



 , Qilong Li 1,2, Yiwei Zhang 1,2, Ning Jiang 1,2, Qijun Chen 1,2,*
, Qilong Li 1,2, Yiwei Zhang 1,2, Ning Jiang 1,2, Qijun Chen 1,2,*
1 Key Laboratory of Livestock Infectious Diseases, Ministry of Education, Key Laboratory of Ruminant Infectious Disease Prevention and Control (East), Ministry of Agriculture and Rural Affairs, College of Animal Science and Veterinary Medicine, Shenyang Agricultural University, 110866 Shenyang, Liaoning, China
2 Research Unit for Pathogenic Mechanisms of Zoonotic Parasites, Chinese Academy of Medical Sciences, Shenyang Agricultural University, 110866 Shenyang, Liaoning, China
Abstract
Parasitic diseases, caused by a diverse array of parasites, remain a substantial threat to global health. Toll-like receptor 3 (TLR3) represents a pivotal element in the innate immune system, distinguished by an ability to signal via the TIR-domain-containing adapter-inducing interferon-β (TRIF)-dependent pathway upon detecting pathogen-derived double-stranded RNA (dsRNA), exosomal RNA (exoRNA), and long non-coding RNA (lncRNA). Predominantly localized on endosomal membranes, TLR3 is extensively expressed in neurons, immune cells, fibroblasts, and epithelial cells. Upon activation, TLR3 engages adaptor molecules such as TRIF, facilitating the phosphorylation of TANK-binding kinase 1 and the subsequent activation of interferon regulatory factors. This signaling cascade triggers the production of type I interferons (IFN-α/β) and proinflammatory cytokines such as interleukin (IL)-6, IL-8, IL-12, and tumor necrosis factor-alpha, which are crucial for effective immune defense against infections. Recent findings highlight the essential role of TLR3 in parasitic infections by detecting nucleic acids from damaged cells to activate dendritic and natural killer cells. TLR3 also functions with other receptors, such as TLR2 and TLR4, to enhance cytokine production and improve parasite clearance. However, TLR3 overactivation can induce excessive, harmful inflammation and tissue damage, highlighting its dual role in balancing immune defense. This review comprehensively examines the TLR3 signaling pathway and its multifaceted role in various parasitic infections, including those caused by Plasmodium spp., Leishmania spp., Clonorchis sinensis, Schistosoma japonicum, Trichinella spiralis, and Neospora caninum.
Keywords
- Toll-like receptor 3
- parasitic disease
- immune defense
- inflammation
Parasitic infections pose a substantial global health burden, affecting millions of individuals annually and causing significant morbidity and mortality, particularly among vulnerable populations such as children and pregnant women [1, 2, 3, 4]. These infections are caused by a diverse range of pathogens, including Plasmodium spp., Leishmania spp., Clonorchis sinensis, Schistosoma japonicum, Trichinella spiralis, and Neospora caninum [5, 6, 7, 8, 9, 10]. A key factor in the persistence and severity of these diseases is the ability of these parasites to evade host immune surveillance, ensuring their survival and replication within the host [11]. The host’s innate and adaptive immune systems are critical for combating parasitic infections, maintaining immune homeostasis by recognizing and eliminating pathogens [12, 13]. Among the pattern recognition receptors of the innate immune system, the Toll-like receptor (TLR) family plays a pivotal role in initiating immune responses upon detecting pathogen-associated molecular patterns [14, 15]. TLRs activate downstream signaling pathways that lead to the production of cytokines and other inflammatory mediators essential for effective immune responses [16].
