



 , Quan Liu 2, Zhihao Luo 1, Qian Hu 1, Li Wang 1, Zifen Guo 1,*
, Quan Liu 2, Zhihao Luo 1, Qian Hu 1, Li Wang 1, Zifen Guo 1,*
1 Institute of Pharmacy and Pharmacology, School of Pharmaceutical Science, Hengyang Medical School, University of South China, 421001 Hengyang, Hunan, China
2 Department of Laboratory Medicine, Huazhong University of Science and Technology Union Shenzhen Hospital (Nanshan Hospital), Shenzhen University, 518052 Shenzhen, Guangdong, China
Abstract
Breast cancer (BC) is the second leading cause of death among women worldwide. Immunotherapy has become an effective treatment for BC patients due to the rapid development of medical technology. Considerable breakthroughs have been made in research, marking the beginning of a new era in cancer treatment. Among them, various cancer immunotherapies such as immune checkpoint inhibitors (ICIs), cancer vaccines, and adoptive cell transfer are effective and have good prospects. The tumor microenvironment (TME) plays a crucial role in determining the outcomes of tumor immunotherapy. Tumor-associated macrophages (TAMs) are a key component of the TME, with an immunomodulatory effect closely related to the immune evasion of tumor cells, thereby affecting malignant progression. TAMs also significantly affect the therapeutic effect of ICIs (such as programmed death 1/programmed death ligand 1 (PD-1/PD-L1) inhibitors). TAMs are composed of multiple heterogeneous subpopulations, including M1 phenotypes macrophages (M1) and M2 phenotypes macrophages (M2). Furthermore, they mainly play an M2-like role and moderate a variety of harmful consequences such as angiogenesis, immunosuppression, and metastasis. Therefore, TAMs have become a key area of focus in the development of tumor therapies. However, several tumor immunotherapy studies demonstrated that ICIs are effective only in a small number of solid cancers, and tumor immunotherapy still faces relevant challenges in the treatment of solid tumors. This review explores the role of TAMs in BC immunotherapy, summarizing their involvement in BC development. It also explains the classification and functions of TAMs, outlines current tumor immunotherapy approaches and combination therapies, and discusses the challenges and potential strategies for TAMs in immuno-oncology treatments.
Keywords
- tumor-associated macrophages
- breast cancer
- immunotherapy
Breast cancer (BC) is the leading cause of cancer-related deaths among women worldwide [1]. The tumor microenvironment (TME) comprises cancer cells and host-derived normal cells, such as lymphocytes, fibroblasts, and macrophages [2]. Macrophages are involved in homeostasis, inflammation, and progression. Tumor-associated macrophages (TAMs) play a significant role in cancer progression [3] by facilitating tumor metastasis and contributing to poor clinical outcomes. High levels of TAMs in BC are considered a poor prognostic factor [4, 5]. Increased infiltration of M2 phenotypes macrophages (M2) in BC is associated with shorter survival times, whereas higher infiltration of M1 phenotypes macrophages (M1) is linked to longer lifespans across various BC subtypes (Fig. 1). These findings imply that modifying the function of TAMs might offer a prospective target for BC therapy. Immunotherapy of BC has attracted much attention recently [6], particularly for systemic treatments that involve targeting programmed death ligand 1 (PD-L1) [7]. However, immune-based cancer treatment, particularly immune checkpoint inhibitors (ICIs) ((PD-1/PD-L1) inhibitors), is efficient in only a minority proportion of patients with solid cancers. Studies reported [8, 9, 10] that TAMs influence the care process of PD-1/PD-L1 inhibitors, thus limiting the effect of the current treatment strategies for advanced malignancies. Thus, targeting TAMs could be an emerging area of interest, as these strategies may synergize with existing immunotherapies.
