



 , Xi Wang 2,†, Peter G Alexander 1, Peng Feng 1,3,4,*
, Xi Wang 2,†, Peter G Alexander 1, Peng Feng 1,3,4,* , Jianying Zhang 1,*
, Jianying Zhang 1,*1 Department of Orthopaedic Surgery, University of Pittsburgh, Pittsburgh, PA 15213, USA
2 Department of Trauma Orthopedics, The First Affiliated Hospital of Xinjiang Medical University, 830011 Urumqi, Xinjiang, China
3 School of Medicine, China Academy of Chinese Medical Sciences, 100700 Beijing, China
4 Department of Trauma Orthopedics, Wangjing Hospital of China Academy of Chinese Medical Sciences, 100102 Beijing, China
†These authors contributed equally.
Abstract
Adenosine 5′-monophosphate (AMP)-activated protein kinase (AMPK) is an energy homeostasis controller that regulates various metabolic pathways to promote adenosine triphosphate (ATP) generation and suppress energy expenditure, thereby restoring energy homeostasis. As a co-factor in many enzymes, iron is an essential mineral for maintaining ATP levels in our bodies. Ferroptosis is an iron-dependent mode of cell death that occurs in various pathological processes, including cancer, metabolic disorders, and autoimmune diseases, by regulating iron metabolism, lipoperoxidation, and anti-oxidation functions. Ferroptosis is triggered by oxidative and energy stress, both controlled by cancer-associated signaling pathways. Emerging studies have demonstrated that AMPK directly influences ferroptosis by modulating lipid metabolism, redox homeostasis, and iron transport. Cancer cells exhibiting elevated baseline AMPK activity demonstrate resistance to ferroptosis, whereas AMPK suppression enhances their susceptibility to this regulated form of cell death. While the precise mechanistic details are yet to be fully elucidated, accumulating evidence suggests that AMPK-mediated ferroptosis regulation may contribute to cancer development and therapeutic responses. This review summarizes recent advances in understanding the interplay between AMPK and ferroptosis in cancer biology and discusses the potential of targeting the AMPK-ferroptosis axis for innovative anticancer strategies.
Keywords
- AMPK
- ferroptosis
- cancer
- energy metabolism
- iron homeostasis
Adenosine 5′-monophosphate (AMP)-activated protein kinase (AMPK) is a critical regulator of energy status in cells, playing a key role in maintaining cellular energy balance1 [1]. When cells experience energetic stress (e.g., adenosine triphosphate (ATP) depletion and elevated AMP levels), AMPK is activated [2], AMPK activation both downregulates anabolic activities that can deplete a cell’s ATP store while concurrently promoting catabolic activities that also generate ATP. Together, these activities ensure cell viability by restoring energy homeostasis [3]. However, under prolonged or severe energy stress, adaptive responses may fail to restore homeostasis, leading to unresolved energy stress and ultimately resulting in cell apoptosis. Beyond its traditional role in energy metabolism, AMPK has been increasingly recognized as a key player in various cellular stress responses, including oxidative stress, autophagy, and cell death regulation.
Cancer is a leading cause of death globally, thus there is an urgent need for innovative therapeutic strategies for the prevention and treatment of cancer. Traditional antitumor therapies typically attempt to eliminate cancer cells by inducing apoptosis, but many malignant tumors develop resistance to apoptosis-inducing treatments [4], making it essential to explore new cell death mechanisms. Ferroptosis is a recently discovered mechanism of cell death triggered by metabolic and redox imbalance that is observed in a wide variety of pathological processes including tumor growth, immunologic evasion, neuronal loss, muscular dystrophy, and ischemia-reperfusion injury [5]. Recent research has identified numerous regulators of ferroptosis that not only inhibit tumor growth but also enhance immunotherapy responses and overcome drug resistance [6], defining a new frontier in oncology research and offering new pathways for the treatment of ferroptosis-related diseases.
Recent studies have begun to uncover direct regulatory links between AMPK and ferroptosis, highlighting AMPK as a potential modulator of ferroptotic pathways in cancer [7, 8, 9, 10, 11, 12, 13]. AMPK has been shown to regulate ferroptosis through multiple mechanisms, including modulating lipid metabolism, oxidative stress responses, and iron homeostasis [14]. The precise molecular mechanisms through which AMPK regulate ferroptosis remain to be fully elucidated, and whether AMPK predominantly acts as a ferroptosis promoter or suppressor may depend on the specific tumor microenvironment and genetic landscape. Given the increasing recognition of ferroptosis as a key determinant of tumor progression and therapeutic response, further exploration of the AMPK-ferroptosis interplay is warranted. This review aims to summarize the latest advancements in this field, providing insights into the potential of targeting AMPK-regulated ferroptosis for novel cancer therapies.
Mammalian AMPK is a heterotrimeric complex consisting of three subunits: a
catalytic
The heterotrimeric structure of AMPK and the synergistic interactions among its subunits form a unique complex, offering the potential for differential design as a drug target and providing an essential structural basis for developing novel small-molecule activators. The structure of AMPK is shown in Fig. 1 (Ref. [26, 27]).
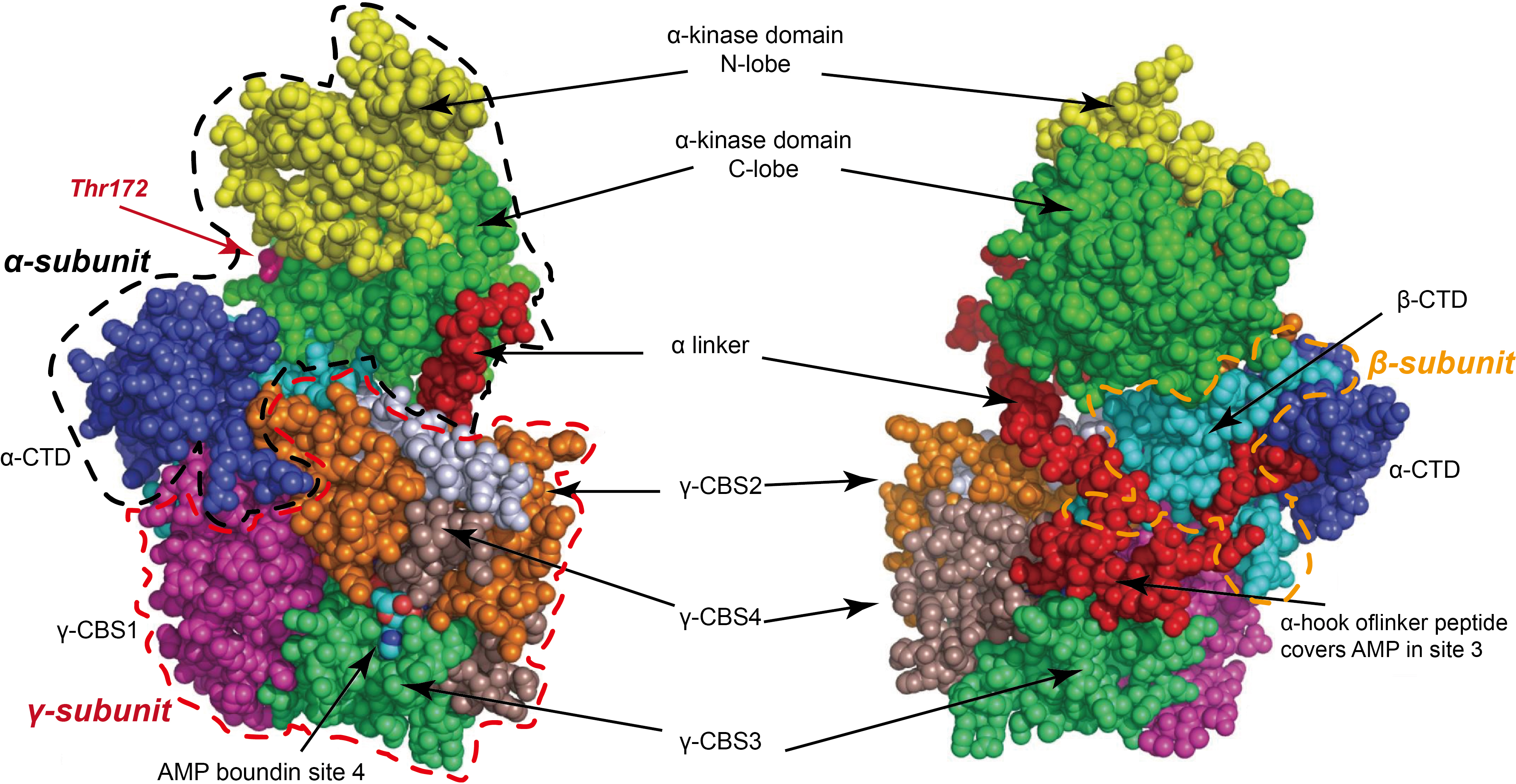 Fig. 1.