Toll-like receptor 3 (TLR3), a key member of the TLR family, uniquely signals
through the TIR-domain-containing adapter-inducing interferon-
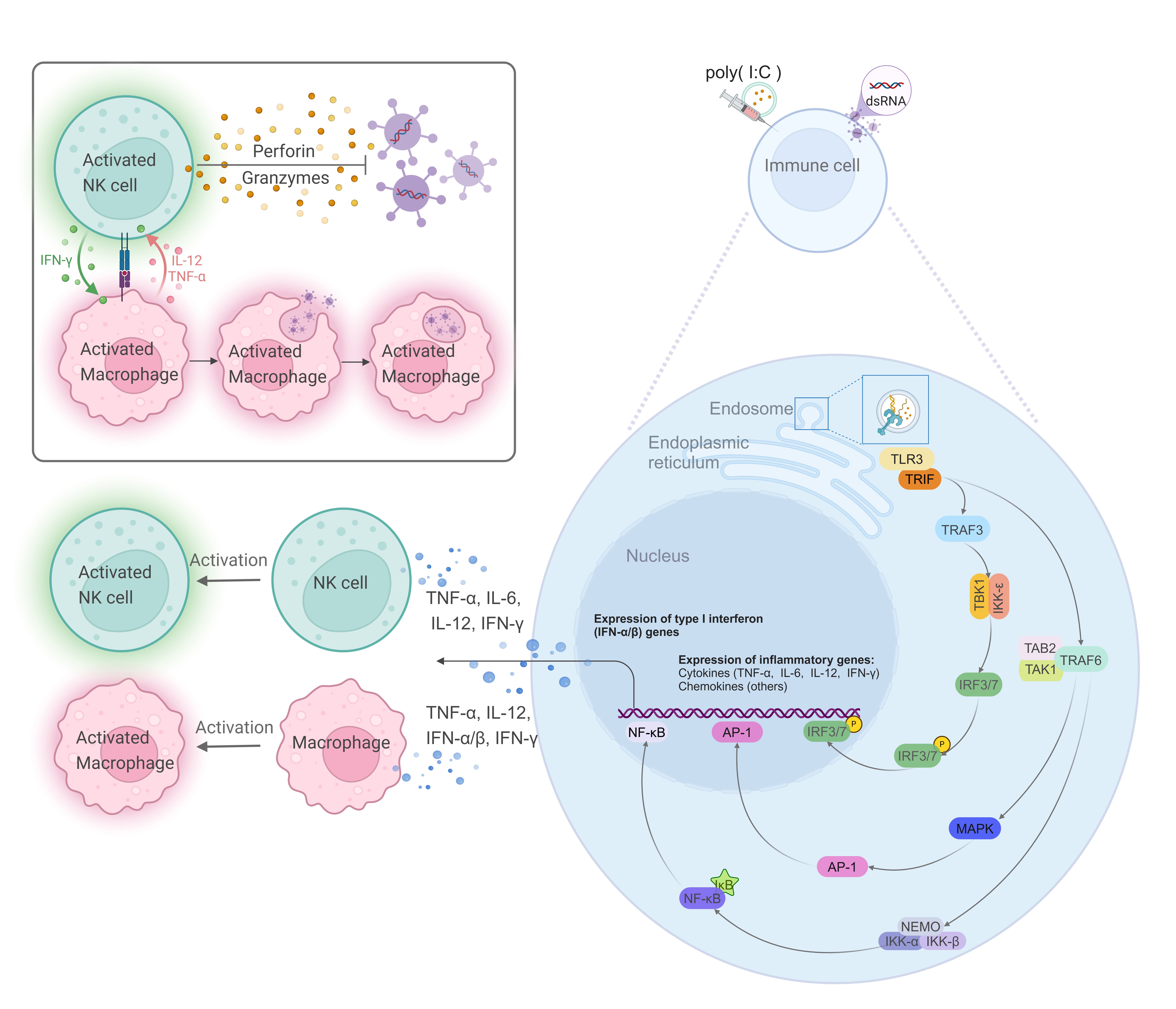 Fig. 1.
Fig. 1.
The signaling pathway of TLR3 in responses to double-stranded
RNA (dsRNA). TLR3, located on endosomal membranes, recognizes dsRNA or the
synthetic analog poly(I:C), thereby activating the TLR3 signaling pathway. The
adaptor protein TRIF is recruited, which subsequently activates TRAF3 and TRAF6.
TRAF3, in conjunction with TBK1 and IKK-
The present review provides a comprehensive overview of the TLR3 signaling pathway and its critical role in various parasitic infections. It aims to identify potential targets for immunotherapy and vaccine development to improve host resistance and global health outcomes.
