.1 channels exhibited membrane potential hyperpolarization without the occurrence of abnormal pacemaker activities. The negatively charged residues (E224, D255, D259, and E299) located at the cytoplasmic domain of Kir2.1 channels determine the strength of inward Ik1 rectification [22]. In line with our studies, Moreno-Manuel et al. [23] recently reported that the rectification-deficient Kir2.1-E299V mutation prevented arrhythmias in the ventricles of a mouse model. Other ionic currents may also contribute to the abnormal spontaneous pacemaker activities induced by low [K+]e in cardiomyocytes. Both inward leak Na+ and Ca2+ currents have been suggested to contribute to the low [K+]e-induced membrane depolarization. Wiggins and Cranefield [24] reported that Ca2+ currents induced membrane potential depolarization of cardiomyocytes in K+ free external solutions. Sheu et al. [25] reported that depolarized membrane potential induced by low [K+]e was abolished when extracellular Na+ was removed, suggesting that Na+ influx is essential for this phenomenon. Therefore, the membrane potential depolarization induced by Na+ and Ca2+ influx may also account for the abnormal spontaneous pacemaker activities induced by low [K+]e in cardiomyocytes.
The present study was performed in hiPSC-CMs, which may not be the ideal model
to examine our hypothesis. hiPSC-CMs are known to exhibit immature
electrophysiological, structural, and metabolic features that significantly
differ from adult ventricular cardiomyocytes. Their lack of structural
organization and underdeveloped sarcomere alignment can influence action
potential characteristics and susceptibility to arrhythmias. Due to the lack of
expression of the Kir2.1 channels, hiPSC-CMs have lower resting membrane
potential levels and show spontaneous pacemaker activities. However, previous
studies have documented that the maturation of electrophysiological profiles in
hiPSC-CMs can be achieved through enhanced expression of Kir2.1 channels [19, 20].
In the present study, the Kir2.1-overexpressing hiPSC-CMs had a resting membrane
potential of –73.7
In summary, this study demonstrates that Kir2.1 channel-mediated membrane potential depolarization contributes to hypokalemia-induced abnormal spontaneous pacemaker activities of cardiomyocytes and that the inward rectification of Kir2.1 channels plays a critical role in this process.
The datasets used and analyzed during the current study are available from the corresponding author on reasonable request.
JL, YJ and JZ conducted experiments. DZ designed the research study, performed the research, wrote the manuscript, and analyzed the data. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
For animal studies, all procedures involving animals were conducted in accordance with the guidelines established by Animal Welfare Act and the hospital, and approved by the Institutional Animal Ethics Committee at Southwest Medical University (Approval No. 20240903-003).
We would like to express our gratitude to all those who helped me during the writing of this manuscript and thanks to all the peer reviewers for their opinions and suggestions.
This work was supported by the Sichuan Science and Technology Program: 2024NSFSC2103; Southwest Medical University Project: 2024ZKY110.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.