Fig. 1.
Mammalian adenosine 5′-monophosphate
(AMP)-activated protein kinase (AMPK) is a heterotrimeric complex composed of
three subunits: a catalytic
AMPK activity is tightly regulated by various hormones and metabolic signals, and its activation can occur through classical AMP-dependent and non-classical pathways [28]. The classical pathway relies on AMP binding as an allosteric activation mechanism. During energy stress, elevated AMP and/or adenosine diphosphate (ADP) levels and reduced ATP levels activate AMPK to restore energy balance. Phosphorylation of Thr172 is pivotal for AMPK activation by upstream kinases including liver kinase B1 (LKB1) and calcium/calmodulin-dependent protein kinase kinase 2 (CaMKK2) [20, 29]. LKB1 activates AMPK following its binding with AMP. Studies on LKB1-knockout mice reveal that under energy stress, LKB1 predominantly mediates AMPK activation across various mammalian tissues, including the kidney, heart, and liver [30, 31, 32]. In vitro experiments demonstrate that in response to low-energy states and mitochondrial dysfunction, AMPK activation is entirely dependent on LKB1 [33]. Additionally, other stimuli, such as glucose deprivation, physical activity, and mitochondrial toxins, also contribute to AMPK activation [34].
The non-classical pathway, however, does not depend on AMP or ADP levels but
involves responses to nutrient sensing (e.g., metabolites, small molecules,
glucose, amino acids, and fatty acids) and cellular damage (e.g., inflammatory
factors, lysosomal and nuclear DNA damage) [35, 36]. Metformin can directly
activate the AMPK/sirtuin 1 (SIRT1) pathway, subsequently stimulating the
activation of the nuclear factor-kappa B (NF-
AMPK is an intracellular energy sensor, and its downstream signaling pathways involve multiple biological processes such as metabolic regulation, autophagy and inflammation and immune regulation. AMPK acts on a variety of key substrates through phosphorylation to regulate cellular metabolic homeostasis:
(1) The Effect of ACC in Fatty Acid Metabolism
Acetyl-CoA carboxylase (ACC) is a critical enzyme in the metabolism of fatty
acids, and it is encoded by two different genes: ACC1 and ACC2.
ACC1 primarily functions in cytoplasmic fatty acid synthesis, while
ACC2 is closely associated with mitochondrial
(2) Autophagy Regulation by ULK1
ULK1, a core regulatory factor in autophagy, is activated by AMPK-mediated multi-site phosphorylation (e.g., S555, S637) and cooperates with FAK family-interacting protein of 200 kDa (FIP200) and autophagy-related protein 13 (ATG13) to initiate autophagosome formation [43, 44, 45]. AMPK activation of ULK1 not only maintains protein homeostasis but also promotes mitophagy under stress conditions to protect cell survival [46]. Additionally, the secretory factor Metrnl triggers autophagy via the LKB1/AMPK/ULK1 pathway while inhibiting cyclic guanosine monophosphate-adenosine monophosphate (cGMP-AMP) synthase-stimulator of interferon genes (cGAS-STING) signaling, providing protective effects against myocardial hypertrophy and cardiac toxicity [47, 48].
(3) mTOR Signal Inactivation
Although mammalian target of rapamycin (mTOR) and AMPK are involved in distinct signaling pathways, their pathways converge at critical junctions to coordinate key homeostatic functions such as autophagy and cellular metabolism. AMPK downregulates mTORC1 activity through multiple mechanisms [49]. It phosphorylates and activates tuberous sclerosis complex 2 (TSC2) [50], a key inhibitor of mTORC1, while also phosphorylating and inhibiting Raptor, a crucial mTORC1 subunit [51]. Conversely, mTOR exerts a suppressive effect on autophagy, partly by phosphorylating ULK1, thereby inhibiting its activity [45]. Consequently, AMPK facilitates autophagy through two pathways: activating ULK1 directly and counteracting the inhibitory influence of mTORC1 on ULK1. This dual regulation highlights ULK1 as a central point of interaction where AMPK and mTOR exert opposing influences on a shared metabolic pathway [52]. By targeting overlapping regulatory nodes, these two pathways integrate signals to fine-tune cellular responses, emphasizing the complexity of their interaction. AMPK activation and signaling pathways are shown in Fig. 2 (Ref. [28, 37, 38, 42, 43, 44, 45, 47, 48, 50, 51, 53, 54, 55, 56, 57, 58, 59, 60]).