In parasitic infections, the timely recognition and activation of the host immune system are critical for regulating parasite proliferation and determining disease outcomes. Emerging evidence underscores the indispensable role of TLR3 in the recognition and regulation of parasitic infections [8, 46, 47, 51, 52]. TLR3 can mediate immune responses to parasitic infections through several distinct mechanisms. Parasite-derived nucleic acids are recognized by TLR3, which then activates downstream immune signaling pathways and induces a strong pro-inflammatory response [22, 37, 53]. This immune activation not only aids in parasite clearance but also regulates immune homeostasis [36]. The first mechanism involves TLR3 recognizing parasite-derived exoRNA released from damaged host cells, promoting cytokine secretion, and enhancing host defense, which is critical for initiating an effective immune response against parasitic infections. The second mechanism is the regulation of immune cell functions by TLR3, which involves regulating dendritic cells (DCs) and NK cells, boosting their activity, and enhancing the host’s resistance to parasites. The third mechanism is the synergistic action of TLR3 with other TLRs, which amplifies immune responses by interacting with these receptors to enhance immune signaling, ultimately facilitating more effective parasite clearance. Understanding these mechanisms is essential for elucidating immune regulation in host-parasite interactions and developing related immunotherapies. These mechanisms will be further elaborated in the subsequent sections of the review.
Once exoRNA released by host cells damaged during parasitic infections is
recognized by TLR3, an immune response is initiated [22, 38]. Based on current
experimental evidence, TLR3 may recognize parasite-derived extracellular RNA
during malaria infection, thereby triggering an immune response in the host. This
recognition induces the production of significant levels of pro-inflammatory
cytokines, including IFN-
Another crucial role of TLR3 in parasitic infections is the modulation of host
immune cell functions [36]. Activation of TLR3 by pretreatment with the agonist
poly(I:C) in Leishmania infections promotes a T helper 1 (Th1)-type
immune response and enhances antigen presentation, thereby improving host
resistance to the parasite [56]. Similar observations have been reported in
Plasmodium falciparum-infected BALB/c mice, where poly(I:C) pretreatment
activates TLR3, enhances the Th1 response, and induces elevated IgG antibody
levels as well as IFN-
TLR3 plays a crucial role in maintaining host immune homeostasis, while also
enhancing immune responses through interactions with other TLRs, thereby
amplifying overall immune activity [61]. During parasitic infections,
