











1 Department of Pathophysiology, College of Basic Medical Sciences, Jilin University, 130021 Changchun, Jilin, China
2 School of Basic Medical Sciences, Hebei University, 071000 Baoding, Hebei, China
3 Department of Pathology and Cancer Research Center, Yanbian University Medical College, 133002 Yanji, Jilin, China
Abstract
Frequent drug resistance seriously limits the therapeutic efficacy of sorafenib in advanced hepatocellular carcinoma (HCC). Strategies to increase the response to sorafenib are limited, and the underlying mechanism to facilitate such an increase is not entirely understood. Homeobox (HOX) transcript antisense intergenic RNA (HOTAIR) expression is high in HCC, promoting the occurrence and progression of HCC. In this study, we explored the mechanism through which HOTAIR knockdown affects the response of HCC cells to the chemotherapeutic sorafenib.
Cell viability and apoptosis were assessed using MTT assay, flow cytometry, and nuclear staining. Mitochondrial function and isolation were determined using flow cytometry and a mitochondrial isolation kit. Glycolysis was measured by glucose and lactic acid assay kits. The underlying mechanisms were explored through western blotting, quantitative reverse-transcription polymerase chain reaction (qRT-PCR), and chromatin immunoprecipitation (ChIP).
HOTAIR knockdown increased sorafenib-induced apoptosis in the HCC cells. HOTAIR and hexokinase 2 (HK2) expression levels were upregulated in human HCC tissues, demonstrating a significant correlation. The knockdown of HOTAIR or HK2 aggravated mitochondrial dysfunction and inhibited glycolysis. Further, HOTAIR knockdown promoted sorafenib-mediated HK2 mRNA downregulation, resulting in decreased HK2 protein levels in cells and mitochondria. This ultimately facilitated the mitochondrial apoptotic pathway. Moreover, it was demonstrated that HOTAIR regulated HK2 via polycomb repressive complex 2 (PRC2)-mediated epigenetic modification of miR-145-5p in HCC.
HOTAIR knockdown increased the sensitivity of HCC cells to sorafenib by disrupting the miR-145-5p/HK2 axis-mediated mitochondrial function and glycolysis, suggesting new strategies for HCC treatment.
Keywords
- HOTAIR
- sorafenib
- mitochondrial function
- HK2
- miR-145-5p
- HCC
Hepatocellular carcinoma (HCC) is a rapidly growing malignant tumor worldwide, and has the third-highest mortality rate among cancers [1]. Surgical intervention is the preferred strategy for early HCC. However, at the time of diagnosis, 70% of patients are in the intermediate and late stages and have missed an optimum occasion for surgical intervention [2]. With few treatments available, the overall prognosis of HCC is poor. Sorafenib, a pan-kinase inhibitor, is a first-line medication for advanced HCC [3, 4]; however, the recurrent emergence of drug resistance significantly undermines the therapeutic efficacy of sorafenib.
Homeobox (HOX) transcript antisense intergenic RNA (HOTAIR) was the first discovered long non-coding RNA (lncRNA) with reverse transcription function, with a length of 2158 nucleotides [5, 6]. The expression of HOTAIR is high in various cancers [7, 8, 9, 10, 11, 12]. The elevated levels of HOTAIR are significantly associated with unfavorable cancer prognosis [5, 10]. While HOTAIR knockdown impedes the progression of HCC [9, 13], the effect of HOTAIR on the response to chemotherapeutics in HCC is unclear.
Mitochondria have long been thought to be double-edged swords. Under normal conditions, mitochondria facilitate cell survival by maintaining cell respiration and regulating complex biological signals. Under excessive stress, mitochondria engage with molecules associated with apoptosis, necrosis, and autophagy, thereby modulating cell death. HCC cells exposed to sorafenib frequently exhibit a mitochondrial adaptive response [14, 15, 16], which includes alterations in mitochondrial biogenesis, enhancement of mitophagy, and adjustments in mitochondrial respiration. These adaptations serve to mitigate cellular damage, ultimately leading to drug resistance. Therefore, understanding how to amplify mitochondrion-related death signals is important for treatment purposes. Morphological changes and functional disorders of mitochondria are observed after HOTAIR knockdown [17]. This suggests that inhibiting HOTAIR may represent a potential strategy for exacerbating mitochondrial damage. However, the mechanism through which HOTAIR modulates mitochondrial function is unknown. Identifying the specific molecules by which HOTAIR regulates mitochondria will help to elucidate the application prospects of mitochondria in HCC treatment.
Hexokinase 2 (HK2) is a key speed-limiting enzyme in glycolysis. The expression of HK2 is elevated in a variety of tumors [18, 19, 20]. The inhibition of HK2 in HCC suppresses glycolysis and enhances the sensitivity to chemotherapeutic agents [18, 21, 22]. While the previous study on HK2 mostly focused on glycolysis, HK2 is observed to affect mitochondrial function [23]. Although HK2 is predominantly located in the cytoplasm, a portion of HK2 associates with the mitochondria through its interaction with the mitochondrial protein voltage-dependent anion channel (VDAC) [24]; this part of HK2 facilitates the rapid transport of adenosine triphosphate (ATP) from the mitochondrial matrix to the cytoplasm [25]. When HK2 depolymerizes from the mitochondria, it promotes the opening of the mitochondrial permeability transition pore and the perforation of Bax on mitochondria, resulting in increased mitochondrial outer membrane permeability and apoptosis [26]. As the localization of HK2 on mitochondria in tumors is associated with tumor resistance to apoptosis [26, 27], reducing this localization may be a method to regulate tumor cell death.
The role of microRNA (miRNA) in tumors is receiving increasing attention. Since 2013, the application of microRNA in clinical treatment of tumors has gradually increased [28]. HOTAIR indirectly controls HK2 by regulating microRNAs such as miR-125 and miR-143 [29, 30]. However, it is still unknown whether HOTAIR regulates HK2 in HCC chemotherapy sensitivity through microRNAs. Furthermore, it remains to be explored whether HOTAIR regulates HK2 through other microRNAs. The 5′ domain of HOTAIR binds polycomb repressive complex 2 (PRC2) to regulate the trimethylation of histone H3 at lysine 27 (H3K27me3) [31]. Thus, HOTAIR may regulate microRNAs and HK2 through epigenetic modifications. Enhancer of zeste homolog 2 (EZH2) is a catalytic component within PRC2. EZH2 is high in a variety of cancerous tissues [32]. Exploring the role of epigenetics regulated by HOTAIR and EZH2 on the response to chemotherapeutics in HCC will open up new ideas for drug discovery.
Human cDNA microarrays (LncDNA-HLivH060PG02) were purchased from ShGnghGi Outdo Biotech Co., Ltd., Shanghai, China. Clinical characteristics of patients corresponding to the human cDNA microarrays are shown in Supplementary Table 1.
DMEM (Gibco Life Technologies, Carlsbad, CA, USA) containing 10% FBS (HyClone, Logan, UT, USA) was applied to culture HepG2 (BNCC339266, BeNa Culture Collection, Beijing, China) and HCC-LM3 (BNCC342335, BeNa Culture Collection, Beijing, China) cells. Both cell lines were validated by short tandem repeat (STR) profiling and tested negative for mycoplasma.