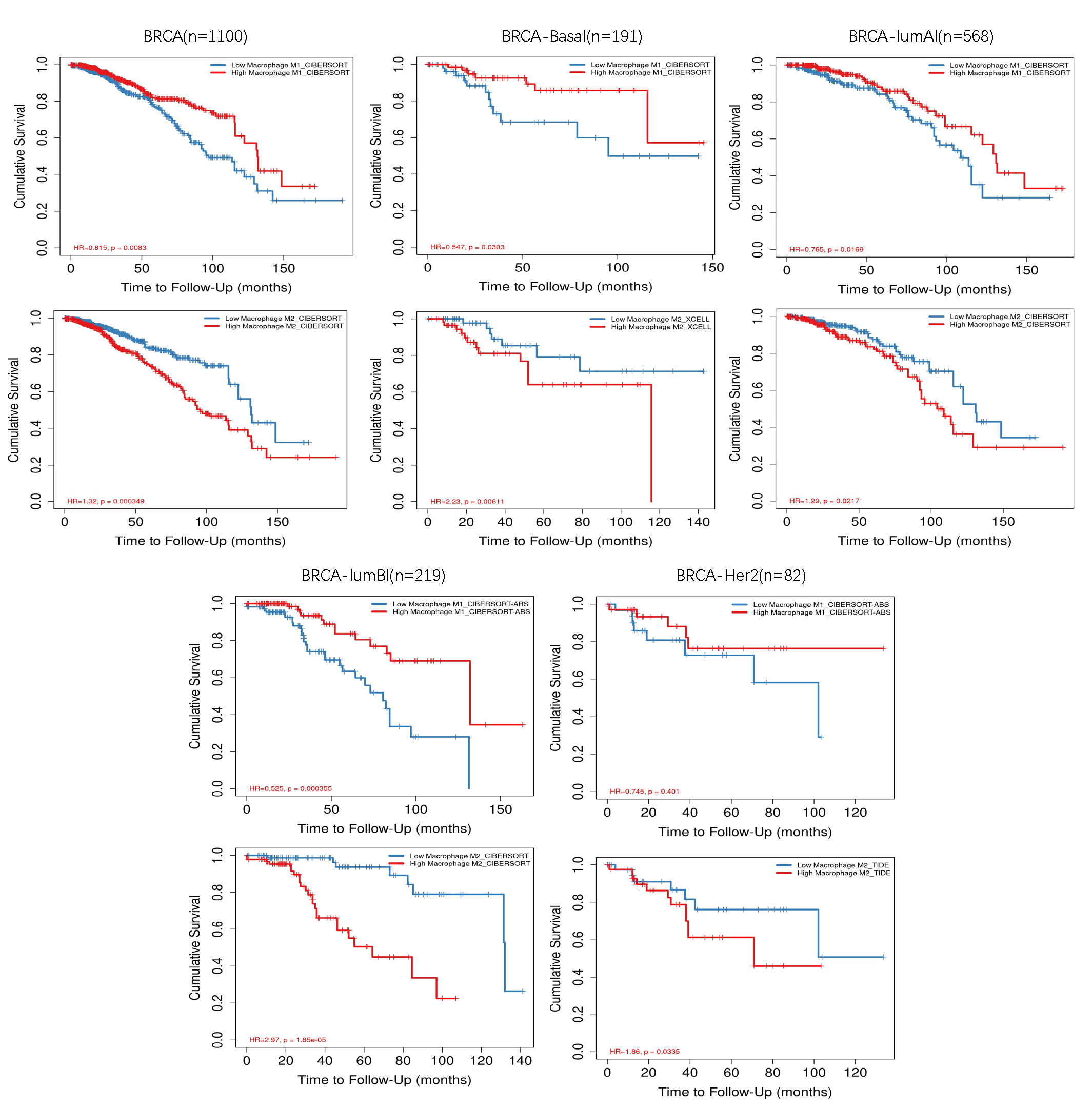 Fig. 1.
Fig. 1.
M1 and M2 infiltration in breast cancer analyzed in the Timer 2.0 database. M1, M1 phenotypes macrophages; M2, M2 phenotypes macrophages; BRCA, breast cancer susceptibility gene; BRCA-basal, BRCA1-mutated basal-like breast cancer; BRCA lumB, BRCA1/2-mutated luminal B breast cancer; BRCA lumA, BRCA1/2-mutated luminal A breast cancer; BRCA HER2+, BRCA1/2-mutated HER2-positive breast cancer; HR, hazard ratio.