cross-regulation between TLR3 and other TLRs, such as TLR2, TLR4, and TLR7, is
essential for orchestrating an effective immune response. In Schistosoma
japonicum (S. japonicum) infections, these TLRs contribute to the
activation of T, B, NK, and
In S. japonicum infections, TLR2, TLR3, TLR4, and TLR7 cooperate in a
tissue-specific manner, particularly within pulmonary lymphocytes, to boost
IFN-
Investigating the temporal dynamics of TLR3 activation and its interplay with other innate immune receptors could significantly advance our understanding of immune homeostasis in the context of persistent parasitic infections. A more profound understanding of the role of TLR may guide the development of innovative vaccine adjuvants, immune modulators, and therapeutic strategies, ultimately improving clinical outcomes for patients suffering from parasitic diseases. Through such targeted investigations, we can move closer to a paradigm in which immune-based therapies can effectively eradicate parasitic infections while minimizing adverse effects, thereby addressing a critical unmet need in global health.
TLR3 recognizes exoRNA, long non-coding RNA (lncRNA), or poly(I:C)
produced by damaged cells in response to parasitic infections (Fig. 2) to
initiate immune responses [22, 37, 66, 67, 68]. TLR3 activates multiple transcription
factors, such as IRF3, IRF7, and NF-
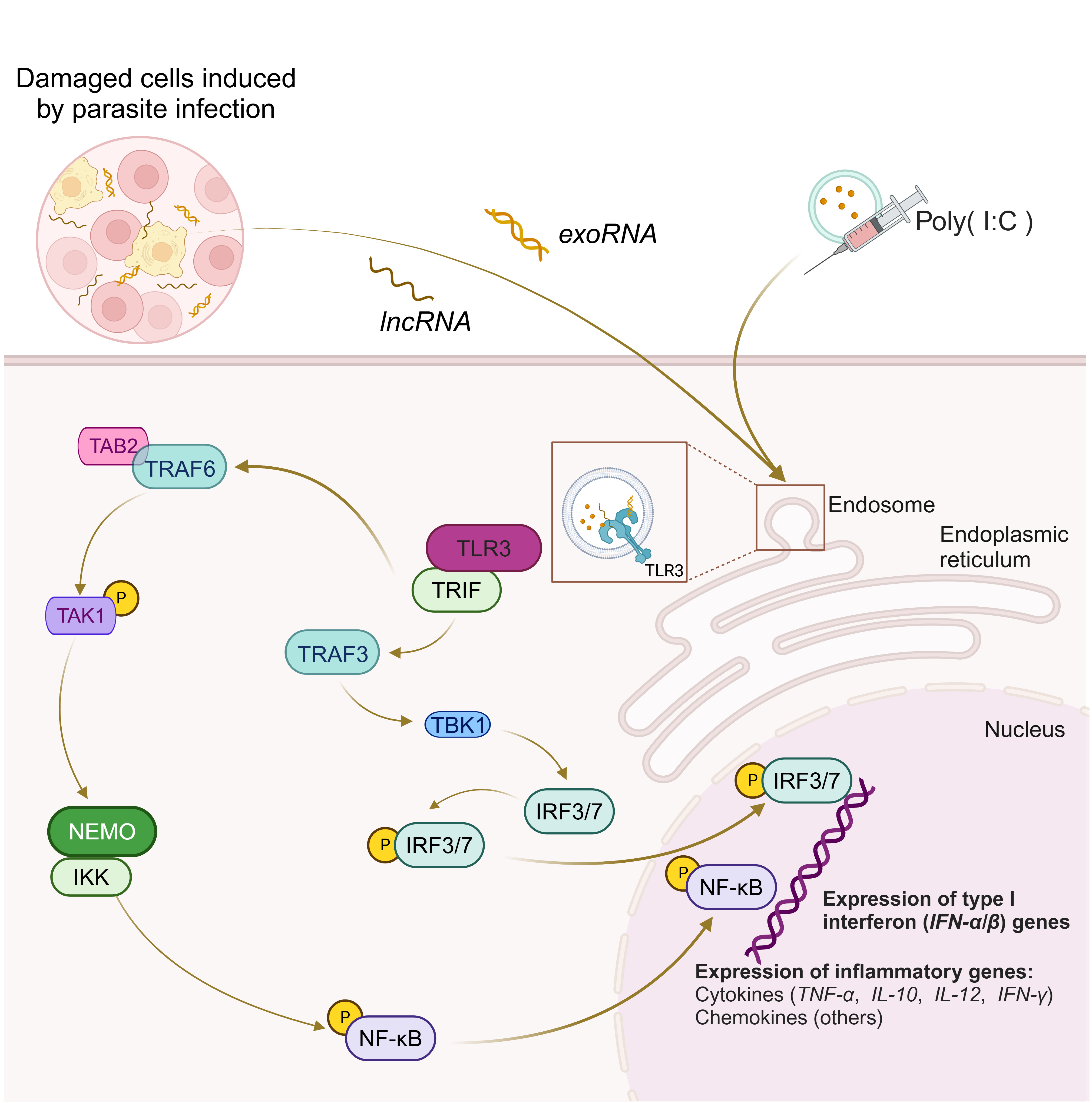 Fig. 2.
Fig. 2.
Signaling pathway of TLR3 in parasite infection. TLR3
recognizes activation of exoRNA, lncRNA or poly(I:C) produced by damaged cells
induced by parasite infection and generates various immune responses. Upon
activation of TLR3, recruitment of TRIF occurs, which initiates signal
transduction through its TIR. This activation subsequently leads to activation of
the serine/threonine kinase TBK1, which then phosphorylates IRF3/IRF7. Another
pathway of TLR3-TRIF is the recruitment of TAB2 via TRAF6 leading to
phosphorylation and activation of TAK1 itself. Phosphorylated TAK1 stimulates IKK
via NEMO, which in turn activates NF-
Malaria is caused by Plasmodium species, which are transmitted to vertebrates through the bite of female Anopheles mosquitoes [5, 80, 81]. Plasmodium infects hundreds of millions of people with malaria each year, with the vast majority of severe malaria cases concentrated in African countries, particularly Nigeria, the Democratic Republic of the Congo, Niger, and the United Republic of Tanzania [82]. Patients with malaria usually exhibit symptoms such as fever, chills, and acute anemia. In severe cases, malaria can be fatal, causing hundreds of thousands of deaths each year, with children and pregnant women being particularly vulnerable, making it a significant health threat [80, 83].