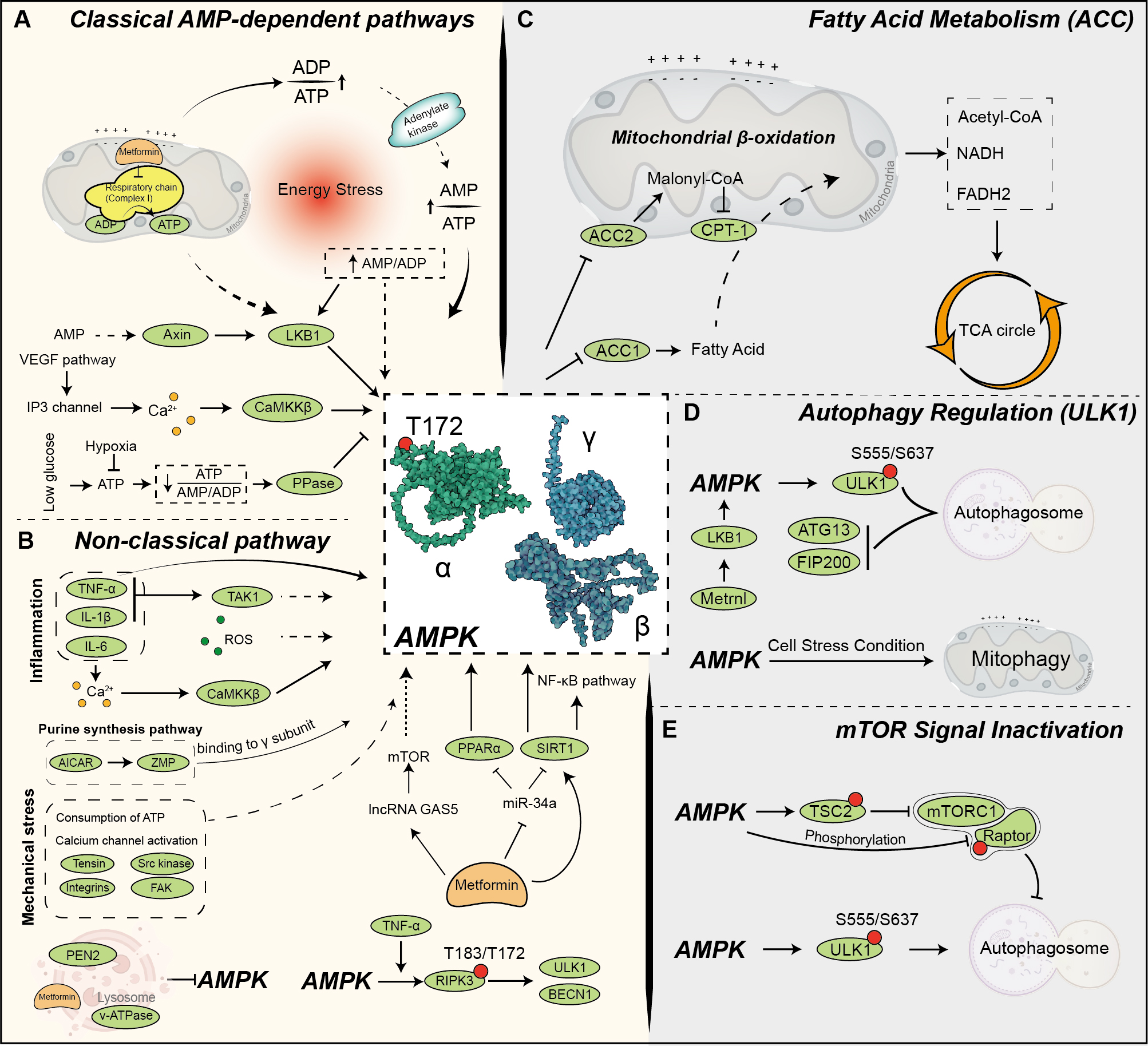 Fig. 2.
Fig. 2.
AMPK activation and signaling pathways. (A) AMPK is activated
through multiple mechanisms, integrating metabolic, stress, and pharmacological
signals to regulate cellular energy homeostasis. Under metabolic stress, AMPK
activation is primarily triggered by an increased AMP/adenosine triphosphate
(ATP) or adenosine diphosphate (ADP)/ATP ratio, indicating cellular energy
depletion [28]. This activation is mediated by upstream kinases such as liver
kinase B1 (LKB1), calcium/calmodulin-dependent protein kinase kinase beta
(CaMKK
Iron is an essential trace element in living organisms and participates in a variety of key physiological processes, including the production of hemoglobin and myoglobin, the maintenance of mitochondrial function, the regulation of cytochrome enzyme activity, and DNA synthesis [61]. After intestinal absorption, a portion of iron is stored as ferritin, while the rest enters the bloodstream via the ferroportin (FPN) and is transported to various organs with the help of transferrin (TF) to fulfill its functions [62].
The maintenance of iron homeostasis in the body primarily depends on the liver-secreted hormone hepcidin [63]. When iron load increases (e.g., high iron stores or elevated serum iron) or during infections and chronic inflammation, hepcidin levels rise, binding to FPN, inducing its internalization and degradation, thereby reducing intestinal iron absorption and limiting excessive iron entry into circulation [64, 65]. Under hypoxic conditions, hypoxia-inducible factor (HIF) downregulates hepcidin expression, promoting iron release to meet the body’s oxygen demands [66]. Iron balance within the body is essential for numerous life activities, including erythropoiesis, muscle energy metabolism, cell cycle regulation, hormone synthesis, immune function, DNA replication and repair, brain development, aging modulation, and cytochrome formation. Iron deficiency can result in impaired erythropoiesis, reduced immune function, and disrupted energy metabolism, while iron overload may cause oxidative stress and tissue damage [67]. Iron homeostasis is a critical regulatory point for maintaining normal physiological functions, and its imbalance can have profound effects on health. Therefore, understanding the mechanisms of iron absorption and regulation is of great significance for maintaining health and treating iron-related diseases.
Ferroptosis is an iron-dependent form of cell death characterized by iron-dependent lipid peroxidation, enhanced oxidative stress, and depletion of antioxidant defenses [5, 62, 68]. This type of cell death is distinct from apoptosis, autophagy, and pyroptosis, exhibiting unique biochemical and morphological characteristics [67]. The occurrence of ferroptosis is mainly regulated by iron homeostasis, redox balance, and lipid metabolism, with its core mechanisms centered on lipid oxidation and oxidative stress [69, 70].
Lipid peroxidation plays a critical role in ferroptosis. When iron overload
occurs, levels of non-transferrin-bound iron (NTBI) increase, generating large
amounts of hydroxyl radicals (
Key enzymes such as acyl-CoA Synthetase Long Chain Family Member 4 (ACSL4) and lysophosphatidylcholine acyltransferase 3 (LPCAT3) play important roles in lipid oxidation by incorporating PUFA into phospholipids, enhancing the oxidizability of membrane lipids [70, 72]. The study found that LPCAT3, in collaboration with ACSL4 and Yes-associated protein (YAP), collectively determines the sensitivity of lung adenocarcinoma (LUAD) cells to ferroptosis. Overexpression of LPCAT3 and ACSL4 enhances LUAD cell susceptibility to ferroptosis, whereas knockout of these genes exerts the opposite effect. Therefore, the combined expression of LPCAT3, ACSL4, and YAP may serve as a potential indicator for assessing ferroptosis sensitivity in LUAD cells [73]. Studies have shown that when monounsaturated fatty acids (MUFAs) are supplied extracellularly, they are actively taken up by cells and catalyzed by ACSL3 to form acyl-CoA, which is subsequently incorporated into membrane phospholipids. The integration of MUFAs reduces the proportion of highly oxidizable PUFAs in membrane phospholipids. In contrast, ACSL4 preferentially facilitates the incorporation of PUFAs. This shift in membrane lipid composition decreases the susceptibility of the cell membrane to oxidative damage, reduces lipid peroxidation, and ultimately effectively inhibits ferroptosis [74]. Therefore, inhibiting ACSL4 or regulating LPCAT3 expression may be potential strategies for treating ferroptosis-related diseases [75].