Globally, BC is the most commonly diagnosed female cancer and the leading cause of cancer-related deaths, accounting for 23% of all female cancers [11]. The treatment selected is contingent upon the disease’s stage and includes surgery, radiation therapy, and chemotherapy. Chemotherapy is designed to target cancer cells that are rapidly dividing; however, its toxicity can also harm healthy cells, leading to unwanted side effects. Though hormone therapy is a widely used unspecific treatment for BC, its effectiveness is limited to tumors that are responsive to hormones, and it is associated with significant side effects [12]. Thus, researchers are exploring potential treatment strategies, including immunotherapy. Immunotherapy has been seen as a promising treatment method aimed at a specific protein found in cancer cells, holding great promise for the treatment of BC. Recent findings have uncovered the potential clinical efficacy of PD-1 and PD-L1 inhibitors in a subset of individuals with BC. For example, PD-L1 is the most commonly utilized biomarker for BC. In the phase II study (NCT02447003), monotherapy with pembrolizumab showed potential anti-tumor activity and a favorable safety profile in patients with PD-L1+ BC [13]. A phase III clinical trial (NCT02819518) revealed that the addition of pembrolizumab to conventional chemotherapy may enhance progression-free survival (PFS) in individuals with BC, as opposed to those who underwent chemotherapy alone [14]. Moreover, in the phase I trial (NCT01375842), atezolizumab as a single-agent treatment showed a tolerable safety profile and provided lasting therapeutic benefits for patients [15]. Atezolizumab combined with nab-paclitaxel demonstrated an extension of recurrence-free survival in patients when compared to the subgroup treated solely with nab-paclitaxel in a phase III clinical trial (NCT02425891) [16]. The effects of atezolizumab and pembrolizumab are long-lasting in patients with BC, indicating that these therapies may improve the quality of life for those who respond positively. Antibodies targeting PD-1, including cemiplimab and nivolumab; those targeting PD-L1, including avelumab and durvalumab; and those targeting cytotoxic T-lymphocyte-associated protein 4 (CTLA-4), including tremelimumab and ipilimumab, have shown promise in therapy and have been cleared for treating a range of cancers, encompassing solid tumors. While ICIs like PD-1 and PD-L1 blockers are commonly used in clinical settings [17], there are also emerging therapies aimed at novel targets such as LAG3, TIM3, and ICOS that are in the developmental phase [18], which are suppressive receptors that negatively regulate T cell function and suppress immune cells, thereby promoting tumor escape immune surveillance; Challenges for effective use of ICIs persist such as the identification of definitive biomarkers and effective combinatorial strategies, inherent or acquired resistance to ICIs and some therapeutic side effects [19]. Given the remarkable clinical success of ICIs in many solid tumors, immunotherapy has become a new therapeutic approach and one of the significant landmarks in modern cancer research. In addition, ICIs synergized with chemotherapy have shown notable effectiveness in both early-stage as well as metastatic breast cancer (mBC), which has sparked interest in immune-based BC treatment and prevention strategies.