Understanding the interplay between TLR3 and immune responses is crucial for
elucidating the mechanisms through which the host combats Plasmodium
infection. Notably, TLR3 plays a key role in regulating Plasmodium
infection [46]. In wild-type (WT) mice, activation of the TLR3-TRIF signaling
axis induces the nuclear translocation of phosphorylated IRF3 and IRF7,
triggering an early surge of pro-inflammatory cytokines such as IFN-
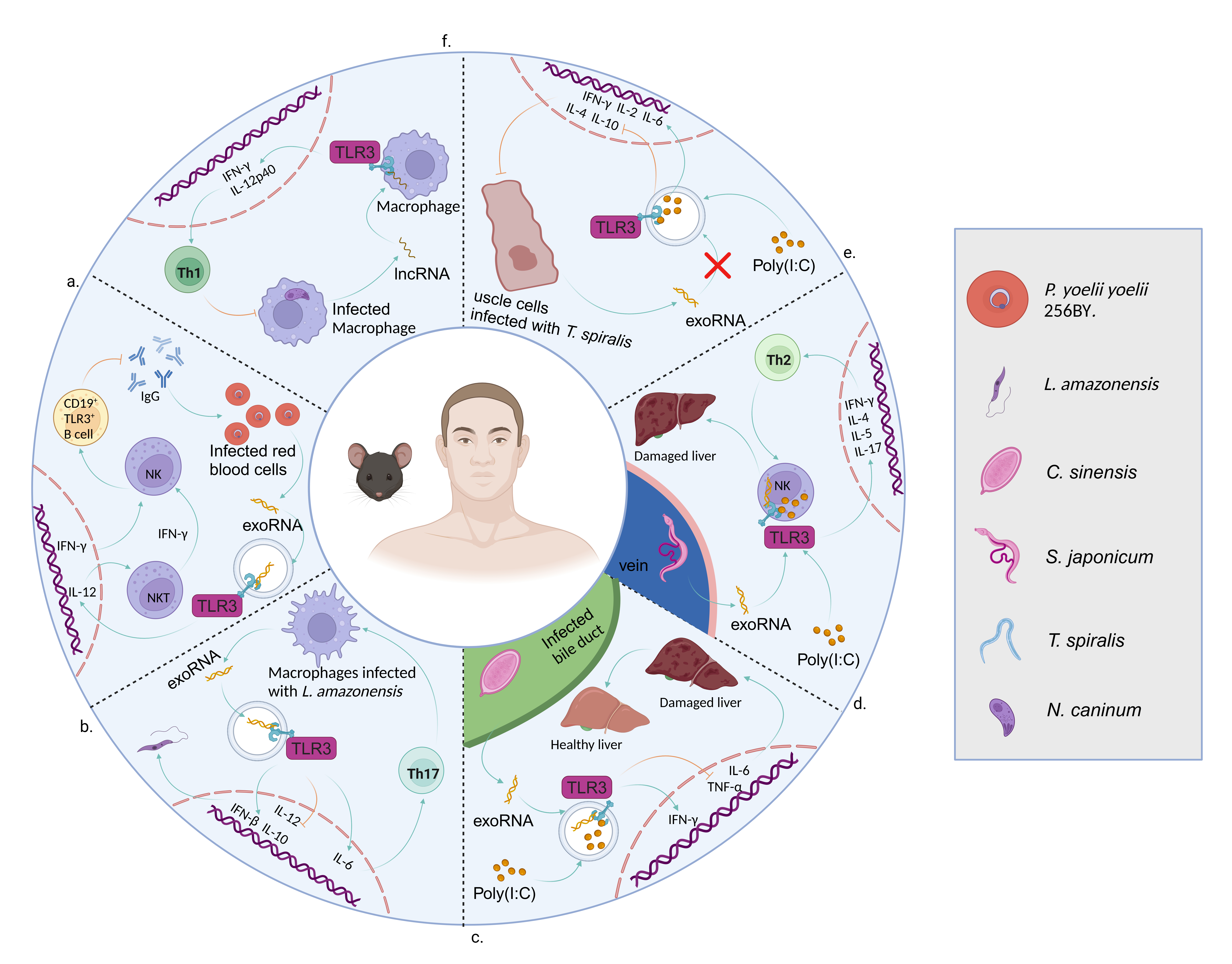 Fig. 3.
Fig. 3.