Mitochondria are the primary source of intracellular ROS and play a central role in ferroptosis [76]. Ferroptosis is induced by an imbalance between enhanced oxidative stress and depleted antioxidant system function. Oxidative stress plays a crucial role in the excessive peroxidation of PUFAs, leading to the accumulation of lipid peroxides and ultimately triggering cell death. The intracellular antioxidant system, including glutathione peroxidase 4 (GPX4) and coenzyme Q10 (CoQ10), plays a vital role in inhibiting lipid peroxidation and preventing ferroptosis. When these antioxidant defense mechanisms fail or are inhibited, cells become more susceptible to ferroptosis. Therefore, a close relationship exists among oxidative stress, the antioxidant system, and ferroptosis, and modulating these systems may provide new therapeutic strategies for related diseases [62].
GPX4 is a lipid peroxidase that reduces lipid peroxides to non-toxic lipid alcohols, thereby preventing the formation of iron-dependent ROS derived from lipid peroxidation. When GPX4 function is inhibited, the accumulation of lipid peroxides leads to the induction of ferroptosis. Therefore, GPX4 plays a crucial role in preventing lipid peroxidation, maintaining cell membrane integrity, and regulating ferroptosis [77]. System Xc– is a cystine/glutamate antiporter that imports cystine into cells, which is essential for the synthesis of glutathione (GSH). GSH is a critical antioxidant that helps maintain the cell’s redox balance and protects against oxidative damage. The decline in GSH levels compromises the activity of GPX4, leading to the accumulation of lipid peroxides and subsequently promoting ferroptosis. This further underscores the critical role of the System Xc–/GSH/GPX4 axis in the regulation of ferroptosis [5]. Therefore, enhancing GSH synthesis or preserving GPX4 activity may be crucial pathways for mitigating ferroptosis.
AMPK regulates lipid metabolism in a variety of ways, including phosphorylating key substrates, reducing lipid storage, promoting fatty acid oxidation, and inhibiting fatty acid and cholesterol synthesis. The synthesis of fatty acids and cholesterol both rely on acetyl-CoA, a core substance in the interplay between free radicals and lipid metabolism during ferroptosis. Studies indicate that AMPK regulates ferroptosis via the ACC signaling pathway, playing a significant role in nonalcoholic fatty liver disease (NAFLD) and diabetes-related conditions [28].
Guo et al. [78] found that liraglutide alleviates type II
diabetes-associated NAFLD and inhibits ferroptosis by activating the AMPK/ACC
pathway. Similarly, Zhang et al. [79] found that gallic acid, a plant
compound, alleviated NAFLD by regulating lipid metabolism and mitochondrial
function through the AMPK-ACC-peroxisome proliferator-activated receptor alpha
(PPAR
Iron overload induces oxidative stress, with non-transferrin-bound iron (NTBI)
being its main mediator. When the binding capacity of serum transferrin is
saturated, NTBI forms and penetrates cell membranes, inducing free radical
generation and cellular damage [81]. The study shows that AMPK activation through
energy stress effectively inhibits ferroptosis, demonstrating significant
therapeutic effects, especially in pathological conditions such as
ischemia-reperfusion injury (IRI) [82]. AMPK inhibits the biosynthesis of PUFA
and other fatty acids by phosphorylating ACC, reducing lipid peroxidation and
preventing ferroptosis. Metformin, a first-line drug for diabetes, not only halts
the progression of NAFLD but also alleviates hepatic iron overload (HIO) and
ferroptosis via the AMPK-FPN pathway [83]. Yue et al. [83] demonstrated
that metformin activates AMPK to reduce lysosomal degradation of FPN,
upregulating its expression, thereby alleviating iron overload and associated
cellular damage. This suggests that targeting AMPK effectively regulates
oxidative stress and improves ferroptosis-related pathological conditions.
Moreover, iron overload is a significant risk factor for diabetes. High dietary
iron intake increases diabetes risk, while iron overload leads to
Reprogramming of energy metabolism is a hallmark feature of cancer, with profound implications for tumorigenesis, progression, and treatment [85]. As a transducer of cellular energy status, AMPK participates in tumor metabolic adaptation by regulating metabolic pathways. Studies have shown that increased AMPK expression and activity are associated with improved survival rates in various tumor types, suggesting its critical role in cancer [86]. For example, mutations in tumor suppressor genes (e.g., phosphatase and tensin homolog (PTEN), LKB1, p53) and oncogenes (e.g., rat sarcoma viral oncogene (RAS), MYC proto-oncogene, bHLH transcription factor (MYC)) often result in decreased AMPK activity, exacerbating metabolic imbalances in tumor cells [87, 88].
Li et al. [89] investigated the role of AMPK in ferroptosis within renal cancer cells. They discovered that energy stress activates AMPK, which in turn modulates the janus kinase 2 (JAK2)/signal transducer and activator of transcription 3 (STAT3)/p53 signaling pathway. This modulation leads to the downregulation of GPX4, a key inhibitor of ferroptosis, thereby promoting ferroptotic cell death. These findings highlight a significant link between AMPK activation and ferroptosis in renal cancer, suggesting that targeting this pathway could offer new therapeutic strategies. Liu et al. [9] investigated the role of TP53-induced glycolysis and apoptosis regulator (TIGAR) in ferroptosis resistance in colorectal cancer (CRC) cells and found that TIGAR modulates ROS levels to activate AMPK, which subsequently upregulates stearoyl-CoA desaturase 1 (SCD1) expression. This regulation maintains lipid homeostasis and enhances resistance to ferroptosis. Notably, inhibition of TIGAR or SCD1 significantly increases ferroptosis sensitivity, highlighting the ROS/AMPK/SCD1 pathway as a key mechanism underlying ferroptosis resistance in colorectal cancer. Schneider et al. [8] find that AMPK plays a crucial role in pancreatic ductal adenocarcinoma (PDAC) survival and metastasis by promoting ferroptosis resistance through metabolic regulation. Inhibiting AMPK can disrupt tumor metabolic defenses, enhancing ferroptosis sensitivity and providing a potential therapeutic approach for pancreatic cancer treatment. A study [90] investigated the effects of 3,5-di-O-caffeoylquinic acid (3,5-diCQA) on colorectal cancer cells. They found that 3,5-diCQA induces ROS production, which activates the AMPK pathway and inhibits mTOR signaling. This leads to mitochondrial dysfunction and promotes ferroptosis, thereby inhibiting cancer cell proliferation. These findings suggest that targeting the ROS/AMPK/mTOR axis with compounds like 3,5-diCQA could be a potential therapeutic strategy for colorectal cancer.
AMPK enhances tumor cell antioxidant capacity by upregulating the pentose phosphate pathway (PPP), promoting nicotinamide adenine dinucleotide phosphate (NADPH) production, and regulating glutamine and folate metabolism. This metabolic adaptation helps cancer cells resist oxidative stress and improve survival rates, especially in harsh microenvironments [91]. Regulation of ferroptosis has potential value in cancer immunotherapy, as inducing ferroptosis or increasing tumor sensitivity to ferroptosis can enhance the efficacy of immunotherapy. For example, degradation of GPX4 not only promotes ferroptosis but also significantly enhances antitumor immune responses [91, 92]. Additionally, non-GPX4-dependent pathways (e.g., Smad ubiquitination regulatory factor 2 (SMURF2)-mediated Glutathione S-transferase P1 (GSTP1) degradation) can also accelerate tumor cell ferroptosis [93]. These studies indicate that ferroptosis is a critical mechanism for regulating tumor metabolism and immune responses, offering new insights for optimizing cancer treatment strategies.