Under the guidance of chemokine ligand 2 (CCL2), chemokine ligand 18 (CCL18),
chemokine ligand 20 (CCL20), colony-stimulating factor-1 (CSF-1), and vascular
endothelial growth factor (VEGF)—cytokines and growth factors—monocytes from
the blood convert into macrophages. A variety of factors alter the phenotypes and
functions of macrophages. Lipopolysaccharide (LPS), tumor necrosis factor alpha
(TNF-
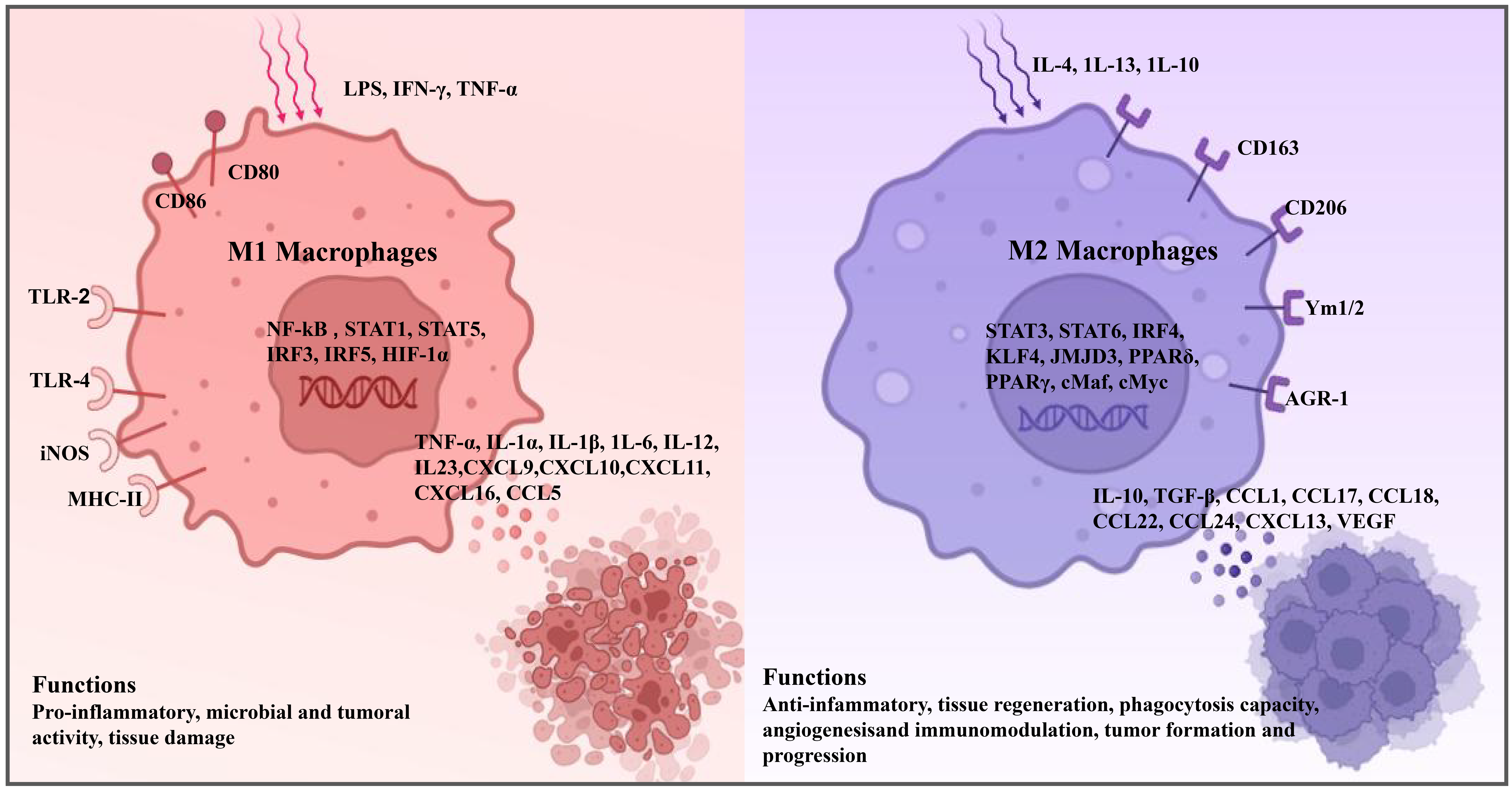 Fig. 2.
Fig. 2.
TAM subtype: function of M1 and M2 macrophages. TAM,
tumor-associated macrophage; LPS, lipopolysaccharide; IFN-
In contrast, M2 macrophages, similar to TAMs in phenotype, facilitate cancer progression and metastasis and correlate with an adverse prognosis [20]. They contribute to the promotion of cancerous expansion, invasion, metastasis, and angiogenesis through the secretion of cytokines and growth factors. Moreover, M2 macrophages secrete immunosuppressive cytokines that suppress the action and metabolic activity of T cells [23]. They suppress T helper 1 cell (Th1) immunity, which is fundamental for healing injured tissues, thereby accelerating cancerous spread and neovascularization [24]. Furthermore, TAMs directly impede the expansion of CD8+ T cells by metabolizing L-arginine through arginase 1 (ARG-1), nitric oxide synthase 2 (Nos2), oxidative free radicals, or nitrogen-based compounds. They also lure Tregs with chemokine ligand 22 (CCL22), further restricting T cell tumor-inhibiting actions [25, 26].
