





 , Hui Du 1,†, Zhi Liu 1, Yuxi Lei 1, Huizhi Hu 1, Junwen Zheng 1, Pu Yang 1,2,*
, Hui Du 1,†, Zhi Liu 1, Yuxi Lei 1, Huizhi Hu 1, Junwen Zheng 1, Pu Yang 1,2,* , Dongchi Zhao 1,2,*
, Dongchi Zhao 1,2,*1 Department of Pediatrics, Zhongnan Hospital of Wuhan University, 430071 Wuhan, Hubei, China
2 Children’s Digital Health and Data Center of Wuhan University, 430071 Wuhan, Hubei, China
†These authors contributed equally.
Abstract
As pivotal immunoregulatory sentinels in pulmonary defense systems, alveolar macrophages (AMs) play dual roles in mediating inflammatory responses and tissue repair processes during various phases of inflammatory cascades. The present investigation focuses on elucidating the regulatory influence of Notch pathway activation within AM populations on the pathophysiological mechanisms underlying acute lung injury (ALI) development.
To investigate the regulatory roles of Notch intracellular domain (NICD) and C-C chemokine receptor type 5 (CCR5) in pulmonary inflammation, an ALI model was established through lipopolysaccharide (LPS) administration. Complementary studies used macrophage-specific Notch1 knockout mice and immortalized bone marrow-derived macrophages (iBMDMs). Molecular profiling of CCR5 and inflammatory mediators was performed through real-time quantitative reverse transcription PCR (qRT-PCR) and immunofluorescence staining. Functional assessments of macrophage migration were carried out using scratch wound healing assays and transwell migration assays.
In the LPS-induced ALI model, pulmonary tissues exhibited elevated expression of both NICD and CCR5. Conversely, Notch1 knockout mice attenuated CCR5 expression, reduced macrophage infiltration and downregulated transcription of pro-inflammatory mediators compared to wild-type controls (p < 0.05). Lung injury was milder in the Notch1-deficient mice model compared to wild mice (p < 0.05). In vitro experiments demonstrated that inhibiting the Notch pathway in macrophages reduced CCR5 expression and attenuated CCL5-induced macrophage migration.
Notch signaling regulates macrophage infiltration and the inflammatory response by modulating CCR5 expression in ALI induced by LPS.
Graphical Abstract
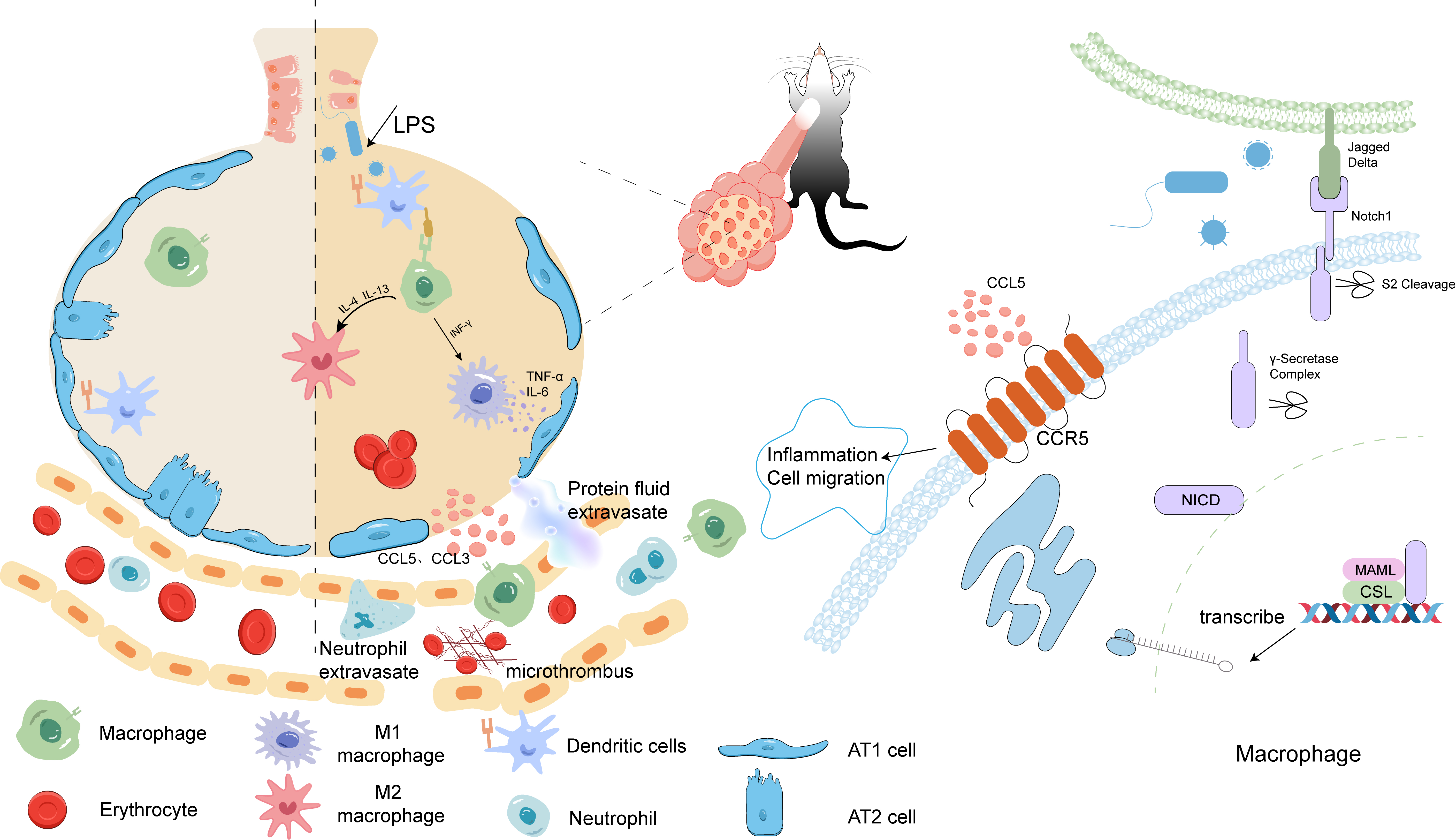
Keywords
- acute lung injury
- alveolar macrophages
- Notch
- CCR5
- cell migration
Acute lung injury (ALI) is a severe inflammatory lung disease, characterized by hypoxemia and progressive respiratory distress, which may progress to acute respiratory distress syndrome (ARDS) in clinically severe presentations [1]. The activation of alveolar macrophage (AMs) leading to excessive inflammatory responses is a primary cause of damage to alveolar cells, which results in heightened alveolar-capillary permeability, fibrin exudation into the alveoli, diffuse interstitial edema, reduced lung compliance, ultimately, impairment of pulmonary gas exchange [2].