TLR3-mediated immune responses in parasite infections. (a)
ExoRNA released by plasmodium-infected red blood cells activates TLR3,
thereby promoting the transcription of cytokines such as IFN-
| Parasite | Detailed information | Reference | |
| Plasmodium yoelii yoelii 265 BY (P. yoelii yoelii 265 BY) | Host: | 6–8 weeks old C57BL/6jRj mice and TLR3-knockout mice | [46] |
| Study Organ: | Liver and Spleen | ||
| Cellular and Cytokine changes: | CD19+TLR3+ | ||
| Result: | Compared to WT mice, TLR3-knockout mice exhibited a significant increase in parasite-specific antibodies when infected with P. yoelii yoelii 265 BY. These mice demonstrated better control over the growth of Plasmodium and cleared the parasite earlier during infection. In WT mice infected with P. yoelii yoelii 265 BY, there was an elevated frequency of CD19+ and TLR3+ B cells, a decrease in overall IgG levels, and an increase in TNF- | ||
| Leishmania amazonensis (L. amazonensis) | Host: | 6–8 weeks old C57BL/6 mice and TLR3-knockout mice | [43] |
| Study Organ: | Skin | ||
| Cellular and Cytokine changes: | IFN- | ||
| Result: | This study concluded that L. amazonensis enhanced its survival within host macrophages by activating the TLR3 signalling pathway, promoting the expression of IFN- | ||
| Clonorchis sinensis (C. sinensis) | Host: | 6–8 weeks old C57BL/6 mice and TLR3-knockout mice | [47] |
| Study Organ: | Liver and Bile Ducts | ||
| Cellular and Cytokine changes: | IFN- | ||
| Result: | TLR3-knockout mice exhibited significantly lower body weights, higher mortality rates, and increased parasite loads compared to WT mice. The absence of TLR3 further exacerbated liver injury and inflammatory responses, leading to increased hepatic fibrosis in infected mice. | ||
| Schistosoma japonicum (S. japonicum) | Host: | C57BL/6 mice | [8, 90] |
| Study Organ: | Liver and Spleen | ||
| Cellular and Cytokine changes: | IL-4 | ||
| Result: | The percentage of cells producing IFN- | ||
| Trichinella spiralis (T. spiralis) | Host: | 6–8 weeks old female C57BL/6 mice with poly(I:C) treatment | [52] |
| Study Organ: | Spleen | ||
| Cellular and Cytokine changes: | IFN- | ||
| Result: | Compared to WT mice, treatment with poly(I:C) led to a significant increase in Th1-type cytokines, including IFN- | ||
| Neospora caninum (N. caninum) | Host: | 6–8 weeks old C57BL/6 mice, TRIF-knockout mice and TLR3-knockout mice | [44, 91] |
| Study Organ: | Spleen, Lung and Brain | ||
| Cellular and Cytokine changes: | IL-12p40 | ||
| Result: | When WT mice TLR3 recognize N. caninum, inducible IL-12p40 secretion promotes IFN- | ||
Note: activation, stimulation of secretion or increased expression
Leishmaniasis is caused by Leishmania spp., which is transmitted to humans, mammals (such as dogs and rats), and certain reptiles (such as lizards) through the bite of a sandfly [92]. Leishmaniasis is a zoonotic parasitic disease endemic to numerous regions, including parts of North America such as the United States and Canada, posing a significant risk to human health [93]. Leishmaniasis exhibits a range of clinical manifestations, from skin lesions to potentially fatal visceral involvement [92, 94].
The immune responses elicited during Leishmania infection are pivotal
in determining disease outcomes, particularly through mechanisms such as TLR3
signaling, which shapes cytokine profiles and regulates immune cell activity [45, 95]. TLR3 initiates a cascade of host immune responses by recognizing exoRNA
released from damaged cells following Leishmania infection [22, 96].
Infection with Leishmania amazonensis (L. amazonensis) disrupts
the host immune response and suppresses macrophage function [97, 98]. This
disruption includes alterations in cytokine production, such as restricted
IFN-
In conclusion, the complex immune modulation observed during Leishmania infection, particularly via TLR3-mediated pathways, underscores the parasite’s capacity to manipulate host immune responses to support its intracellular survival and propagate disease. Further research focusing on these immune pathways could provide crucial insights into developing novel therapeutic strategies to control leishmaniasis progression and severity.