AMPK is not only a key molecule in metabolic regulation but also acts as a tumor
suppressor involved in immune modulation [94]. Solute carrier family 7 member 11
(SLC7A11) is a major upstream regulator of ferroptosis and is closely associated
with tumor cell resistance to ferroptosis and drug resistance [95]. AMPK inhibits
the activity of SLC7A11 by phosphorylating BECN1, a key regulator with distinct
roles in cell ferroptosis [96, 97]. Studies have found that overexpression of
SLC7A11 drives the development of various cancers (e.g., liver [98], gastric
[99], and lung cancer [100]), while its downregulation significantly increases
tumor cell sensitivity to ferroptosis [6, 101, 102]. Feng et al. [103]
found that low doses of decitabine (DAC) and the ferroptosis inducer
RAS-selective lethal 3 (RSL3) synergistically promote ferroptosis in acute
myeloid leukemia (AML) cells. DAC increases the expression of MAGE family member
A6 (MAGEA6) by reducing its promoter methylation, leading to the degradation of
AMPK. This degradation results in decreased expression of SLC7A11 and reduced
GPX4 enzyme activity, thereby enhancing ferroptosis. These findings suggest that
targeting the MAGEA6-AMPK-SLC7A11-GPX4 signaling pathway could be a potential
therapeutic strategy for AML. Zhong et al. [104] demonstrated that under
energy stress, the AMPK/Forkhead box O3 (FoxO3a) signaling pathway is activated,
suppressing the expression of ferroptosis inducers (e.g., erastin)-related genes
and alleviating lipid peroxidation and mitochondrial dysfunction. Additionally,
FoxO3a activators (e.g., trifluoperazine (TFP)) reduce cerebral
ischemia-reperfusion (CIR) injury by downregulating hypoxia inducible
factor-1 (HIF-1
A study [105] investigating the role of hydroxycarboxylic acid receptor 1 (HCAR1)/monocarboxylate transporter 1 (MCT1) in regulating ferroptosis in tumor cells found that lactate activates HCAR1/MCT1, leading to AMPK activation and subsequent upregulation of SCD1. This process maintains lipid homeostasis, thereby enhancing resistance to ferroptosis. Notably, inhibition of HCAR1/MCT1 or SCD1 increases ferroptosis sensitivity, indicating that the lactate-mediated AMPK-SCD1 pathway plays a crucial role in ferroptosis resistance in tumor cells.
Wang et al. [106] investigated the role of arachidonate 5-lipoxygenase (ALOX5) in regulating ferroptosis in melanoma through the AMPK/mTOR signaling pathway. The results of the study suggest that ALOX5 activation enhances AMPK signaling and subsequently, inhibits mTOR, promoting autophagy-dependent ferroptosis. This mechanism underscores AMPK as a key regulator in ferroptosis-related cancer cell death. By linking metabolic stress and lipid peroxidation with autophagy and ferroptosis, the research highlights a potential therapeutic target in melanoma. Modulating the ALOX5-AMPK-mTOR axis may offer new avenues for cancer treatment by enhancing ferroptosis sensitivity and overcoming tumor resistance.
These studies reveal AMPK’s potential as a tumor suppressor, highlighting its role in regulating ferroptosis-related factors (e.g., SLC7A11) and mitochondrial function, significantly impacting tumor metabolism, oxidative stress, and the immune microenvironment.
The ubiquitination of target proteins is a crucial preliminary step for the
proteasome system to exert its biological functions, playing a significant role
in ferroptosis homeostasis [107]. The study has shown that MAGEA6 promotes
AMPK
Regarding deubiquitinases (DUBs), ubiquitin-specific peptidase 10 (USP10)
specifically removes ubiquitin marks from AMPK
The role of ubiquitin-like modifications in maintaining tumor ferroptosis
homeostasis is equally important. The study suggests that AMPK phosphorylates
UFM1-specific ligase 1 (UFL1) at Thr536, inhibiting programmed cell death protein
1 (PD-1) ubiquitin-fold modifier 1 (UFM1) modification, thereby enhancing T-cell
activity and improving the tumor immune microenvironment [117]. In certain
cancers, MAGEA3 and MAGEA6 form a ubiquitin ligase complex with TRIM28 to
ubiquitinate and degrade the AMPK
Autophagy is a highly conserved evolutionary mechanism that maintains intracellular homeostasis through self-degradation processes [119], Autophagy and its regulation have been recognized as novel therapeutic targets in cancer treatment [120, 121]. AMPK is considered one of the key regulators of autophagy, triggering it by activating ULK1 (the autophagy initiation complex) or inhibiting the mTOR signaling pathway [45]. Studies show a complex interaction between autophagy and ferroptosis, with selective autophagy capable of inducing ferroptosis [122]. Autophagy plays a vital role in ferroptosis by selectively degrading iron-sulfur cluster proteins or iron transport proteins, thereby regulating intracellular iron levels and affecting ferroptosis sensitivity [123]. Meanwhile, AMPK further influences ferroptosis by regulating ferritinophagy (autophagy of ferritin). Ferritinophagy releases free iron by degrading ferritin, leading to lipid peroxidation and triggering ferroptosis. Studies have found that AMPK-activated autophagy accelerates iron accumulation in tumor cells through ferritinophagy, thereby enhancing the anticancer effects of ferroptosis [68].
Tang et al. [124] investigated the combined effects of olaparib and
arsenic trioxide (ATO) on platinum-resistant ovarian cancer cells. They found
that the combination treatment significantly inhibited cell proliferation and
colony formation, increased DNA damage, and enhanced apoptosis. Additionally, the
combined treatment increased lipid peroxidation, leading to ferroptosis.
Mechanistically, the combination activated the AMPK
Sun et al. [12] examined the effects of nicotinamide mononucleotide (NMN) on hepatocellular carcinoma (HCC) cells. They discovered that NMN treatment elevated nicotinamide adenine dinucleotide (NAD⁺) levels and activated the AMPK/mTOR signaling pathway, leading to increased autophagy and ferroptosis. This suggests that AMPK activation plays a crucial role in mediating NMN-induced cell death in HCC, highlighting the potential of targeting the AMPK pathway to induce ferroptosis as a therapeutic strategy against liver cancer.
While the interplay between autophagy and ferroptosis in cancer cells is intricate and context-specific. In healthy cells, autophagy serves as a defense mechanism, preventing the buildup of damaged proteins and organelles that could contribute to cancer onset [125]. Conversely, in certain scenarios, autophagy supports cancer cell proliferation and survival by supplying essential nutrients and energy to sustain their metabolic needs [125]. Notably, autophagy can facilitate ferroptosis in cancer cells by breaking down and recycling iron-storage proteins like ferritin. This process releases iron, which drives lipid peroxidation and increases oxidative stress, highlighting the dual and context-dependent roles of autophagy in cancer biology [125].