The excessive activation of inflammation caused by infection is one of the key pathological foundations of ALI. AMs play a crucial role in recognizing and phagocytosing pathogens and necrotic cells while also maintaining the balance of the local microenvironment [3]. During the acute phase of inflammation, activated AMs undergo phenotypic polarization, accompanied by significant functional alterations. These polarized macrophages recruit additional inflammatory cells to migrate to the lungs and release a variety of inflammatory mediators, thereby amplifying the inflammatory response. Macrophage polarization can be initiated through the recognition of inflammatory cells or stimulation by cytokines, with the Notch signaling pathway playing a pivotal role in this process. This pathway regulates macrophage polarization by binding to its ligands (Jagged/Delta) and subsequently initiating nuclear transcription [4]. In addition to cytokine release, polarized AMs also participate in intercellular signaling through Notch receptors, altering their immune phenotype and exerting both pro-inflammatory and anti-inflammatory effects at different stages [5, 6]. Therefore, Notch deficiency can lead to incomplete immune responses.
C-C chemokine receptor type 5 (CCR5) is a receptor for
This study investigated the regulatory relationship between macrophage Notch1 signaling and CCR5 expression dynamics in the ALI murine model. The results showed that macrophage Notch1 signaling deficiency reduced CCR5 expression, inhibited cell migration, and suppressed the release of inflammatory mediators, thereby exerting lung-protective properties against ALI progression in experimental mice.
Myeloid-specific Notch1 knockout mice (C57BL/6J background) were generated by
targeted gene deletion, and the knockout mice (Notch1cre+) and wild-type
control mice (Notch1flox+) were produced by Cyagen Biotechnology (Suzhou,
China) and referred to as Cre and Flox mice respectively. Male mice (4–6 weeks
old, 14–18 g) were selected for the experiments. The experimental mice were
maintained under specific pathogen-free (SPF) conditions, with temperature
regulated at 22
The ALI models were established following previously described methods [13]. Notch1flox mice were divided into three experimental groups (n = 5 per group): the control group (saline), the lipopolysaccharide (LPS) group (LPS), and the LPS+Mvc group (LPS + Maraviroc). Specifically, mice were anesthetized with sodium pentobarbital (1%, 70 mg/kg, i.p.) and then administered LPS via tracheal injection. Notch1cre+ mice were randomly assigned to two groups (n = 5 per group): the saline control group and the LPS group. For the ALI model, mice were given 3 mg/kg LPS (S1732, Beyotime, Shanghai, China) via intratracheal injection. A dose of 10 µg/g Maraviroc (CCR5 antagonist, HY-13004, MedChemExpress, Monmouth County, NJ, USA) was administered 30 minutes prior to LPS induction. After 24 hours, all mice were humanely euthanized by CO2 asphyxiation (20% chamber volume per minute) to minimize suffering, followed by cervical dislocation as a secondary confirmatory method. Peripheral blood was collected via retro-orbital venous plexus puncture. The thoracic cavity was rapidly accessed through a midline sternotomy, and bilateral lung tissues were resected en bloc. The harvested specimens were used for subsequent histopathological evaluation, real-time quantitative reverse transcription PCR (qRT-PCR), and western blotting analysis.
Post-fixation, lung tissue specimens underwent sequential ethanol dehydration
followed by paraffin embedding to generate histological sections. After the
Hematoxylin and eosin (G1120, Solarbio, Beijing, China) staining of lung tissue
sections, the slides were observed under an optical microscope (Leica, Wetzlar,
Germany) for histopathological examination and quantitative lung injury scoring
[14]. For each tissue slide, five different areas were selected and examined at
high magnification (200
Fresh lung tissue was carefully blotted with absorbent paper to remove surface moisture and blood, and its wet weight was recorded. Then, the samples were incubated in a 60 °C constant-temperature incubator for 72 hours. Following complete desiccation, the tissue mass was recorded. The gravimetric ratio was determined to assess pulmonary edema severity.
500 µL of chilled phosphate-buffered saline (PBS) was injected through intratracheal instillation and gently aspirated to obtain BALF. This lavage procedure was iterated three times to maximize fluid recovery. The pooled BALF was subjected to centrifugation and the supernatant was aliquoted for protein quantification. The remaining cellular fraction was washed twice with PBS before resuspension for quantitative analysis.