Clonorchiasis is caused by Clonorchis sinensis (C. sinensis), a liver fluke from the family Opisthorchiidae, infecting humans or mammals through the consumption of undercooked freshwater fish containing encysted metacercariae [105]. This disease is prevalent mainly in parts of China and other East Asian countries [106]. Over 15 million people are, reportedly, infected with the parasite, with more than 80% of the infections occurring in China. Consequently, it is considered one of the most widespread and detrimental food-borne parasitic diseases in China [7, 107]. Infection with C. sinensis leads to various pathological changes such as epithelial cell proliferation, degeneration, liver inflammation, cholangitis, and bile duct obstruction. If left untreated, this long-term infection can result in bile duct fibrosis, liver cirrhosis, and even cholangiocarcinoma [108].
A recent study has highlighted the significant role of TLR3 activation in liver
fibrosis associated with C. sinensis, emphasizing its importance as a
target in modulating hepatic stellate cell activation and promoting liver
regeneration [47]. Activation of TLR3 using poly(I:C) promotes the regeneration
of damaged liver tissue due to C. sinensis infection [109]. TLR3 may
potentially detect exocytotic RNA released from host cells damaged by C.
sinensis infection [22, 110]. Compared to WT mice, TLR3-knockout mice exhibit
hyperactivation of pro-inflammatory pathways such as ERK, p38, AKT, and p65,
along with increased expression of cytokines IL-4, IL-6, and TNF-
In conclusion, TLR3 signaling plays a pivotal role in modulating the immune response and hepatic fibrosis during C. sinensis infection, with its activation demonstrating protective effects against liver damage. Future studies should aim to elucidate the dual role of TLR3 in liver diseases to better leverage its therapeutic potential for managing fibrosis and other liver pathologies.
Schistosoma japonicum is a dioecious zoonotic parasite in the family Schistosomatidae, causing schistosomiasis in 78 tropical and subtropical countries, particularly where medical resources are limited, and thus posing a major threat to human health and economic development [116, 117, 118]. The cercariae of S. japonicum are released into freshwater environments by their intermediate host, the Oncomelania snail, contaminating water sources. The end hosts are vertebrates, including humans, who become infected with schistosomiasis through exposure to this contaminated water [119]. The damage caused by S. japonicum to the host, particularly from parasitized eggs, can be severe and varies in magnitude [120, 121]. Schistosomiasis represents a significant public health challenge, causing chronic inflammation and organ damage that leads to genitourinary and intestinal complications. This, in turn, severely impairs the quality of life, especially in affected populations [122].
Upon S. japonicum infection, the host initiates a variety of immune
responses, with NK cells playing a crucial role. These cells, integral to the
innate immune system, respond rapidly to pathogens through cytotoxic mechanisms
and cytokine secretion [123]. Recent studies suggest that TLR3 detects exoRNA
released from damaged host cells during S. japonicum infection [53, 124]. In experimental models, S. japonicum-infected mice showed
increased secretion of IFN-
Trichinellosis is caused by Trichinella spiralis (T. spiralis), which belongs to the nematode order of the phylum Aschelminthes [125]. The life cycle of this species consists of three distinct phases: the myxo-larval, adult, and neonatal larval stages, all of which occur within a common host organism [126]. Poor dietary habits, such as consumption of raw or undercooked pork or wild game contaminated with trichinella larvae, can lead to T. spiralis infections in a wide range of mammals, including humans and pigs [127]. During the acute phase of infection, individuals commonly exhibit symptoms such as increased body temperature; these symptoms can persist for multiple weeks and potentially result in organ dysfunction [128]. In certain cases, the mortality rate of T. spiralis infection may soar up to 30% [129]. As of 2023, the health of people living in the African continent continues to be threatened by T. spiralis [130].
Building on the understanding of immune regulation induced by T.
spiralis, current research is delving into the underlying molecular mechanisms
through which the parasite evades the immune system, aiming to identify novel
molecular targets for therapeutic intervention against T. spiralis. This
pathogen modulates the host immune response by regulating the expression of TLRs,
thereby influencing cytokine expression at various stages of infection [131]. TLR
agonists have a wide range of therapeutic applications, including their use in
signal therapy to accelerate and enhance vaccine-specific immune responses [132].