Recently, the role of the tumor microenvironment (TME) in tumor growth has garnered significant attention [126], as cancer cell metabolism is closely linked to its surrounding microenvironment. Iron, as an essential trace element, not only participates in oxygen transport and energy metabolism but also plays a critical role in the cancer microenvironment by influencing redox balance, with iron homeostasis imbalance being a key feature of the cancer microenvironment. Cancer cells exhibit a heightened demand for iron due to their accelerated proliferation and elevated synthetic and metabolic activities. To meet this requirement, iron uptake mechanisms in cancer cells are upregulated, while iron export is downregulated [127]. AMPK plays a key role in regulating iron metabolism by upregulating the expression of iron transport protein transferrin receptor 1 (TFR1) or iron storage protein ferritin heavy chain 1 (FTH1), helping cancer cells adapt to microenvironmental changes [128]. Transferrin synthesis provides an autocrine mechanism to maintain iron supply, promoting cancer cell growth. Overexpression of TFR1 is frequently observed in malignant tissues and correlates with poorer patient outcomes. Notably, suppressing TFR1 expression significantly impedes tumor progression and metastasis [129, 130]. Additionally, members of the six-segment transmembrane epithelial antigen of prostate (STEAP) protein family, commonly overexpressed in various cancers, enhance iron uptake [131, 132, 133, 134]. The study indicates that AMPK activation can enhance iron-dependent oxidative stress, further promoting ferroptosis under hypoxic conditions [104].
Liu et al. [135] explored the effects of Gboxin on cervical cancer cells in low-glucose environments. They discovered that Gboxin activates the AMPK pathway, leading to increased autophagy. This autophagic activity reduces p62 levels, which in turn diminishes nuclear factor erythroid 2-related factor 2 (NRF2) signaling, thereby decreasing the cells’ antioxidant defenses. The suppression of NRF2 enhances ROS accumulation, resulting in both apoptosis and ferroptosis. These findings suggest that targeting the AMPK-autophagy-NRF2 axis with Gboxin could be a promising therapeutic strategy for cervical cancer under nutrient-deprived conditions. Furthermore, ferroptosis triggers a pro-immunogenic tumor microenvironment, characterized by enhanced antigen presentation, increased immune cell infiltration, and T-cell activation, thereby amplifying anti-tumor immunity. These findings highlight AMPK as a crucial regulator of ferroptosis and suggest that nanoparticle-based modulation represents a promising dual therapeutic strategy: directly inducing ferroptosis in cancer cells while concurrently enhancing immune-mediated tumor clearance.
In the tumor microenvironment, macrophage status is closely related to iron metabolism. Emerging research highlights the role of innate immune cells, such as macrophages and neutrophils, in disrupting iron metabolism within cancer cells. In the tumor microenvironment (TME), these immune cells may act as sources of iron and iron-related proteins or secrete factors that stimulate signaling pathways governing iron metabolism in cancer, further exacerbating dysregulation [127]. AMPK reduces the activity of iron-export proteins (e.g., FPN1), allowing macrophages to retain more iron, thereby promoting their transformation into the antitumor M1 phenotype [128].
Non-coding RNAs (ncRNAs) are key regulators of cancer development and progression, especially in the ferroptosis and AMPK signaling pathways, which play complex and diverse roles. Numerous microRNAs (miRNAs) exert their regulatory influence by directly targeting key genes, such as AMPK or GPX4, thereby modulating ferroptosis and tumor cell survival [136]. For instance, microRNA-451 directly targets calcium-binding protein 39 (CAB39), a binding partner of LKB1, to regulate LKB1-AMPK signaling in glioblastoma cells [137]. Similarly, the long non-coding RNA (lncRNA) neighbor of BRCA1 gene 2 (NBR2) is upregulated by AMPK during energy stress and interacts with AMPK to enhance its activity [138].
It has been shown that miRNAs can promote iron metabolism in tumor cells through the glutamine degradation pathway. For example, miRNA-137 induces ferroptosis by downregulating solute carrier family 1 member 5 (SLC1A5), a key solute carrier family member, leading to cysteine deprivation and impaired glutathione (GSH) synthesis [139]. This disruption results in the activation of ferroptosis. Furthermore, Zhang and colleagues [140] reported that miRNA-522 secreted by cancer-associated fibroblasts could inhibit iron mutations in gastric cancer cells by targeting arachidonic acid 15-lipoxygenase (ALOX15), thereby preventing ROS accumulation. Additionally, lncRNA H19 plays a regulatory role in ferroptosis by influencing miRNA expression via the AMPK-mTOR signaling pathway, impacting the expression of key proteins related to iron metabolism [141]. LncRNAs are also implicated in modulating oxidative stress in cells, which contributes to ferroptosis [142]. Circular RNAs (circRNAs), another class of ncRNAs, modulate ferroptosis through their ability to act as miRNA sponges, thereby indirectly influencing AMPK activity or ferroptosis-related genes [143]. CircRNAs can also regulate gene expression by inhibiting the maturation of target RNAs, further influencing ferroptosis. For example, specific circRNAs have been shown to interfere with the transcriptional machinery, thereby altering the expression of genes critical to ferroptotic pathways [144].
Liang et al. [109] investigated the role of TRIM11 in non-small cell lung cancer (NSCLC). They found that TRIM11 enhances NSCLC cell proliferation by inhibiting ferroptosis through the activation of the AMPK pathway. This suggests that TRIM11-mediated AMPK activation suppresses ferroptosis, contributing to cancer cell growth. Targeting the TRIM11-AMPK axis may offer a potential therapeutic strategy for NSCLC.
In summary, ncRNAs, encompassing miRNAs, lncRNAs, and circRNAs, orchestrate a complex regulatory network that governs ferroptosis and AMPK signaling. Their ability to regulate oxidative stress, iron metabolism and tumor cell survival highlights their importance in cancer biology. These insights highlight lncRNAs as potentially therapeutic targets for the treatment of cancer, providing avenues for intervention through manipulation of iron metabolism and the AMPK pathway.
The tumor microenvironment (TME) is recognized as a complex and dynamic ecosystem, primarily composed of tumor cells, cancer-associated fibroblasts (CAFs), vascular endothelial cells, immune cells, and various stromal cells [145]. The TME is enriched with cytokines and chemokines that not only promote cellular energy metabolism but also play pivotal roles in intercellular signal transduction [145]. In recent years, the focus of cancer therapy research has shifted from directly targeting malignant tumor cells to intervening in the intricate interactions among components within the TME. Notably, tumor-derived exosomes (TDEs) have garnered considerable attention for their role as critical mediators of intercellular communication within the TME [146].
Exosomes are extracellular vesicles ranging from 30 to 150 nm in diameter, containing a variety of bioactive molecules such as proteins, diverse RNAs (including miRNAs and lncRNAs), and lipids. They participate in intercellular material exchange and signal transduction through paracrine mechanisms, playing significant roles in the pathogenesis of various diseases [147]. In cancer, exosomes contribute to angiogenesis, inhibit apoptosis, and enhance tumor cell resistance to radiotherapy and chemotherapy [148]. Current research indicates that exosomes modulate ferroptosis and tumor progression through several mechanisms:
(1) Exosome-mediated Chemoresistance and Ferroptosis Inhibition: Exosomes secreted by tumor cells and CAFs enhance chemoresistance in tumor cells by delivering specific molecular cargo. For instance, Zhang et al. [140] demonstrated that exosomal miR-522 derived from CAFs suppresses the activity of arachidonate 15-lipoxygenase (ALOX15), reducing the accumulation of lipid ROS. This inhibition of lipid peroxidation enhances chemoresistance and suppresses ferroptosis in tumor cells [140].