Immortalized bone marrow-derived macrophages (iBMDMs) were purchased from
Cyagen Bioscience Inc., Guangzhou, China. We validated all cell
lines by Short Tandem Repeat (STR) profiling. All cells were confirmed to be
mycoplasma-free by testing with a PCR-based mycoplasma detection kit (C0301S,
Beyotime, Shanghai, China), ensuring the absence of
contamination. The iBMDM cell line was maintained in high-glucose DMEM containing
10% FBS (Gibco, Carlsbad, CA, USA) under standard culture conditions (37
°C, 5% CO2). For experimental treatments, cells were pre-incubated
with MK-0752 (
After protein extraction, samples were subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) for molecular separation. Subsequently, the resolved proteins were electrophoretically transferred onto polyvinylidene fluoride (PVDF) membranes, which were then incubated with 5% non-fat dry milk for 60 minutes at ambient temperature to prevent non-specific binding. Primary antibodies were applied according to the experimental requirements and incubated overnight at 4 °C: CCR5 (BS77962, 1:1000, Bioworld Biotechnology, Nanjing, China), NICD detection using Cleaved Notch1 (V1744) antibody (4147S, 1:1000, Cell Signaling Technology, Danvers, MA, USA), GAPDH (GB15004, 1:6000, Servicebio Biotech, Wuhan, China). After washing with Tris-Buffered Saline with Tween 20 (TBST), the membrane was incubated with the secondary antibody, Goat Anti-Rabbit IgG (H+L) HRP (A0208, 1:1000, Beyotime, Shanghai, China) at room temperature for 1 hour, followed by washing with TBST. Protein bands were visualized using Enhanced Chemiluminescence (ECL) (A10016, Abmart, Shanghai, China) detection. The quantification of blots was performed by using ImageJ software (version 1.8.0, NIH, Bethesda, MD, USA).
Total RNA isolation from pulmonary tissues and cultured cells was carried out
with Trizol reagent (15596026, Thermo Fisher Scientific, Waltham, MA, USA). RNA
integrity and concentration were assessed spectrophotometrically using a Nanodrop
One instrument (Thermo Fisher Scientific). Subsequently, cDNA was synthesized by
reverse transcription. Quantitative real-time PCR analysis was conducted with
2
| Gene | Sense (5′-3′) | Anti-sense (3′-5′) |
| CCR5 | ATGGATTTTCAAGGGTCAGTTCC | CTGAGCCGCAATTTGTTTCAC |
| TNF- |
GGCTGCCCCGACTACGT | AGGTTGACTTTCTCCTGGTATGAGA |
| IL-6 | CCGGAGAGGAGACTTCACAG | ACAGTGCATCATCGCTGTTC |
| IL-1 |
TCATTGTGGCTGTGGAGAAG | AGGCCACAGGTATTTTGTCG |
| GAPDH | AGGTCGGTGTGAACGGATTTG | GGGGTCGTTGATGGCAACA |
qRT-PCR, real-time quantitative reverse transcription PCR; CCR5, C-C chemokine receptor type 5; TNF-
Fresh lung tissue was prepared for flow cytometry analysis. 1
Enzyme Following the manufacturer’s protocols, the ELISA kits (Nanjing Jiancheng
Bioengineering Institute, Nanjing, China) were used to measure the concentrations
of interleukin (IL)-6 (H007-1), IL-1
Lung tissue sections embedded in paraffin were subjected to deparaffinization and rehydration processes. Antigen retrieval was achieved through microwave-mediated heating. Primary antibody incubation was performed overnight at 4 °C using anti-CCR5 (1:50 dilution, BS77962, Bioworld Biotechnology, Nanjing, China) and anti-F4/80 (1:200 dilution, ab6640; Abcam, Cambridge, UK). Following three washes with PBS, samples were exposed to species-specific secondary antibodies: AF488-labeled Goat Anti-Rabbit IgG (H+L) (1:300 dilution, A0423, Beyotime, Shanghai, China), Cy3-labeled Goat Anti-Rat IgG (H+L) (1:300 dilution, A0507, Beyotime, Shanghai, China) for 1 hour at 37 °C under light-protected conditions. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI, C1005, Beyotime, Shanghai, China) for 10 minutes. Fluorescence imaging was conducted using an inverted fluorescence microscope (Olympus, Tokyo, Japan).
iBMDM cells were cultured in a 37 °C incubator until reaching 80% confluence. A sterile pipette tip was used to create a wound in the cell, and the cells were treated with 100 nM MK-0752 either in the presence or absence of LPS. The relative change in the scratch area over 12 hours was measured using ImageJ software to assess the effect of MK-0752 on LPS-induced cell migration. Images were captured at 0 hours, 6 hours, and 12 hours. The extent of cell migration was represented as the relative change in the scratch area compared to the 0-hour time point.
iBMDM cells were seeded into the Transwell system and were divided into four groups: Control, LPS (1 µg/mL), LPS+CCL5 (10 ng/mL, P6780, Beyotime, Shanghai, China), and LPS+CCL5+MK-0752 (100 nM). A complete culture medium (600 µL) was added to the lower chamber, and 200 µL serum-free cell suspension with or without treatment was added to the upper chamber. LPS and MK-0752 were added to the upper chamber, while CCL5 was added to the lower chamber. After incubation at 37 ℃ for 12 hours, the unmigrated cells in the upper chamber were removed. The rest was stained with 0.1% crystal violet (C0121, Beyotime, Shanghai, China) for 20 minutes.
All data were analyzed and processed using GraphPad Prism
(version 10.0, GraphPad Software, LLC, San Diego, CA, USA) and were presented as
the mean
To investigate the pathological mechanisms, initial analyses were conducted to
evaluate pulmonary tissue alterations along with NICD and CCR5 expression levels
in Notch1flox+ mice subjected to ALI. LPS stimulation destroys lung tissue
architecture, which includes alveolar wall thickening, infiltration of red blood
cells, and inflammatory cells in the alveoli and interstitium (Fig. 1A).