Sorafenib was purchased from Selleck (Houston, TX, USA). MTT was obtained from Sigma-Aldrich (Merck, St. Louis, MO, USA).
HepG2 cells (5
Cells were stained according to the instructions in an Annexin V-FITC Propidium iodide (PI) double staining kit (BD Biosciences, San Jose, CA, USA). Guava easyCyte flow cytometry (Luminex, Austin, TX, USA) was applied to test samples.
The shRNAs and siRNAs were obtained from Sangon Biotech and GenePharma (Shanghai, China), respectively. The sequences are as follows: sh-HOTAIR-1 (5′-GAGACACATGGGTAACCTA-3′), sh-HOTAIR-2 (5′-CTGCAACCTAAACCAGCAA-3′), si-HK2-1 (5′-GGAGGAUGAAGGUAGAAAUTT-3′), si-HK2-2 (5′-GUCGCUUUGAGACC AAAGATT-3′), miR-145 inhibitor (5′-AGGGAUUCCUGGGAA AACUGGAC-3′) (miR20000437-1-5, Ribobio, Guangzhou, China), miR-145 mimics (sence 5′-GUCCAGUUU UCCCAGGAAUCCCU-3′, antisence 5′-CAGGUCAAAAGGGUCCUUAGGGA-3′) (miR10000437-1-5, Ribobio, Guangzhou, China). The ViaFectTM transfection reagent (E4981, Promega, Madison, WI, USA) was applied for transfection.
The method is consistent with our previously published articles [33]. The primers are presented in Supplementary Table 2.
The method is consistent with our previously published articles [33]. HK2 (1:1500, 22029-1-AP), B-cell lymphoma-2 (Bcl-2) (1:1000, 12789-1-AP), Bax (1:1500, 50599-2-Ig), Actin (1:20,000, 66009-1-Ig), COX IV (1:2000, 11242-1-AP) antibodies were all obtained from Proteintech (Rosemont, IL, USA).
Cells were exposed to 4% paraformaldehyde (P0099-100ml, Beyotime Biotechnology, Shanghai, China) at room temperature (RT). After 12 min, paraformaldehyde was aspirated and Hoechst 33342 (C1025, Beyotime Biotechnology, Shanghai, China) was added for 5 min. Chromatin condensation was observed using the Echo-lab Revolve microscope (Echo Laboratories, San Diego, CA, USA).
Cells were incubated with indicator (M36008, Thermo Fisher Scientific, Waltham, MA, USA) at 37 °C. After 10 min, wash away indicator and utilize an Accuri C6 flow cytometer (Becton Dickinson, Franklin Lakes, NJ, USA) for assessment.
Cells were assembled and exposed to JC-1 (C2003S, Beyotime Biotechnology, Shanghai, China) at 37 °C. Avoid light throughout the experiment. After 20 minutes, completely remove JC-1 and maintain the cells with culture medium. Guava easyCyte flow cytometry was utilized to test samples.
The glucose assay and lactic acid kits (Nanjing Jiancheng Bioengineering Institute, Nanjing, China) were applied. The medium gained from treated cells was mixed with the working fluid at 37 °C for 10 min. For glucose, the absorbance was obtained at 505 nm. The glucose intake was obtained by subtracting measured glucose values from the glucose value in blank culture medium (without cells). The termination fluid was added and the absorbance (530 nm) was recorded in lactic acid kits. For two experiments, results were standardized to protein level in cell.
The Enhanced ATP Assay Kit (Beyotime Biotechnology, Shanghai, China) was applied. In short, the supernatant was assembled after cell lysis. ATP detection working solution was added. After 5 min, a luminometer (BMG LABTECH Omage, Ortenberg, Germany) was applied to test luminescence.
The Mitochondrial Isolation Kit (Invent Biotechnologies, Plymouth, MN, USA) was
applied. 3
The SimpleChIP® Plus Enzymatic Chromatin IP Kit (Cell Signaling Technology, Danvers, MA, USA) with anti-EZH2 (#5246, Cell Signaling Technology, Boston, MA, USA), anti-H3K27me3 (ab6002, Abcam, Cambridge, MA, USA) and rabbit immunoglobulin G (IgG) (#2729, Cell Signaling Technology, Boston, MA, USA) was applied. Chromatin immunoprecipitation (ChIP) signals were analyzed by standard PCR and agarose gel electrophoresis. Values in immunoprecipitated samples (percent (%) input DNA) were normalized to values for IgG. Primer sequences for pmiR-145 are 5′-GCTCAGATGCAGCTTCAGAA-3′ (forward) and 5′-CCGGAGCCAAGGTTAGAAGT-3′ (reverse).
All experiments were repeated at least three times. All results are presented as
mean values
qRT-PCR analysis revealed that the mRNA levels of both HOTAIR and HK2 in HCC tissue were significantly elevated compared to those in normal liver tissue (Fig. 1A,B). The mRNA expression level of HK2 exhibited a positive correlation with HOTAIR in HCC tissue (Fig. 1C). Then, the expression of HOTAIR was silenced in HCC cells using short hairpin RNAs (Supplementary Fig. 1A). MTT assay showed that HOTAIR knockdown significantly reduced cell viabilities in the presence of sorafenib (Fig. 1D). The greatest inhibitory effect was observed when the concentration of sorafenib was 5 µM for HepG2 and 10 µM for HCC-LM3 cells, respectively. These doses were applied to all follow-up experiments. Subsequently, HOTAIR knockdown enhanced sorafenib-mediated downregulation of HK2 at mRNA and protein levels (Fig. 1E,F). Further, mitochondria were isolated. HOTAIR knockdown enhanced sorafenib-mediated reduction of HK2 and anti-apoptotic protein Bcl-2 in mitochondria, and augmented sorafenib-mediated elevation of pro-apoptotic protein Bax in mitochondria (Fig. 1G). These data suggest that mitochondria-mediated apoptosis occurs after HOTAIR knockdown and sorafenib treatment. In summary, HOTAIR knockdown augmented the response to sorafenib via dominating HK2.