This section primarily depicts the features and functions of the M1 and M2 states in BC, to inspire tactics to hinder the M2 macrophage polarization and amplify the benefits of immunotherapy (Fig. 2).
The TME is an acidic anoxic environment [27]. Angiogenesis is vital to supply
oxygen and nutrients to the tumor, and TAMs are involved in this process in many
types of malignancies, such as ovarian carcinoma, melanoma, and BC [28]. TAMs
sense intratumor hypoxia and react with vascular endothelial growth factor-alpha
(VEGF-
TAMs promote immunosuppressive changes by secreting immune-modulating
anti-inflammatory factors, including TGF-
The presence of a macrophage population with a predominance of the M2 phenotype often leads to treatment resistance. According to Yang et al. [38], TAMs release IL-10, leading to hyperexpression of B-cell lymphoma 2 (BCL-2) and signal transducer and activator of transcript-3 (STAT3), which in turn triggers the IL-10/STAT3/BCL2 pathway in BC cells and boosts therapeutic barriers. TAMs are correlated with resistance to tamoxifen in postmenopausal women with BC, and they prevent the enlistment of CD8+ cytotoxic T cells, leading to the evolution of multidrug resistance [39]. M2 TAMs are more resistant to radiation therapy than M1 macrophages, yielding a challenge in the treatment of cancer.
The lack of response to PD-1/PD-L1 blockade treatment is partly due to the presence of TAMs. Monoclonal antibodies that target checkpoint ligands can be captured by TAMs due to the expression of these ligands on TAMs, such as PD-L1/2. This interaction can render the antibodies ineffective [40]. Paclitaxel or carboplatin treatment is counteracted by an increased IL-10 secretion by TAMs, leading to diminished IL-12 in DCs and an interruption of CD8+ T cell tumor-inhibiting capacity [41]. Understanding the resistance mechanisms of TAMs to therapy is crucial for crafting treatment approaches to overcome those problems and augment medical treatment potency without the risk of recurrence.
Epithelial cell mesenchymal transition (EMT) enables the transition of cancer
cells from the primary site by entering the bloodstream and infiltrating other
parts of the body, eventually developing tumor metastasis. M2 TAMs are involved
in EMT during the development of cancer. They promote the EMT process through
multiple pathways, for example, toll-like receptor 4 (TLR4) and IL-10pathway, TGF-
On the other hand, M2 macrophages release components that directly affect the
expansion of malignant cells, like TGF-
Macrophages play a key role in shaping tumor immunity,
constituting over half of the solid tumor’s mass and representing the predominant
immune cell type infiltrating these tumors [48]. Meanwhile, exhaustion of T cells
significantly hinders the immune response to tumors. This report demonstrated
that the in vivo removal of TAMs alleviates the exhaustion programs of
intratumoral CD8+ T cells [49]. Reciprocally, the exhaustion programs of Tcells release signals that actively draw monocytes into the TME and influences
their differentiation toward M2 macrophages to inhibit the TME. Therefore,
blocking TAM polarization and relieving immunosuppression may be a future
research direction in cancer therapy. CD8+ T cells act as suppressors of tumor
growth, engaging with cancerous cells and triggering their destruction by
initiating internal signaling pathways within the context of tumor immunity. In
the TME, “immune exclusion” impedes cancer immunotherapy by preventing CD8+ T
cells from accessing the area around tumor cells and suppressing their immune
functions [50]. This leads to a scarcity of CD8+ T cells within the TME and
allows tumor cells to evade immune detection [51, 52, 53]. Li et al. [54]
demonstrated that membrane spanning 4-domains A4A (MS4A4A) blockade treatment on
TAM reshapes the immune TME, to decrease the presence of M2-TAMs while enhancing
the infiltration of active CD8+ T cells. The generation of PD-L1 on macrophages
promotes breast tumor immune evasion. Fang and colleagues [55] found that the
production of M2 markers and PD-L1 was raised in macrophages after the treatment
with progranulin. However, the account of immune cells in progranulin–/– BC
tissue were upregulated, and the infiltration of CD8+ T cells was also
elevated. Furthermore, TAMs release
anti-inflammatory cytokines such as TGF-
Until recently, cancer prevention and intervention measures have primarily centered on factors intrinsic to cancer cells. However, recent research is increasingly focused on targeting various active immune cells like macrophages and neutrophils. Among the various cell types in BC, macrophages are considered the most critical, constituting more than half of the tumor’s volume in many malignancies. Their anti-tumor activities include direct cytotoxicity toward cancer cells and the presentation of tumor antigens to cytotoxic T cells. TAMs also boost tumor development directly and/or indirectly by stimulating tumor angiogenesis and metastasis.