Statistical evaluation demonstrated markedly elevated expression levels of CCR5
and NICD proteins in lung tissue homogenates obtained from LPS-treated animals
relative to untreated control groups (p
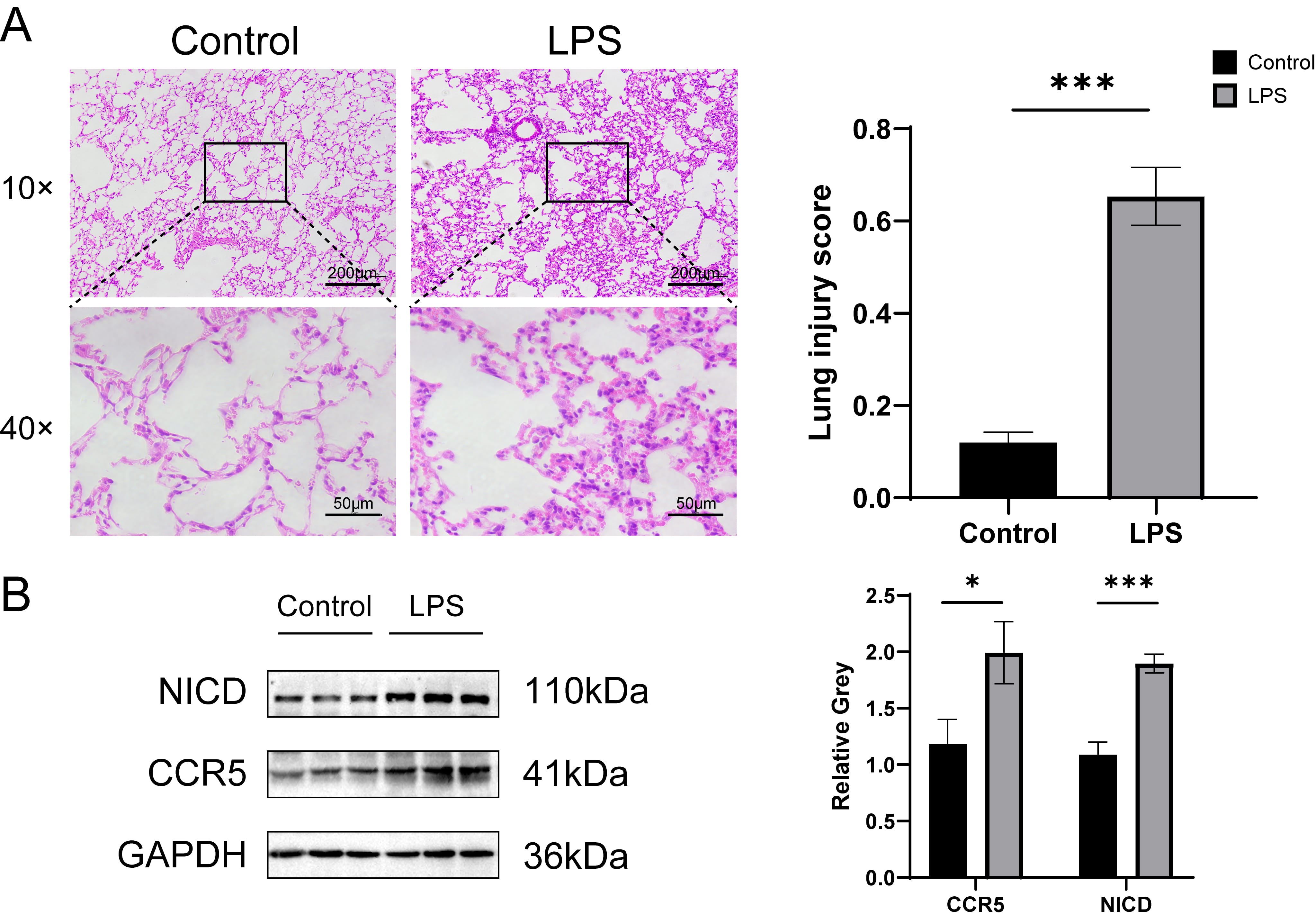 Fig. 1.
Fig. 1.