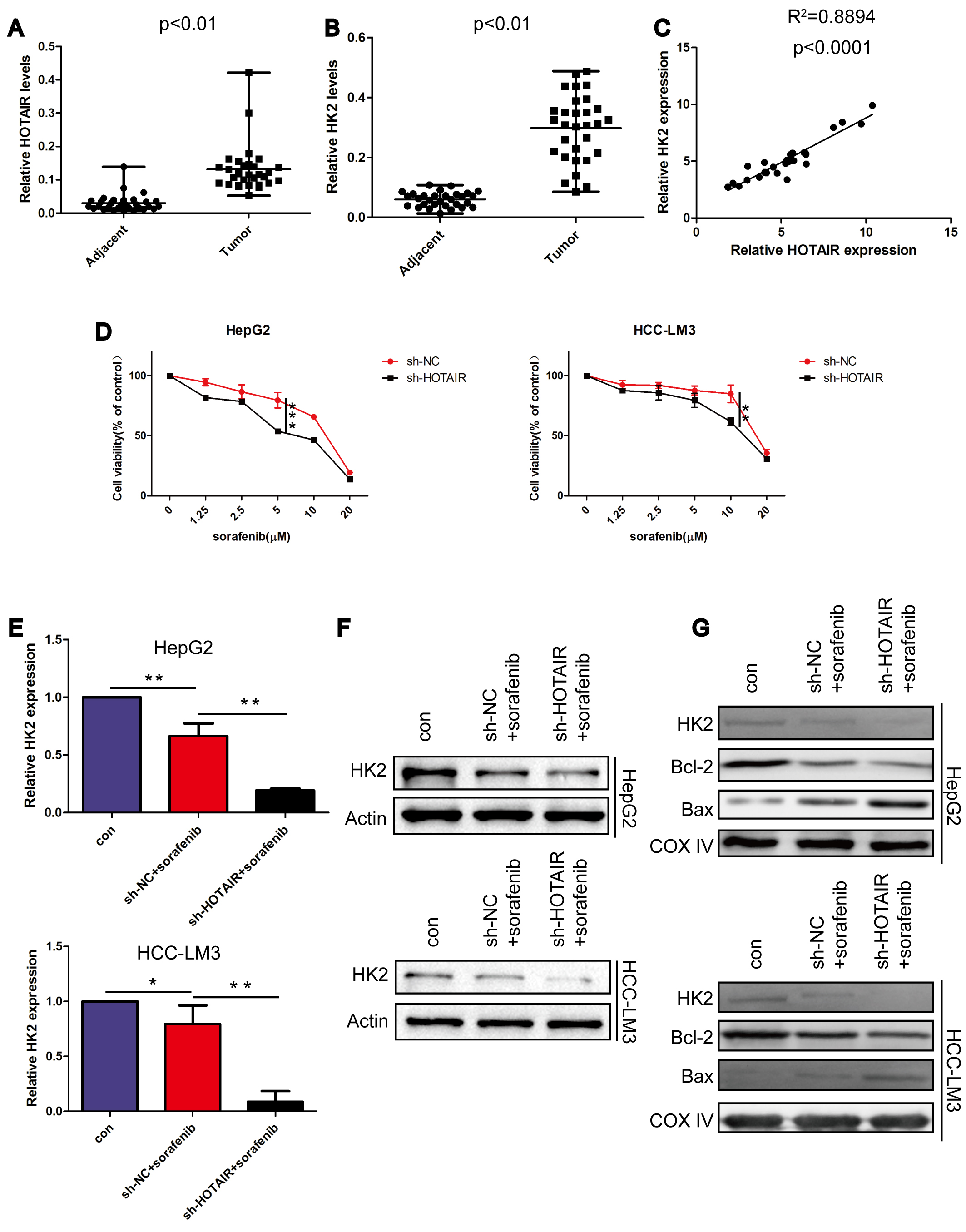 Fig. 1.
Fig. 1.
HOTAIR and HK2 are upregulated in HCC and
HOTAIR knockdown enhanced sorafenib-mediated HK2
downregulation. (A,B) The mRNA levels of HOTAIR and HK2 in
non-cancerous liver and HCC tissues (30 pairs). (C) Correlation between
HOTAIR and HK2 at the mRNA level in HCC. (D) MTT assay assessed
cell viabilities. (E) qRT-PCR assessed the HK2 mRNA level. (F) The protein level
of HK2 in whole cell. (G) The protein level of HK2, Bcl-2, Bax in mitochondria.
*p
Hoechst staining revealed that nuclear fragmentation was induced by sorafenib treatment. Furthermore, when compared to sorafenib treatment alone, the combined treatment of sorafenib and HOTAIR knockdown resulted in an increased level of nuclear fragmentation (Fig. 2A). Annexin V/Propidium iodide (PI) staining showed that apoptosis induced by sorafenib was enhanced by HOTAIR knockdown (Fig. 2B). Combined with data in Fig. 1G, HOTAIR knockdown augmented apoptosis induced by sorafenib.
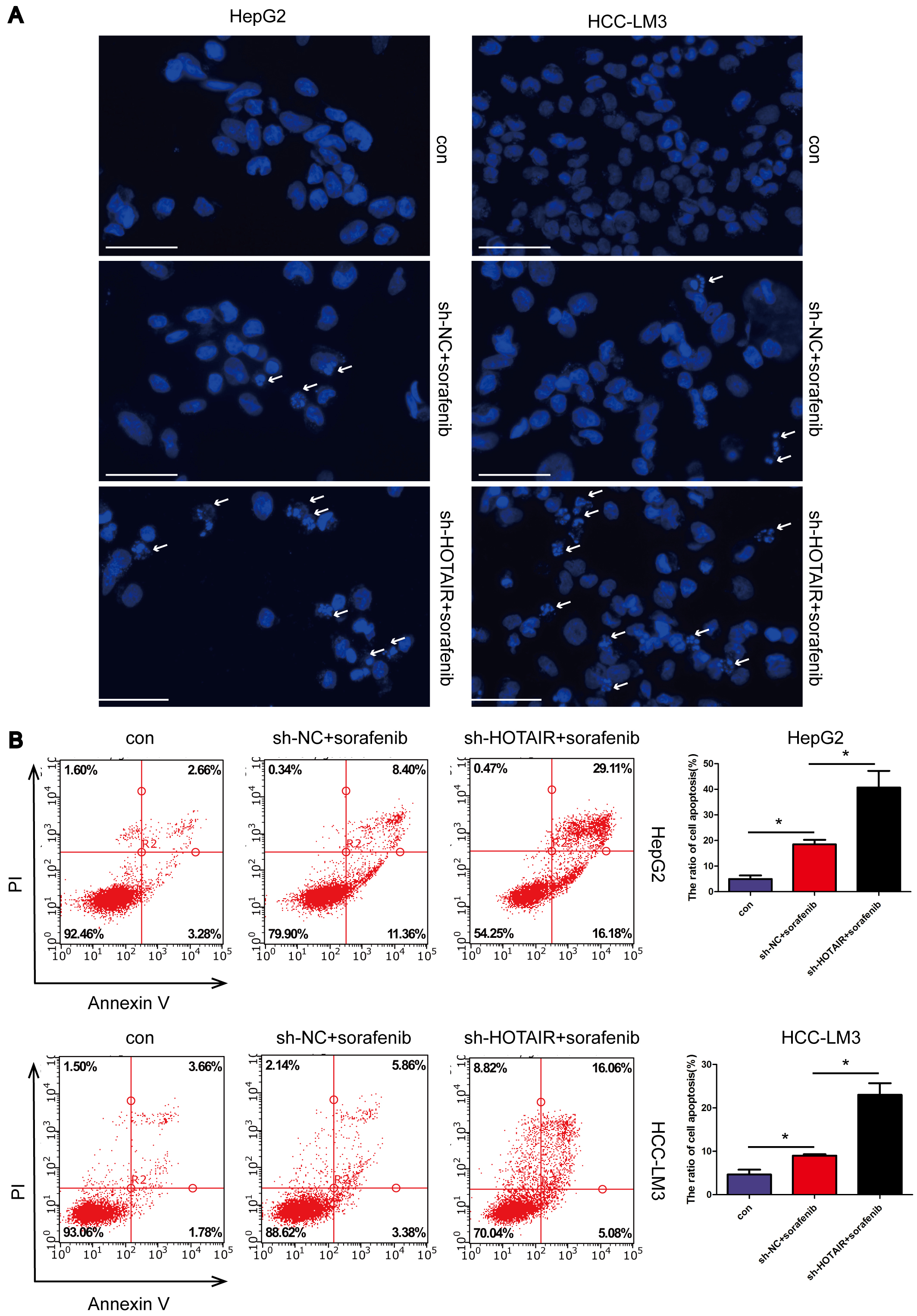 Fig. 2.