(2) miRNA-Containing Exosomes Regulating Ferroptosis and Tumor Cell Proliferation: miRNAs within exosomes play critical roles in ferroptosis regulation. Song et al. [149] reported that exosomal miR-4443 modulates the m6A modification of ferroptosis suppressor protein 1 (FSP1), inhibiting ferroptosis and promoting cisplatin resistance in non-small cell lung cancer (NSCLC) cells. Furthermore, miR-4443 holds potential as a biomarker for predicting the therapeutic response of NSCLC patients to cisplatin treatment [149].
(3) Ferroptosis and Immune Cell Interactions within the TME: Ferroptosis influences tumor progression by modulating immune cell function within the TME. Dai et al. [150] found that the KrasG12D mutant, a common oncogene in human cancers, is released from tumor cells undergoing autophagy-dependent ferroptosis via exosomes. These exosomes are subsequently internalized by macrophages, inducing a shift from the pro-inflammatory M1 phenotype to the pro-tumorigenic M2 phenotype, thereby accelerating tumor progression [151].
(4) Exosome-Induced Ferroptosis Inhibiting Tumor Progression: While exosomes often inhibit ferroptosis to promote chemoresistance, some studies highlight their role in inducing ferroptosis to suppress tumor growth. Jiang et al. [152] demonstrated that exosomal miR-144-3p negatively regulates the expression of zinc finger E-box-binding homeobox 1 (ZEB1), promoting ferroptosis and inhibiting the proliferation, migration, and invasion of osteosarcoma cells. The correlation among AMPK, ferroptosis, and tumors is shown in Fig. 3 (Ref. [14, 128, 140, 149, 152, 153, 154, 155, 156, 157, 158, 159, 160]).
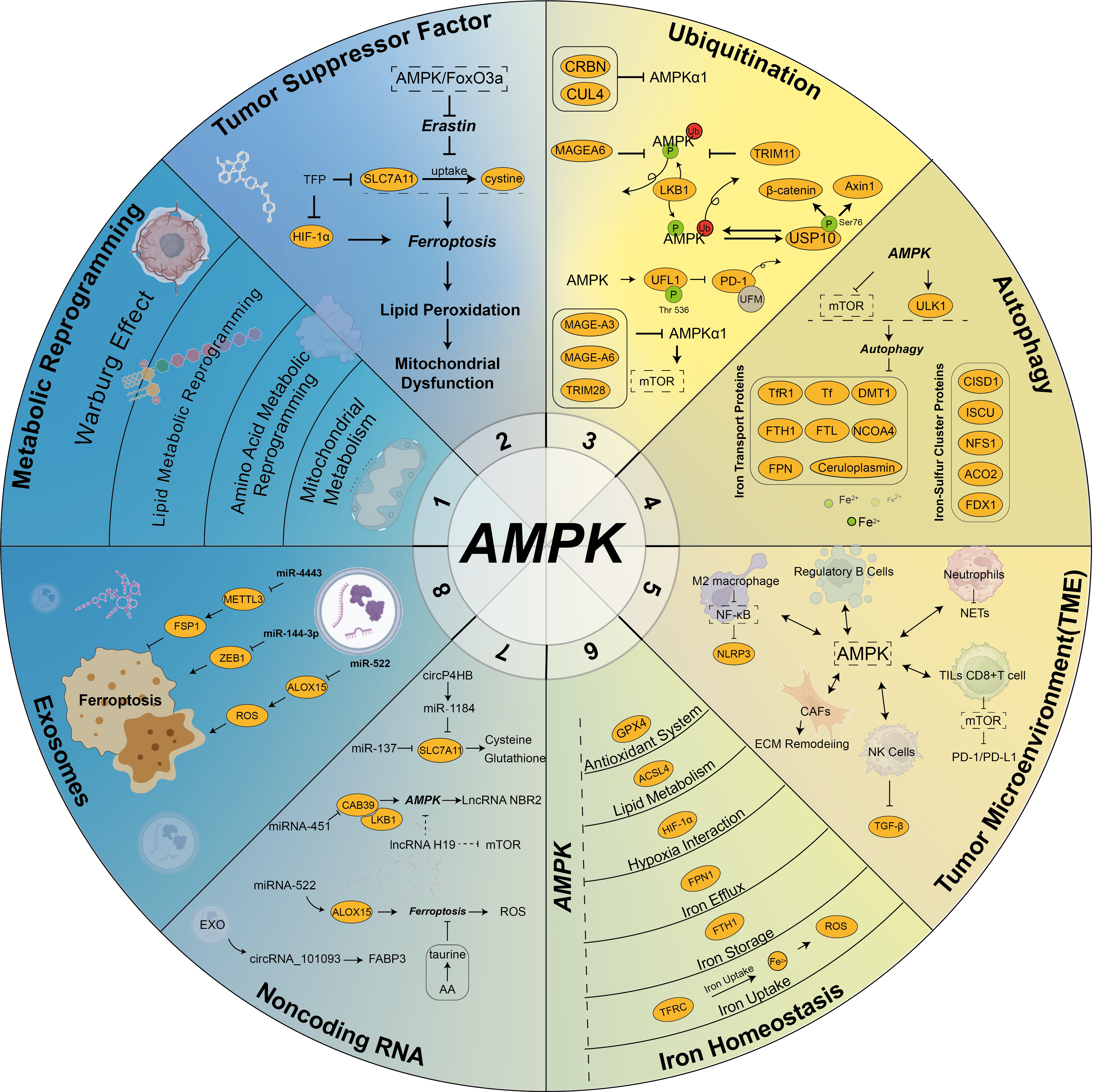 Fig. 3.
Fig. 3.
Interaction of AMPK, ferroptosis, and cancer. AMPK signaling,
as a central energy hub, plays a critical role in the regulation of ferroptotic
homeostasis and the tumor microenvironment in cancer cells. The key aspects
include: (1) AMPK regulates metabolic reprogramming in tumor cells [153, 154]; (2)
AMPK directly or indirectly influences the expression of tumor suppressor factors
[154]; (3) AMPK is modulated by ubiquitination and ubiquitin-like modifications
[155]; (4) AMPK serves as a critical nexus between ferroptosis and autophagy
[128]; (5) AMPK profoundly impacts the tumor microenvironment [14, 156]; (6) AMPK
is intricately linked to iron homeostasis [157]; (7) Non-coding RNAs regulate the
AMPK pathway [158, 159]; (8) Exosomes regulate AMPK activity via noncoding RNA,
thereby affecting ferroptosis [140, 149, 152, 160]. These interactions underscore
the multifaceted role of AMPK in ferroptosis and cancer biology. SLC7A11, Solute
carrier family 7 member 11; FSP1, ferroptosis suppressor protein 1; ZEB1, zinc
finger E-box-binding homeobox 1; ALOX15, arachidonic acid 15-lipoxygenase; TFRC,
transferrin receptor 1; FTH1, ferritin heavy chain 1; FPN, ferroportin;
ACSL4, Long-chain acyl-CoA synthetase 4; GPX4, glutathione peroxidase 4; CAFs,
cancer-associated fibroblasts; TFR1, transferrin receptor 1; NCOA4, nuclear
receptor coactivator 4; MAGEA6, MAGE family member A6; METTL3, methyltransferase
like 3; EXO, exosomes; FABP3, fatty acid binding protein 3; CAB39,
calcium-binding protein39; NBR2, neighbor of BRCA1 gene 2; NLRP3, NLR family
pyrin domain containing 3; NETs, neutrophil extracellular traps; ECM,
extracellular matrix; TGF-
Numerous studies have established that AMPK suppresses tumorigenesis and disease progression by regulating inflammatory and metabolic circuits [161]. However, AMPK may also advance tumor growth due to its capacity to undermine antitumor immunity [162]. Moreover, AMPK activation fosters ferroptosis through autophagy-dependent processes and p53-mediated signaling [14]. Incorporating these intertwined pathways into diagnostic frameworks may facilitate earlier detection of pathological states and better stratify patients for targeted interventions, thereby refining therapeutic approaches in oncology and improving clinical outcomes.