Lung tissue pathology and protein expression in ALI mice. (A)
hematoxylin and eosin (HE) staining of lung tissue sections and quantification of lung injury, n = 5. The
magnification of the objective lens was 10
To investigate the therapeutic potential of CCR5 inhibition, mice were
administered Maraviroc prior to LPS-induced ALI. Histopathological evaluation of
pulmonary tissues demonstrated that Maraviroc pretreatment attenuated
LPS-mediated lung injury, as evidenced by reduced alveolar septal thickening and
inflammatory cell infiltration (Fig. 2A), accompanied by significantly lower
histopathological scores (Fig. 2B, p
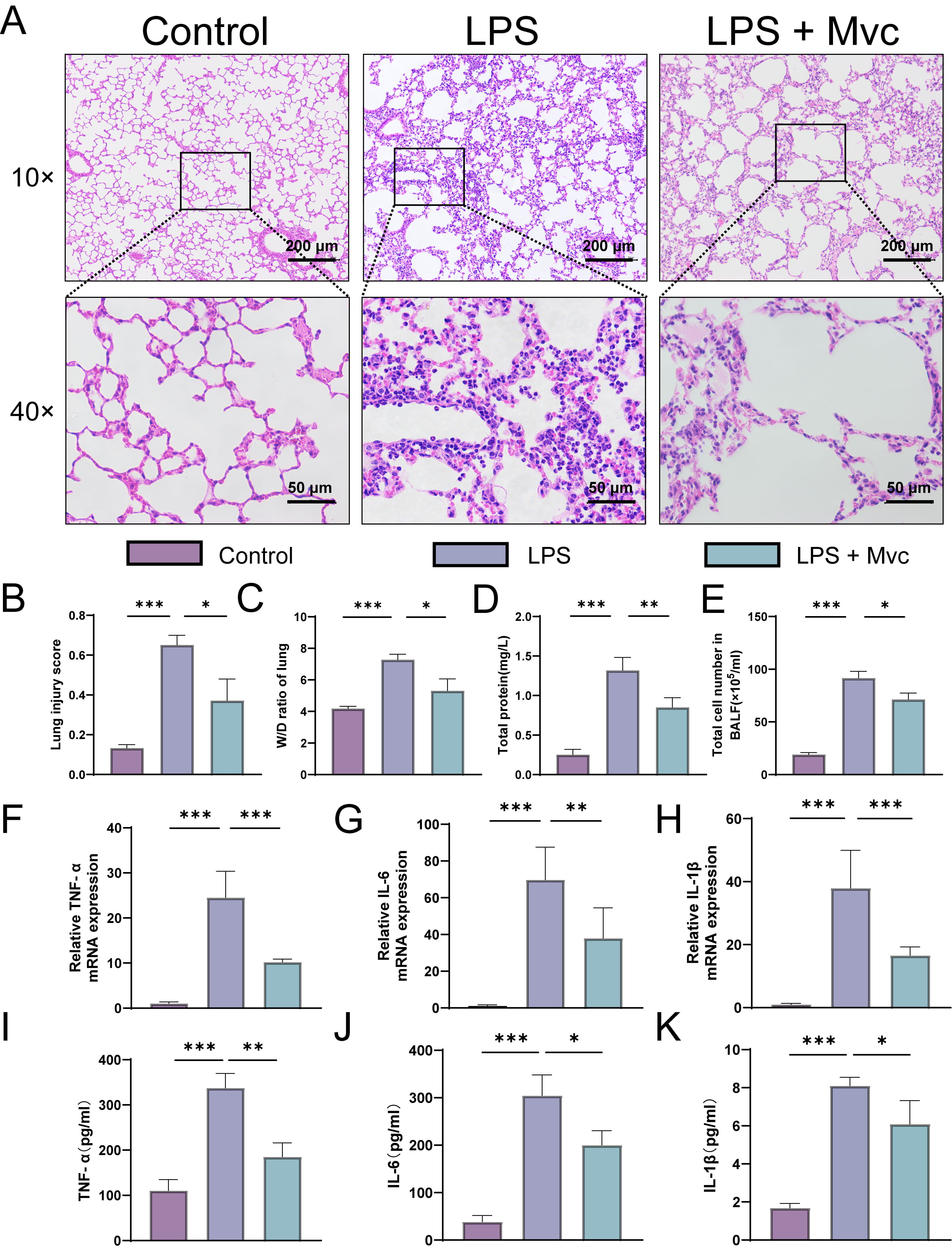 Fig. 2.
Fig. 2.
CCR5 inhibition attenuated pulmonary damage in ALI. (A,B)
Representative hematoxylin and eosin (H&E) lung sections and corresponding histopathological scoring n =
5; The magnification of the objective lens was 10
CCR5, as a receptor for inflammatory cell chemoattractants, is upregulated during the acute phase of ALI, potentially exacerbating secondary alveolar damage. It is therefore speculated that Notch may regulate CCR5 expression through ligand recognition, driving the accumulation of inflammatory cells and contributing to the pathological process.
To verify this hypothesis, we used macrophage Notch1 knockout mice to examine
the effects of Notch1 deficiency on CCR5 activity in ALI. Analysis of BALF from
ALI mice showed that both the total cell counts and total protein level in
Notch1cre+ mice were significantly lower than those of Notch1flox+ mice
(p
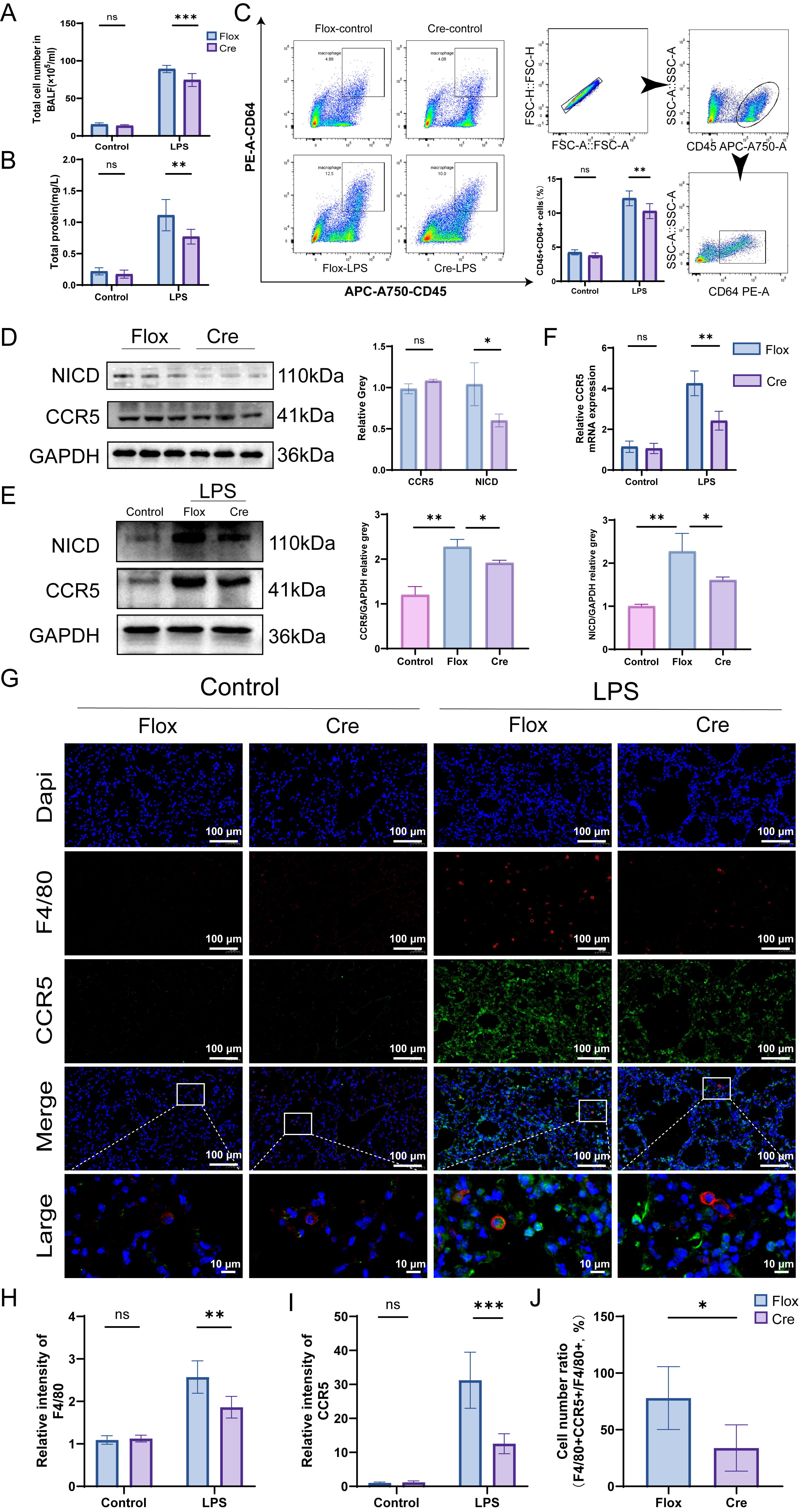 Fig. 3.
Fig. 3.
Notch1-mediated CCR5 anti-inflammatory effect. (A,B) Total cell