TLR3, as a commonly used vaccine adjuvant, plays an important role in host
resistance to pathogen infection [133]. Although T. spiralis infection
induces the release of exoRNA from damaged host cells, this exoRNA either fails
to be recognized by TLR3 or the parasite successfully evades the host immune
response [22, 53, 134]. Notably, the administration of poly(I:C) results in
consistently elevated TLR3 expression and significantly increased levels of serum
IFN-
Neosporosis is caused by the protozoan parasite N. caninum, belonging to the phylum Apicomplexa. It primarily infects intermediate hosts like pigs, sheep, horses, cattle, and rabbits via oral or transplacental transmission, in addition to parasitizing definitive hosts such as dogs and other canids [135]. It is globally distributed and represents a major etiological agent of abortion in pregnant cattle, leading to considerable economic losses in the livestock industry [136]. Although N. caninum has not yet been classified as a zoonotic pathogen, environmental studies have highlighted its potential to pose a significant risk to human health, warranting further investigation [137, 138].
The recognition of N. caninum by host immune receptors, particularly
TLR3, highlights the critical interplay between innate immune sensing and the
subsequent adaptive immune responses, which are essential for controlling
infection and mitigating pathogenic effects. TLR3 may recognize lncRNAs
produced by host cells in response to N. caninum infection [139, 140].
This recognition triggers the production of various cytokines and chemokines,
including IL-12 and IFN-
TLR3 plays a complex role in parasitic infections, contributing to both protective immunity and immune-mediated pathologies through a delicate balance. It recognizes parasite-derived nucleic acids, such as exoRNA and lncRNA, and regulates immune cells like macrophages and NK cells to enhance host defenses. However, its activation can also lead to excessive inflammation, resulting in tissue damage, fibrosis, and exacerbating parasitic diseases. The interplay between TLR3 and other TLRs amplifies immune responses, highlighting its critical role in both innate and adaptive immunity.
In various parasitic infections, TLR3’s effects are host-dependent. For example,
during Plasmodium infection, TLR3 triggers the production of
pro-inflammatory cytokines essential for parasite clearance, but excessive
activation can cause tissue damage. In C. sinensis infections, TLR3
activation is protective, reducing liver fibrosis and injury by modulating
inflammatory and anti-inflammatory cytokines. In contrast, in S.
japonicum infections, TLR3 exacerbates granulomatous inflammation and liver
pathology, with its deficiency mitigating these effects. Similarly, in L.
amazonensis infections, TLR3 skews the immune response towards a Th17
phenotype, promoting parasite survival and disease progression. In T.
spiralis infections, TLR3 activation enhances Th1 responses, helping to
limit parasite burdens. In N. caninum infections, TLR3-mediated
production of IL-12p40 and IFN-
These findings highlight TLR3’s multifaceted roles in parasitic infections, acting both as a defender against pathogens and a contributor to immune-mediated damage. Understanding its dual functions is key to unraveling host-parasite interactions and positions TLR3 as a promising therapeutic target. Targeting TLR3 in parasitic infections holds potential for improving immune defense while minimizing harmful inflammation. Future research should focus on deciphering the precise mechanisms through which TLR3 mediates its diverse roles in different parasitic diseases. Developing specific TLR3 agonists or antagonists tailored to the infection type may offer innovative approaches to disease management. Additionally, understanding the cross-regulation between TLR3 and other immune pathways could pave the way for strategies to fine-tune immune responses. Insights from these studies may contribute to advancing vaccines, immunotherapies, and targeted treatments, ultimately improving patient outcomes and reducing the global burden of parasitic diseases.
C. sinensis, Clonorchis sinensis; DCs, dendritic cells; dsRNA, double-stranded RNA; exoRNA, exosomal RNA; IFN-
YY, QL, YZ, NJ, and QC designed and wrote the manuscript. YY, QL, YZ, NJ, and QC were involved in original draft preparation; YY, QL, YZ, NJ, and QC participated in reviewing and editing. YZ and NJ contributed to the design of the figures. QC finalized the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
We thank the researchers for their wonderful work on TLR3 and apologize to those who are not cited in this manuscript owing to the limited space.
This research was supported by a grant from the National Key Research and Development Program of China (grant number 2022YFD1800200), the National Nature and Science Foundation of China (grant number 82030060), and the CAMS Innovation Fund for Medical Sciences (CIFMS) (grant number 2019-I2M-5-042).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.