Fig. 2.
HOTAIR knockdown aggravated apoptosis induced by
sorafenib. (A) Hoechst 33342 was applied to observe cell apoptotic chromatin
condensation. Arrows indicated apoptotic cells. Scale bar, 70 µm. (B)
Apoptosis rates were analysed by flow cytometry. *p
Mitochondrial dysfunction is the premise of the endogenous apoptosis pathway [34]. To investigate whether sorafenib and HOTAIR knockdown induce apoptosis by targeting mitochondria, mitochondrial function was evaluated using JC-1 staining. Normal MMP is required for maintaining mitochondrial protein import and ATP production [34, 35]. If MMP collapses and cannot be restored after an extended period, mitochondrial homeostasis will be disrupted, leading to apoptosis in the affected cells [34]. The data indicate that HOTAIR knockdown significantly enhanced the decrease in MMP induced by sorafenib (Fig. 3A). Additionally, MitoSOX staining showed that sorafenib-treated cells exhibited increased levels of ROS compared to those in the control group (Fig. 3B). The level of mitochondrial ROS significantly increased following the combined treatment with sorafenib and HOTAIR knockdown. This finding suggests that HOTAIR knockdown exacerbates the mitochondrial oxidative damage induced by sorafenib. These data indicate that HOTAIR knockdown enhanced the mitochondrial dysfunction induced by sorafenib.
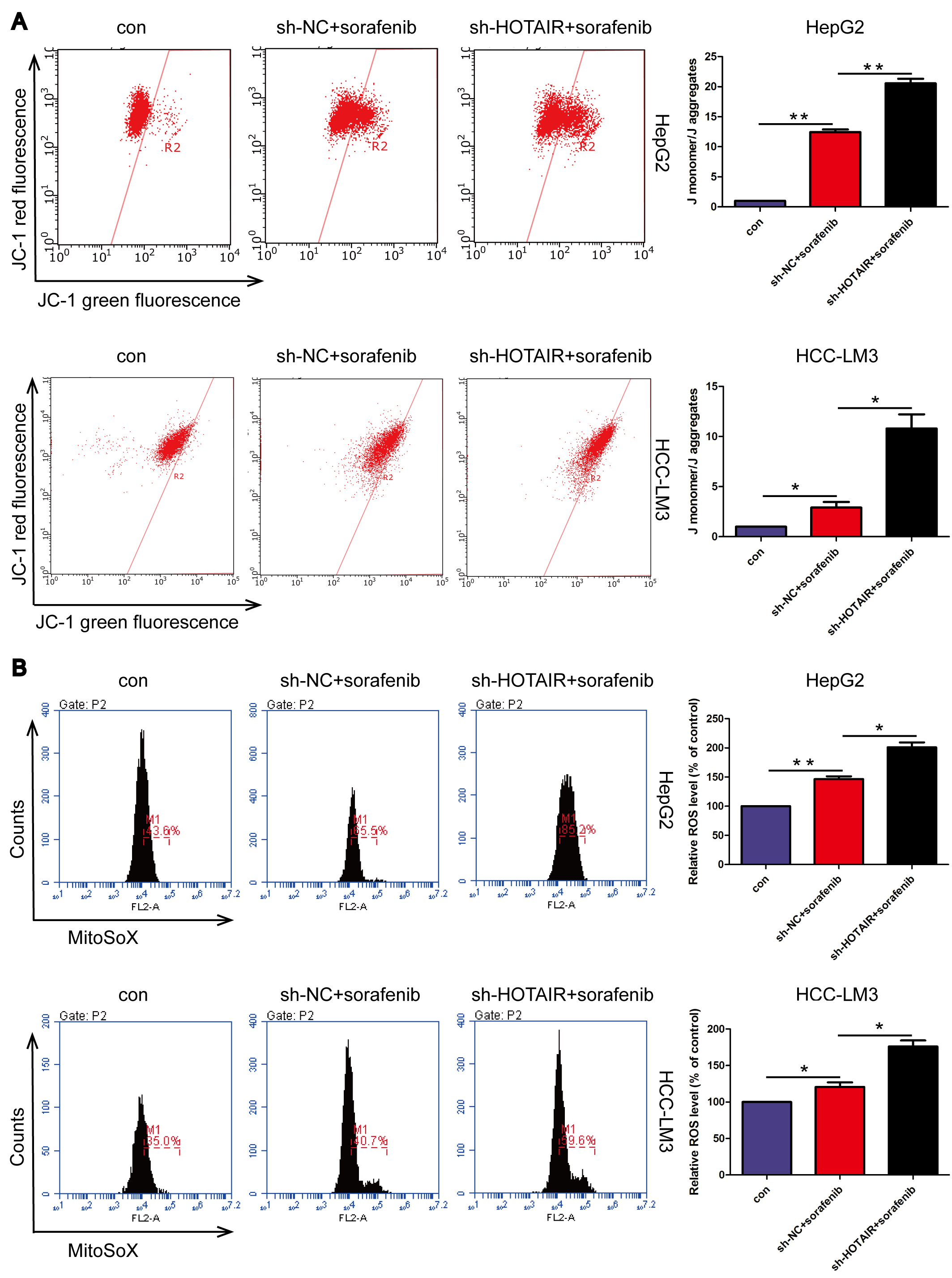 Fig. 3.
Fig. 3.
HOTAIR knockdown aggravated mitochondrial dysfunction
induced by sorafenib. (A) Mitochondrial membrane potential assessment. (B)
Mitochondrial ROS was measured by MitoSOX. *p
Glycolysis is employed by tumors to generate energy, and it also supplies the necessary precursors for macromolecular synthesis within these neoplasms [36]. Therefore, the inhibition of glycolysis often restricts tumor development [18, 37]. Thus, glycolysis levels were examined. HOTAIR knockdown enhanced sorafenib-mediated inhibition on glucose consumption and lactate generation (Fig. 4A,B). These data suggest that HOTAIR knockdown enhanced sorafenib-mediated inhibition of glycolysis. The intracellular concentrations of ATP were also assessed. In comparison to the control group, cells treated with sorafenib exhibited significantly reduced levels of intracellular ATP concentration. Notably, in comparison to the cells treated solely with sorafenib, those subjected to a combination of sorafenib and HOTAIR knockdown exhibited significantly reduced intracellular ATP concentrations (Fig. 4C). Therefore, energy deficiency may contribute to the death of HCC cells.