Ferroptosis is a unique form of regulated cell death (RCD) defined by the iron-dependent accumulation of lethal lipid peroxides in cell membranes [69]. Ferroptotic cells exhibit distinctive features, including abnormally small mitochondria with condensed membranes and reduced cristae [163]. It lacks organelle swelling. Additionally, it does not exhibit hallmark apoptotic features such as chromatin condensation, apoptotic body formation, or cytoskeletal disintegration [164]. Ferroptosis is closely tied to iron metabolism dysregulation, as reflected by altered levels of ferritin, transferrin saturation, and non-transferrin-bound iron (NTBI) [165]. The intracellular and mitochondrial ferrous iron (Fe2+) levels are elevated in ferroptotic cells or tissues and can be detected using biochemical assay kits, Prussian blue staining, or specialized probes [164]. Western blot analysis was employed to assess the protein expression levels regulated by ferroptosis-associated genes, such as ACSL4 and transferrin receptor 1 (TFRC), along with immunohistochemical or immunofluorescence assays to assess their signal intensity and spatial distribution, has been utilized to monitor ferroptotic responses both in vitro and in vivo [164]. Lipid peroxidation and its byproducts’ accumulation represent a crucial biomarker of ferroptosis [166]. The detection of lipid peroxidation end products using biochemical assays and enzyme-linked immunosorbent assay (ELISA) has been widely applied for assessing ferroptosis in both in vitro and in vivo samples [167]. Additionally, reduced GPX4 activity and expression correlate with increased ferroptosis susceptibility and may serve as indicators of therapeutic response [101, 168, 169]. The peroxiredoxin 3 (PRDX3) protein has recently been demonstrated to serve as a novel and specific marker of ferroptosis [170]. Due to its unique characteristics, a robust panel of biomarkers and functional assays has been developed to distinguish ferroptosis from other RCD types [62]. Assessing these biomarkers in tumor tissues, serum, or biofluids can improve diagnostic accuracy and provide insights into ferroptosis’s pathophysiological roles.
Ferroptosis has recently emerged as a promising avenue in cancer treatment
research. Preclinical evidence indicates that ferroptosis modulators, including
SLC7A11 inhibitors (e.g., erastin, imidazole ketone erastin (IKE)) and GPX4
inhibitors (e.g., JKE-1674), demonstrate strong antitumor efficacy in diverse
animal models [171, 172]. For example, Hsieh et al. [156] demonstrated
that zero-valent iron nanoparticles (ZVI-NP) remodel the immunosuppressive
microenvironment by inducing mitochondrial dysfunction and lipid peroxidation
while triggering iron metabolism degradation of NRF2 via AMPK/mTOR signaling in
lung cancer cells. This dual-use nanomedicine provides a novel approach to
combine iron metabolism and immunotherapy [156]. Furthermore, Jiang et
al. [173] designed biomimetic magnetic nanoparticles (Fe3O4-SAS@PLT) capable of
inducing tumor-specific ferroptosis and repolarizing macrophages from the M2
immunosuppressive phenotype to the antitumor M1 phenotype. These cutting-edge
methods boost cancer treatment specificity and introduce novel pathways for
integrating immunotherapy with ferroptosis [173]. Given the dual roles of
ferroptosis in both promoting and inhibiting cancer progression, the
identification and monitoring of ferroptosis-related pathological states within
the body are of paramount importance. Enhancing the specificity and sensitivity
of in vivo detection methods for ferroptosis remains a significant
challenge. Moreover, while several compounds have been identified that can induce
ferroptosis, issues persist regarding their target specificity and low
bioavailability [162]. Therefore, future research should focus on developing more
precise and safer ferroptosis-inducing agents that selectively target cancer
cells. The tumor suppressor protein p53 is traditionally known for its role in
inhibiting cancer progression [14]. AMPK plays an important role in the
activation of p53 by phosphorylation [174]. On the other hand, AMPK is activated
by LKB1, and under this sustained activation state, AMPK can induce p53-related
cellular senescence [174, 175]. P53 has been demonstrated to induce ferroptosis in
cancer through a GPX4-independent pathway [176, 177]. However, a study revealed
that the simultaneous deletion of AMPK
Future research should prioritize: (1) Designing nanomaterials for localized induction of ferroptosis or precise iron ion delivery. (2) Investigating the interactions between ferroptosis and immune responses to optimize combination therapies. (3) Developing targeted therapies that exploit the heightened ferroptosis sensitivity of cancer cells, followed by rigorous preclinical evaluation. (4) Establishing highly specific and sensitive ferroptosis detection methods suitable for clinical application. (5) Elucidating the upstream and downstream signaling pathways of AMPK-mediated ferroptosis in cancer and other diseases.
While some studies suggest that AMPK activation enhances ferroptosis by modulating oxidative stress and lipid metabolism, others indicate that AMPK promotes ferroptosis resistance through antioxidant pathways and metabolic adaptations. This context-dependent duality underscores the need for further investigation into the AMPK-ferroptosis axis. Despite recent progress, many knowledge gaps remain regarding the precise molecular mechanisms through which AMPK differentially regulates ferroptosis. The intricate interplay between AMPK, p53, and key ferroptosis regulators such as GPX4 and SLC7A11 requires further elucidation. Additionally, optimizing ferroptosis-based therapies remains a major challenge due to issues of target specificity, bioavailability, and tumor heterogeneity.
Given its fundamental role in cancer metabolism, AMPK represents a crucial target for ferroptosis regulation. Further studies are essential to refine AMPK-targeted ferroptosis strategies, improve detection methods, and explore clinical applications of AMPK-modulating agents in ferroptosis-based cancer therapy. A deeper understanding of the AMPK-ferroptosis-cancer axis may pave the way for more effective, personalized cancer treatments, ultimately improving patients.
ACC, acetyl-CoA carboxylase; ACO2, aconitase 2; ACSL4, long-chain acyl-CoA synthetase 4; ADP, adenosine diphosphate; AICAR, 5-Aminoimidazole-4-carboxamide1-
TZ and JZ conceptualized the manuscript, defining its theme, direction, and framework. XW contributed to the conception and design of the manuscript. TZ and XW drafted the manuscript. PGA reviewed and revised the manuscript for language, grammatical structure, and logical coherence, collected relevant literature, and offered valuable suggestions for improving certain aspects of the manuscript design. PF created all the figures. PF and JZ contributed to the review, editing, and revision of the manuscript. All authors contributed to editorial modifications of the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the study.
Not applicable.
We also extend our gratitude to Ying Che for her contribution to the creation of the mechanism diagram. The figures were created using Adobe Illustrator and BioRender.
This research received no external funding.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.