count and protein content in BALF, n = 5. (C) Flow cytometric analysis of lung
tissue from mice and representative gating plots; Bar graphs represent the
percentage of CD45+ and CD64+ macrophages of total cells. (D) Western blot
analysis assessing NICD and CCR5 expression in lung tissue under normal
conditions, n = 3. (E) Western blot analysis assessing NICD and CCR5 expression
in lung tissue of wild-type and Notch1 knockout mice with LPS treatment, n = 3.
(F) qRT-PCR detection of CCR5 mRNA levels in lung tissue of LPS-induced ALI mice,
n = 5. (G) Immunofluorescence detection of CCR5 expression (green) in lung tissue
macrophages (red). F4/80, red; CCR5, green; DAPI, blue; white box in each image
indicates magnified views in the inset; magnification 40
Immunofluorescence was used to detect CCR5 expression in alveolar macrophages,
with anti-F4/80 (red) and anti-CCR5 (green) antibodies marking the macrophages to
analyze CCR5 expression in macrophages of lung tissue. We found that under
physiological conditions, the proportion of F4/80 and CCR5 double-positive cells
in the lung tissue was low, with no significant difference between
Notch1flox+ and Notch1cre+ mice (p
The results indicate that although CCR5 expression in the lung tissue of Notch1cre+ mice increased after LPS stimulation, it was lower than that in Notch1flox+ mice. Additionally, the rare presence of F4/80 and CCR5 double-positive cells in Notch1cre+ mice suggests that Notch1 deficiency in alveolar macrophages impedes the expression of CCR5 on their cell membrane during LPS-induced ALI.
To elucidate the regulatory role of the Notch signaling pathway on CCR5