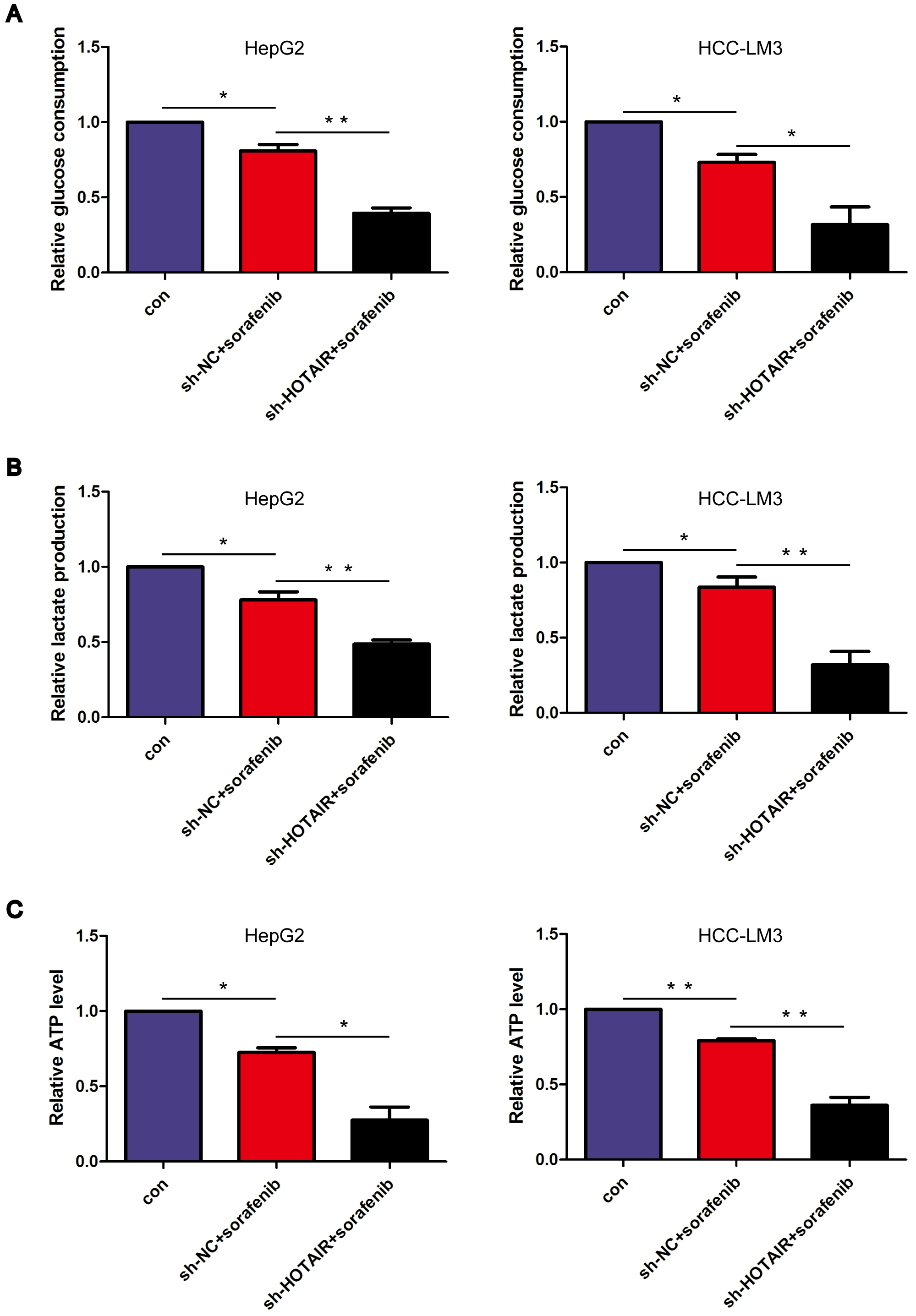 Fig. 4.
Fig. 4.
HOTAIR knockdown enhanced sorafenib-mediated glycolysis
inhibition. (A) Relative glucose intake in treated cells. (B) Relative lactate
level in treated cells. (C) Relative ATP generation in cells. *p
The expression of HK2 was silenced in HCC cells (Supplementary Fig. 1B). MTT assay showed that HK2 knockdown enhanced the sorafenib-mediated decrease in cell viability, suggesting that HK2 knockdown enhanced the response to sorafenib (Supplementary Fig. 2). JC-1 staining showed that HK2 knockdown enhanced the sorafenib-mediated decrease in MMP (Fig. 5A). The level of mitochondrial ROS was elevated following exposure to sorafenib treatment, and this increase was further amplified after the combined treatment with sorafenib and HK2 knockdown (Fig. 5B). These data indicate that HK2 knockdown aggravated sorafenib-regulated mitochondrial dysfunction.
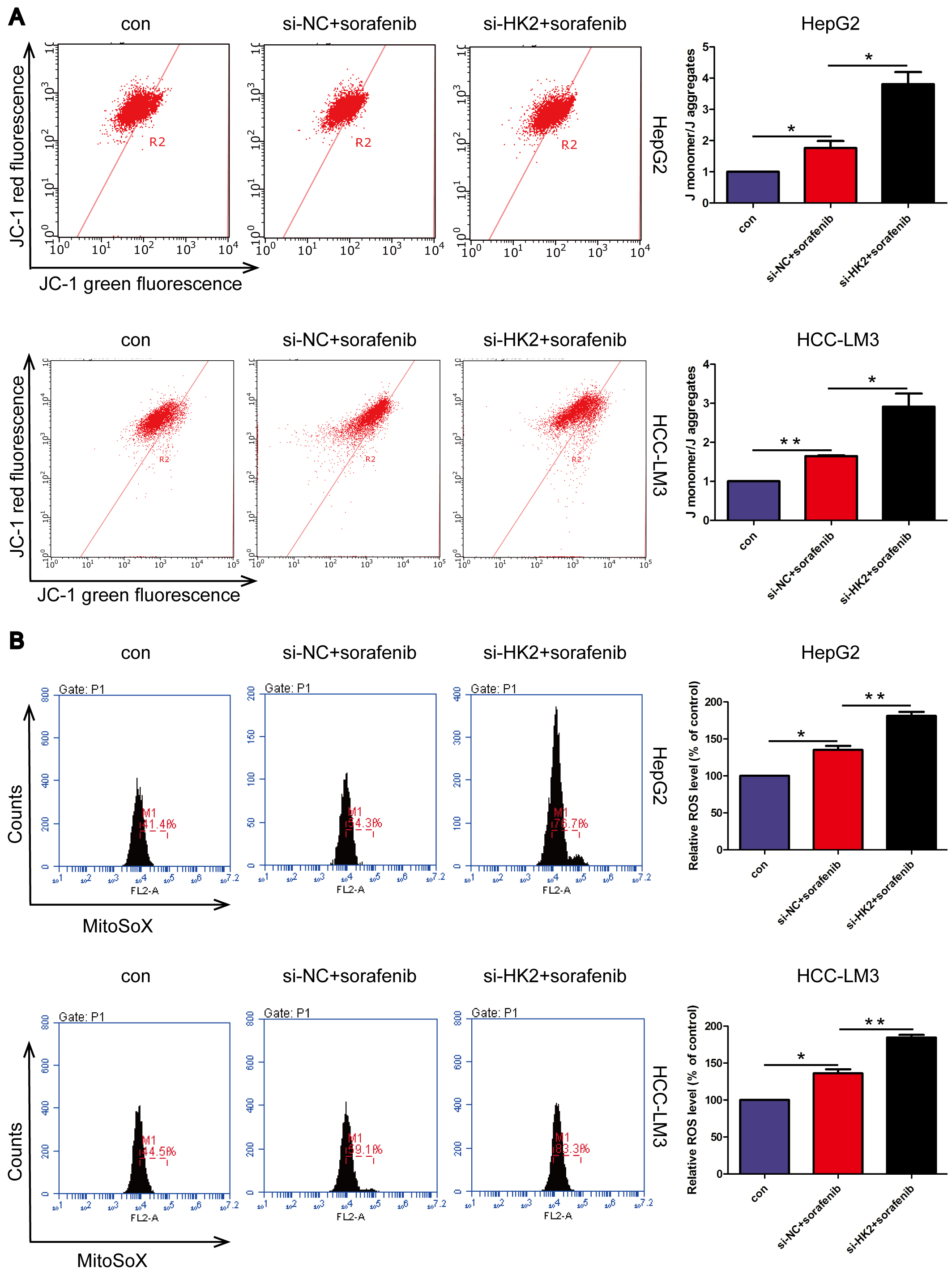 Fig. 5.
Fig. 5.
HK2 knockdown aggravated mitochondrial dysfunction
induced by sorafenib. (A) Mitochondrial membrane potential assessment. (B)
Mitochondrial ROS was measured by MitoSOX. *p
Similar to the effects observed with HOTAIR knockdown, HK2 knockdown further augmented the sorafenib-induced reduction in glucose utilization and lactate levels (Fig. 6A,B). Furthermore, the intracellular concentrations of ATP were significantly reduced following the combined treatment with sorafenib and HK2 knockdown (Fig. 6C). These data indicate that HK2 knockdown enhances the inhibition of glycolysis and exacerbates energy deficiency in HCC cells induced by sorafenib.
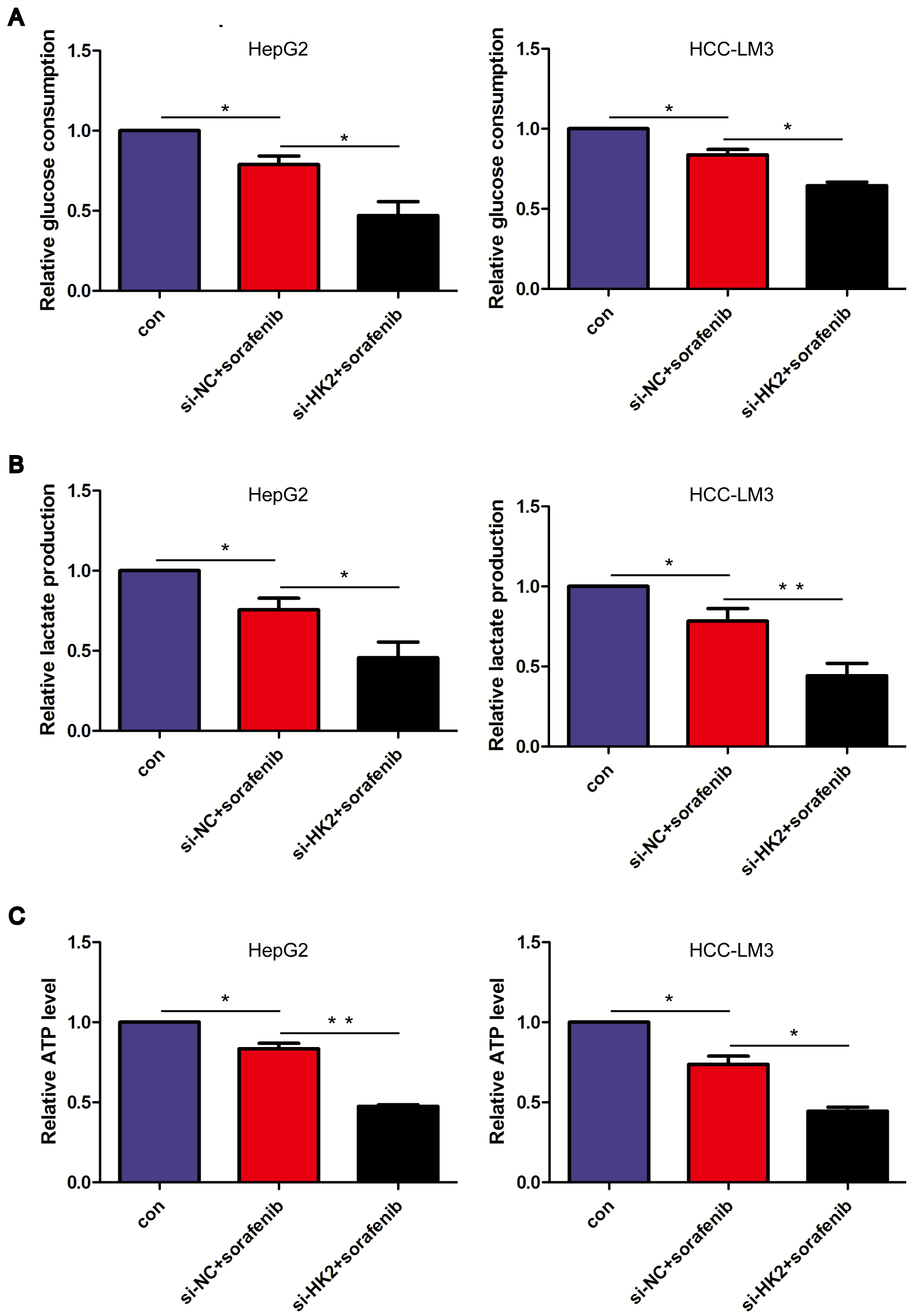 Fig. 6.
Fig. 6.
HK2 knockdown enhanced sorafenib-mediated glycolysis
inhibition. (A) Relative glucose intake in cells. (B) Relative lactate
production in treated cells. (C) Relative ATP production in cells. *p
HOTAIR regulates downstream genes through miR-125, miR-143, and miR-145 [29, 38]. To further elucidate the mechanism by which HOTAIR regulates HK2, these miRNAs were detected. HOTAIR knockdown enhanced the sorafenib-regulated increase in miR-145, while having no effect on miR-125 and miR-143 (Fig. 7aA). Based on the report indicating that miR-145 directly binds to HK2 [39, 40], it is speculated that HOTAIR may regulate HK2 through miR-145. We observed that miR-145 mimics enhanced sorafenib-mediated decrease in HK2, which is consistent with the effects observed following HOTAIR knockdown (Fig. 7aB,C). Further, miR-145 inhibitor reversed the regulatory effects of HOTAIR on HK2 expression and cell viability (Fig. 7aD–F). The ChIP assays demonstrated that the enrichment of H3K27me3 at the promoter region of miR-145 was reduced following sorafenib treatment, with a further decrease observed after the combined intervention of sorafenib and HOTAIR knockdown (Fig. 7bA). Consistently, the enrichment of EZH2, a key component of PRC2 responsible for activating H3K27me3, was found to decrease in response to sorafenib treatment and the combined intervention of sorafenib and HOTAIR knockdown (Fig. 7bB). These data indicate that HOTAIR modulates HK2 and the cellular response to sorafenib through epigenetic modification of miR-145-5p.