expression under in vitro conditions, we employed MK-0752, a specific
As illustrated in Fig. 4A, LPS stimulation markedly enhanced CCR5 fluorescence
intensity on the cellular membrane, a phenomenon that was substantially inhibited
by MK-0752 administration. Consistent with these findings, western blot analysis
demonstrated a concomitant increase in both CCR5 and NICD protein levels upon LPS
challenge, which were prominently downregulated in iBMDM cells pretreated with
MK-0752 prior to LPS exposure (p
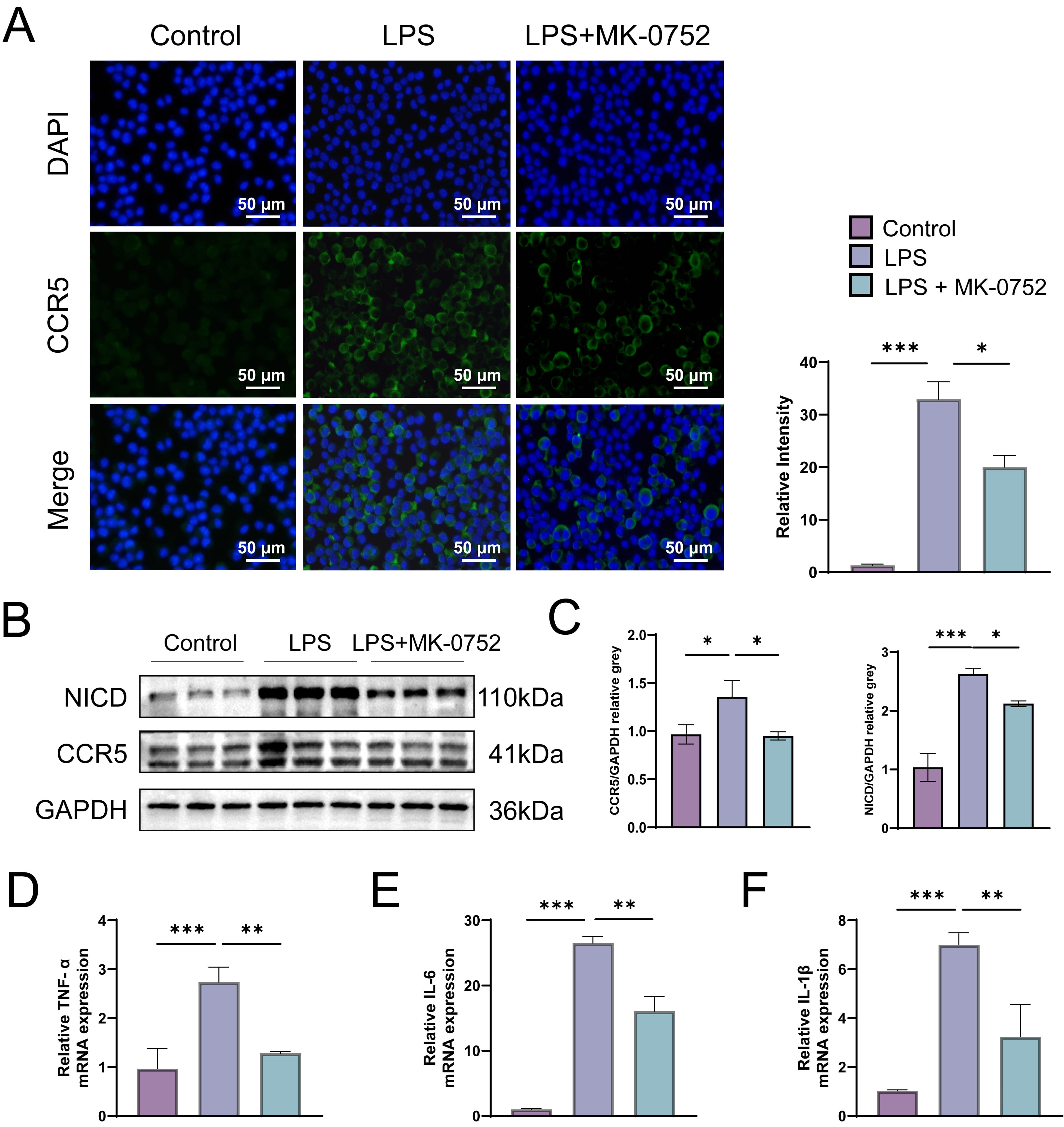 Fig. 4.
Fig. 4.
Inhibition of the Notch pathway reduces CCR5 transcription and
pro-inflammatory cytokine expression in LPS-induced iBMDM cells. (A)
Immunofluorescence was used to detect CCR5 expression (green) on iBMDM cells,
with DAPI staining (blue) for nuclei. The fluorescence intensity was quantified.
Magnification: 40
The activation of Notch receptors on macrophages, upon binding with ligands from neighboring cells, triggers the release of chemokines that orchestrate the recruitment and migration of immune cells to the lungs, thereby exacerbating lung injury [15]. CCR5, as a chemokine receptor, plays an important role in regulating macrophage migration [16, 17]. Therefore, we investigated whether Notch signaling affects macrophage migration through CCR5 expression. Since primary macrophages in vitro are adherent and difficult to track for migration efficiency, this part of the experiment utilized LPS-induced iBMDM cell migration for analysis.
The wound healing assay showed that under MK-0752-mediated
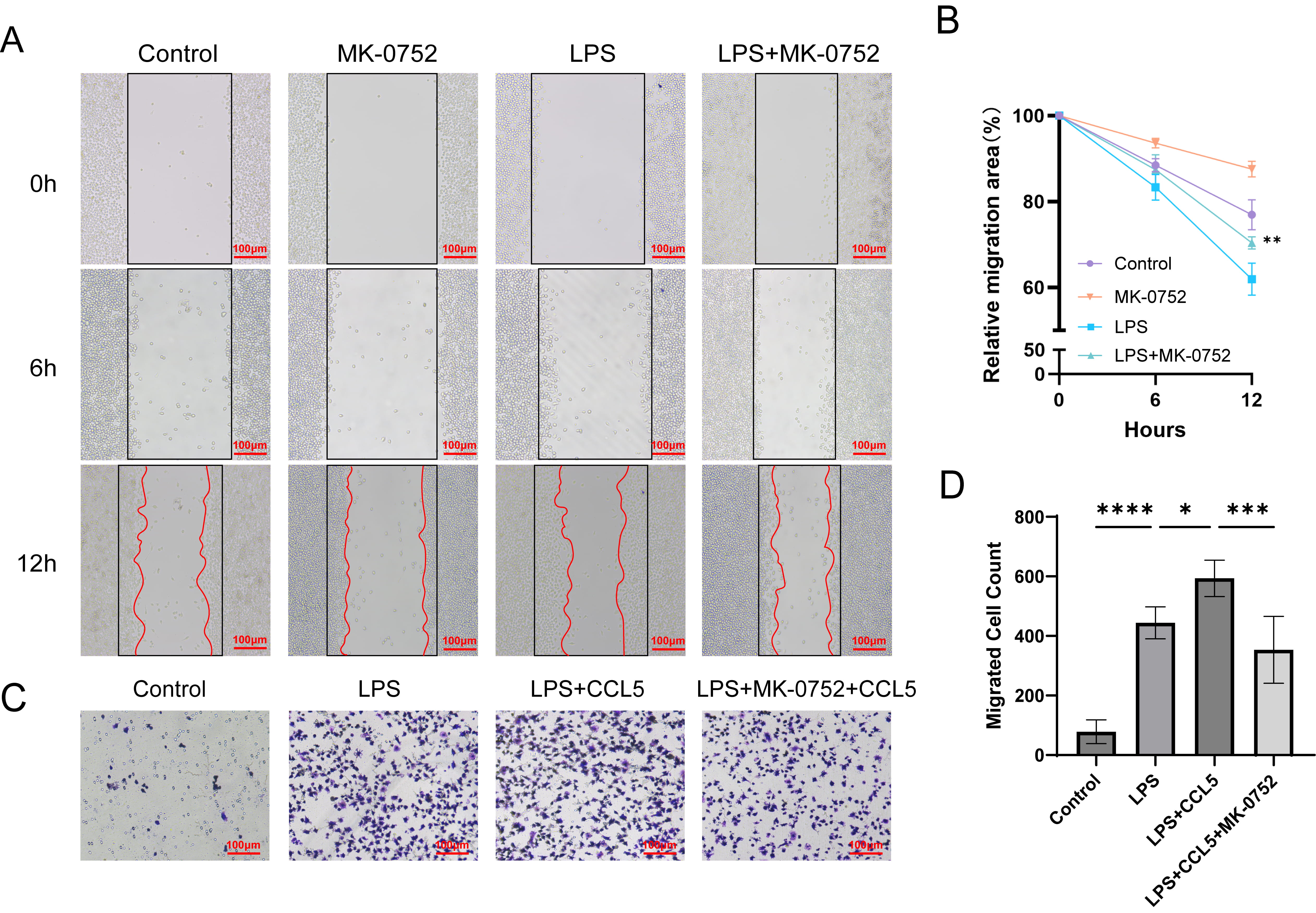 Fig. 5.