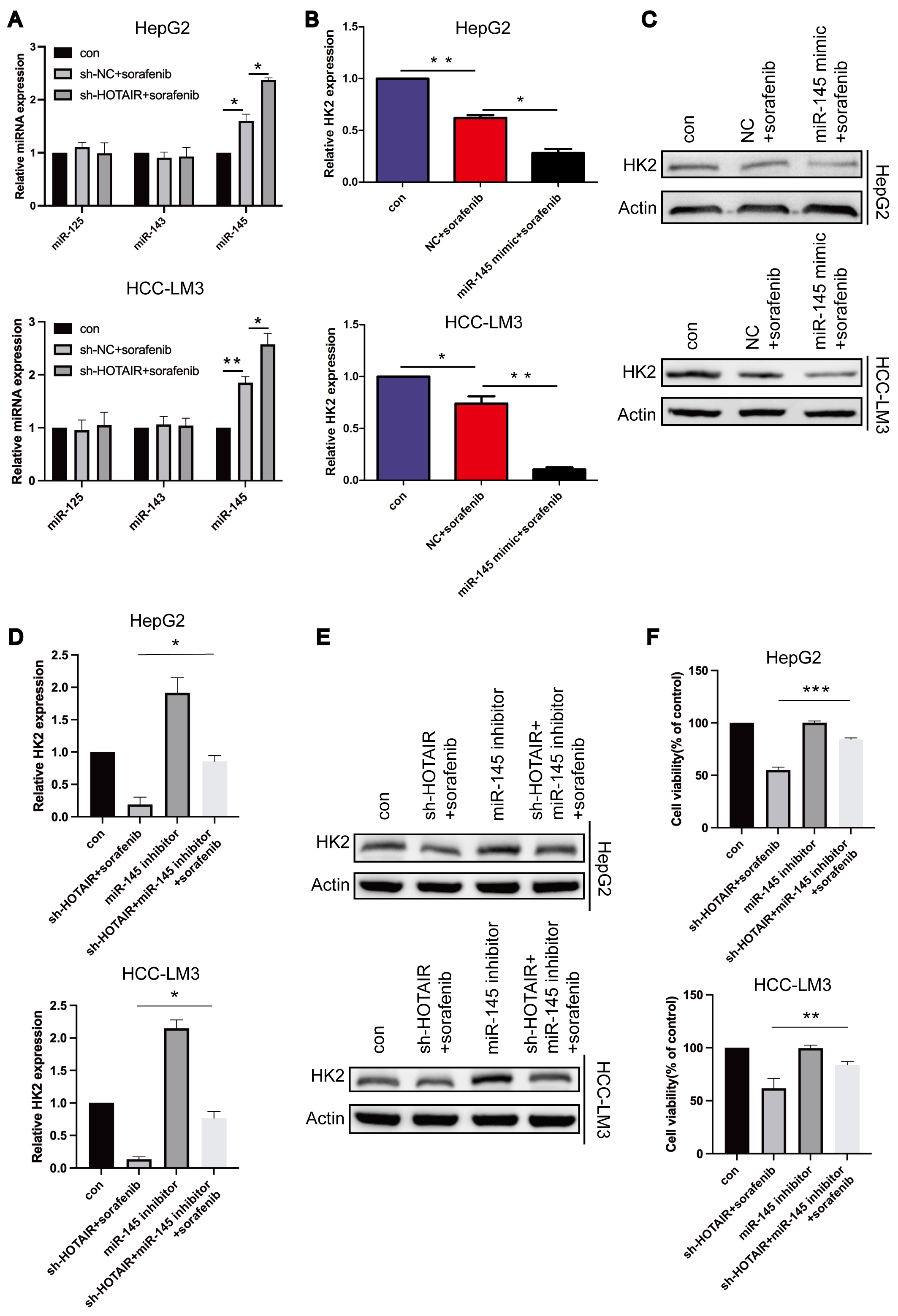 Fig. 7a.
Fig. 7a.
HOTAIR regulated HK2 by miR-145-5p. (A) Relative
microRNA (miRNA) level was measured by qRT-PCR. (B) Relative mRNA level of
HK2. (C) The protein level in whole cell. (D) Relative mRNA level of
HK2. (E) The protein level in whole cell. (F) Cell viabilities of
treated cells. *p
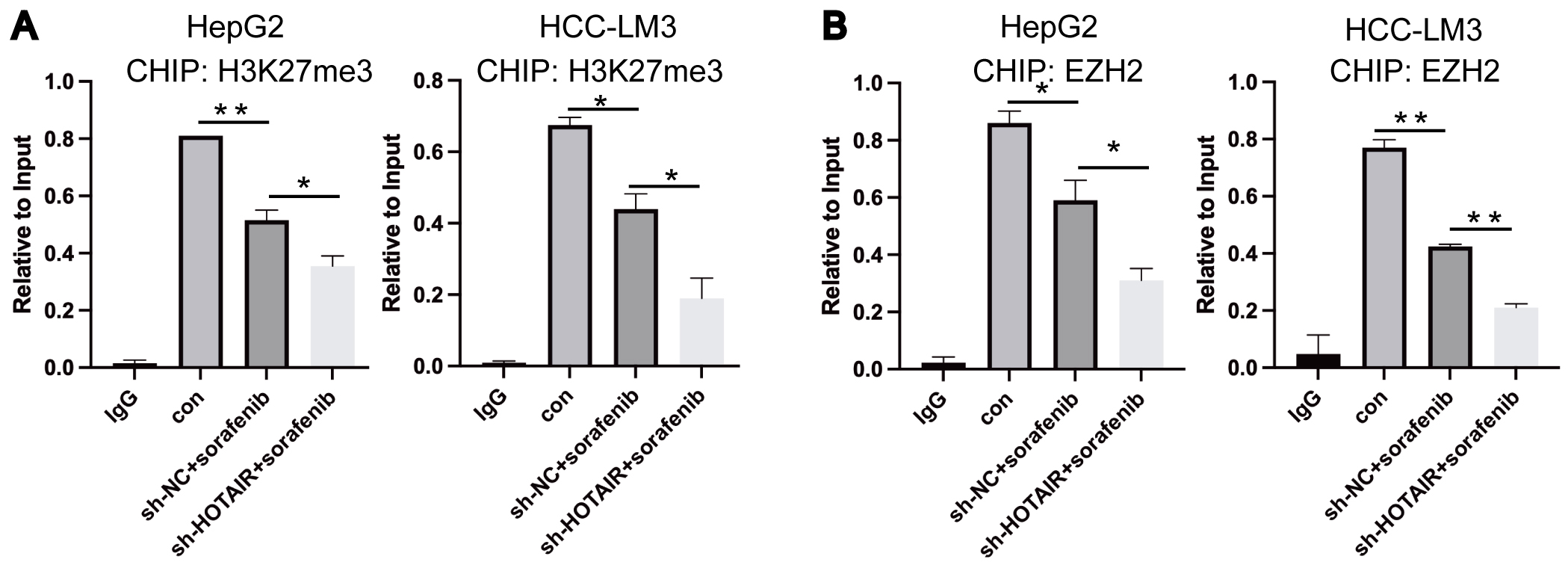 Fig. 7b.
Fig. 7b.
HOTAIR regulated HK2 by epigenetic modification of
miR-145-5p. (A,B) ChIP assays were conducted on miR-145-5p promoter regions.
*p
Sorafenib has a curative effect for about 1 year in HCC [14]. Increasing the dosage of sorafenib does not significantly improve the curative effect and may instead bring about greater toxicity and side effects. In this study, HOTAIR and HK2 were upregulated in human HCC, suggesting that targeting HOTAIR and HK2 may better treat HCC and reduce their effects on normal tissues. Substantial clinical data support that monotherapies triggers chemoresistance [41]. Combination treatments may defeat resistance. Currently, Combination approaches for HCC are scarce. We discovered that the combination of HOTAIR knockdown and sorafenib demonstrates efficacy in HCC, offering a novel reference for combination therapy. In the future, it is anticipated that HOTAIR knockdown will be incorporated as an adjunct to standard treatment. High heterogeneities frequently trigger HCC therapy failure. Some patients may exhibit insensitivity to HOTAIR knockdown. HOTAIR knockdown may be particularly beneficial for patients exhibiting high levels of HOTAIR. By integrating nanodelivery systems, the therapeutic specificity of HOTAIR knockout can be further improved. The research on specific inhibitors targeting HOTAIR will contribute to the clinical application of HOTAIR [42]. HOTAIR manipulates immune cells and signaling pathways to facilitate immunoescape in breast cancer and gliomas [43, 44]. HOTAIR knockdown combined immunotherapy has a promising application in HCC for supports. Nevertheless, the therapeutic application of HOTAIR knockdown necessitates comprehensive clinical trials for validation.
Mitochondria represent a primary target for addressing the chemoresistance associated with HCC [45, 46]. Our experiments showed that HOTAIR knockdown enhanced the sorafenib-induced reduction in MMP and elevation of mitochondrial ROS, thereby causing mitochondrial damage and ultimately triggering mitochondria-mediated apoptosis. Our findings confirmed that HOTAIR knockdown enhanced the sorafenib-mediated downregulation of HK2 expression at the mRNA level. This, in turn, led to a reduction in total HK2 protein levels, which indirectly resulted in decreased mitochondrial HK2. In tumor cells, a part of HK2 is situated within the mitochondria. When HK2 depolymerized from the mitochondria, it promoted the opening of mPTP, leading to an increase in mitochondrial outer membrane permeability and subsequent cell death [26]. The combination of sorafenib and HOTAIR knockdown did not directly influence the binding of HK2 to VDAC; however, it indirectly resulted in a reduction of mitochondrial HK2 at the protein level and initiated mitochondria-mediated apoptosis. This finding aligns with the results reported by Kim et al. [47], who demonstrated that miR-181a downregulated HK2 RNA levels, consequently leading to a reduction in total HK2 protein levels. This process indirectly resulted in decreased mitochondrial HK2 levels, ultimately contributing to cell death [47]. This indicates that the subcellular localization of HK2 within cells is highly significant for its functional role.
Mounting evidence shows that lncRNAs regulate key enzymes or molecules involved in glycolysis [48, 49]. HCC is characterized by aerobic glycolysis, a process that plays a crucial role in regulating angiogenesis, immune escape, and chemoresistance [18, 50]. The overexpression of HOTAIR enhances Glucose Transporter 1 (GLUT1) expression, thereby promoting glycolysis and facilitating the proliferation of HCC [51]. Our study observed that HOTAIR knockdown enhanced sorafenib-mediated downregulation of HK2 expression. This alteration led to a reduction in glycolysis levels and cellular energy production, ultimately resulting in increased damage to HCC cells. This presents new evidence regarding the regulation of glycolysis by lncRNA in HCC. Furthermore, the reduction in lactate production resulting from the knockdown of HOTAIR and HK2 led to a diminished weak acid environment essential for tumor growth, thereby potentially inhibiting tumor progression. Solely inhibiting glycolysis or activating oxidative phosphorylation to disrupt cellular metabolism can impede the progression and chemoresistance in HCC [18, 52]. Our study showed that the simultaneous inhibition of mitochondrial function and glycolysis exerted a significant anticancer effect. This finding aligns with the results reported by Rodríguez-Hernández et al. [14], who demonstrated that a sustained therapeutic dose of sorafenib can concurrently induce mitochondrial damage and inhibit glycolysis. Our prior research demonstrated that the inhibition of glycolysis interferes with mitochondrial quality control, consequently leading to an increase in mitochondrial damage [53]. Therefore, glycolysis inhibition caused by HOTAIR or HK2 knockdown may amplify mitochondrial death signals.
We further investigated the detailed mechanism by which HOTAIR regulated HK2. In recent years, numerous studies have indicated that HOTAIR modulates downstream gene expression and biological processes via miRNAs [54, 55]. Here, we observed that HOTAIR regulated HK2 through miR-145-5p. Inhibiting miR-145 reversed the low HK2 expression induced by HOTAIR knockdown. Additionally, miR-145 mimics were found to inhibit HK2. The findings indicate that HOTAIR plays a role in chemotherapy responsiveness via the miR-145/HK2 pathway. The report that miR145 directly binds HK2 provides strong evidence for our results [39, 40]. Further exploration revealed that HOTAIR knockdown resulted in a diminished enrichment of EZH2 at the promoter region of miR-145. This reduction subsequently led to a decrease in H3K27 trimethylation, thereby exacerbating cell death induced by sorafenib. The results suggest that the interaction between HOTAIR and PRC2 is of great significance in tumor chemotherapy. Zhao et al. [56] designed multiple inhibitors to obstruct EZH2 and HOTAIR, reversing the resistance of glioblastoma to temozolomide. These findings suggest that the epigenetic regulation mediated by HOTAIR holds significant promise in addressing tumor chemotherapy resistance.
Our results showed that HOTAIR knockdown downregulated HK2 by PRC2-mediated epigenetic modification of miR-145-5p, resulting in mitochondrial dysfunction and glycolysis inhibition. We explained the molecular mechanism by which HOTAIR knockdown disrupted mitochondrial function. Our observations indicate that HOTAIR knockdown can simultaneously target both mitochondria and glycolysis, thereby enhancing the response to sorafenib in HCC. This provides new strategies and theoretical bases for HCC treatment.
All data contained in the study are available from the corresponding author on reasonable request.
MC and NL designed the research study. MC performed the research. BW, LD, YJ and WZ actively participated in the experimental procedures and data analysis. MC wrote the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
The work was supported by National Natural Science Foundation of China (No. 81702793, 82173310); Fundamental Research Funds for the Central Universities (No. 2132020KJC017); Norman Bethune Program of Jilin University (2022B24); Outstanding Young Teachers Training Program of Jilin University; High Level Talent Research Launch Project of Hebei University (521100223221); Medical Science Foundation of Hebei University (2023B07); Foundation of President of Hebei University (XZJJ202320).
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/FBL37368.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.