Fig. 5.
Inhibition of the Notch pathway reduces the migration efficiency
of iBMDM cells. (A) Migration of iBMDM cells under Notch inhibition was assessed
using the wound healing assay. (B) Quantification of the migration rate of iBMDM
cells at 6 and 12 hours. LPS group VS LPS+MK-0752 group **p
Sepsis is one of the leading causes of ALI and ARDS, which can progress rapidly, even resulting in death in severe cases. This investigation provides evidence that NICD and CCR5 expression undergo significant upregulation within pulmonary tissues during ALI in a murine model. In addition, the pharmacological blockade of CCR5 signaling demonstrates therapeutic potential in mitigating LPS-induced pulmonary tissue pathology, indicating a crucial involvement of CCR5 in the molecular mechanisms underlying ALI pathogenesis. The absence of Notch1 signaling or inhibition of the intracellular domain significantly reduces CCR5 expression in ALI lung macrophages, alleviates lung tissue damage, and weakens macrophage recruitment and migration mediated by CCR5. This study provides a theoretical foundation for immune modulation therapies targeting the Notch pathway.
The proliferation, differentiation, and functional activation
of myeloid and lymphocyte populations are widely regulated by the Notch signaling
cascade [18]. The Notch receptor family consists of four subtypes (Notch1-4),
which are activated upon binding to external ligands (Delta/Jagged). This
proteolytic activation results in
The directed migration of macrophages to the damaged areas is a key factor
driving acute lung injury and exacerbating the inflammatory response. LPS can
activate macrophages and polarize them into the M1 phenotype, producing
pro-inflammatory cytokines such as RANTES (CCL5) [24, 25], which recruit more
inflammatory cells to the affected lung, thereby intensifying the inflammatory
response in ALI. A previous study has suggested that monocyte accumulation in
lung injury is mediated by CCR2 [26]. Nevertheless, accumulating experimental
data suggest that CCR5 contributes significantly to this biological mechanism. In
sepsis models, monocytes with high CCR5 expression show increased migration into
the circulatory system, contributing to disease progression [27]. Moreover, the
activation of CCR5 is not limited to promoting the migration of immune cells; it
also affects their activation state and function, thereby controlling the release
of inflammatory mediators. The chemokine CCL5 mediates activation of both MAPK
and NF-
The recombination signal binding protein for immunoglobulin kappa J region
(RBP-J
Undeniably, there are some limitations in our research. To be specific, we only validated the role of Notch and CCR5 in macrophages. Nevertheless, the pathogenesis of acute inflammatory responses involves complex interactions among multiple immune cell populations and cytokine networks through diverse signaling cascades. Further studies are needed to complement and expand on these findings. Additionally, our study is based solely on animal and cell models, lacking corresponding clinical data. Further comprehensive investigations are warranted to elucidate the interplay between Notch signaling and CCR5 pathways in ALI pathogenesis, which may facilitate the development of novel therapeutic strategies for clinical translation.
Collectively, our experimental findings demonstrate that Notch signaling pathway modulation attenuates LPS-induced ALI progression through CCR5-mediated mechanisms, leading to reduced macrophage infiltration and inflammatory cascade activation. These findings identify the Notch-CCR5 signaling axis as a potential therapeutic target for the treatment of ALI.
The original contributions made in this study have been incorporated into the article and Supplementary Materials. For any further inquiries, please direct them to the corresponding author.
RYZ and DCZ conceptualized the study design. HD, YXL, HZH, JWZ, PY and DCZ wrote the initial drafts of the manuscript. All authors read and approved the final report and have no conflicting issues with the contributions listed herein. RYZ, HD: Designed the experiments; Performed the experiments; Collected and analyzed data; Wrote the paper. ZL, YXL, HZH, JWZ: Participated in experiments; Made figures; Provided technical support. PY, DCZ: Designed the experiments, Supervision, and Funding acquisition. All authors approved the final version of this manuscript. All authors contributed to editorial changes in the manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
This study was approved by The Animal Ethics Committee of Wuhan University (NO. WP20240088), adhered to the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines 2019.
Not applicable.
This study was funded by the National Natural Science Foundation of China (81670007).
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/FBL37430.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.