Immunotherapy strategies targeting the immune system are promising for BC treatment, even though BC is not generally considered highly responsive to immune-based interventions. Currently, a range of cancer immunotherapy approaches are being developed to target TAMs. Most research methods fall into four categories: The initial method aims to block TAMs from accumulating at the tumor’s location. Pharmaceuticals, including carlumab [59], Bindarit [60], and thalidomide [61], prevent monocytes from being drawn to the tumor, halting their development into TAMs and their contribution to cancerous development. Trabectedin [62] also hamper carcinoma development by either destroying TAMs or lowering their survival rates through a range of mechanisms. To impede TAM-driven tumorigenesis, one can also aim to decrease the differentiation and polarization of macrophages. Compounds such as LPS and TLR agonists [63] can invert the differentiation of macrophages, steering them to resort to the M1 state that is adverse to tumors, while triterpenoid compounds [64] can prevent the early polarization of M1 into the M2 state. In conclusion, blocking the growth of new blood vessels is another potential method to address tumor progression. Combining anti-VEGF antibodies with treatments such as Avastin, bevacizumab [65], or other neutralizing antibodies makes it possible to inhibit the transition of macrophages into the tumor area and the formation of new blood vessels that tumors require. For example, TAMs in solid tumors regulate the angiogenesis process; therefore, their elimination by the medication clodronate [66] leads to a slump in the density of blood vessels within the tumor [67]. Additionally, elevated levels of CSF-1 recruit more TAMs. However, silencing the CSF-1 receptor (CSF-1R) using siRNA reduces angiogenesis and inhibits macrophage migration [68] (Table 1, Ref. [59, 60, 61, 62, 63, 64, 65, 66]). However, the immune response varies according to the BC subtype, and not all patients may experience beneficial effects from the same immunotherapeutic strategy. Thus, new therapeutic strategies to inhibit tumor growth are needed, including anti-tumor vaccine, combined immunotherapy, and nano immunotherapy to enhance anti-cancer effects. This review mainly focuses on the role of TAMs in the development of BC, the challenges and coping strategies faced in tumor immunotherapy, and summarizes the current methods of tumor immunotherapy and combined therapy in detail.
| Drug | Type | Adverse drug reactions and toxicity | Anti-tumor function | Reference |
| Carlumab | Human immunoglobulin G1 kappa monoclonal antibody with high binding affinity and specificity for CCL2 | Shown to possess a favorable safety profile in previous phase 1 and phase 2 clinical studies when administered at up to 15 mg/kg | By binding with CCL2, the content of serum is reduced, thereby reducing the infiltration of CD68+ macrophages/monocytes in tumor tissues, thus slowing tumor growth and angiogenesis | [59] |
| Bindarit | Selective inhibitor of monocyte chemotactic protein with anti-inflammatory activity | Its safety and efficacy have been verified by phase II clinical trials in patients with lupus nephritis and coronary stent restenosis | Inhibits macrophage infiltration and activation | [60] |
| Thalidomide | Immunosuppressor | Severe teratogenicity in the infant | Immunomodulatory and anti-inflammatory effects. Inhibition of angiogenesis and anti-tumor effects: cytokines, such as VEGF and fibroblast factor, are angiogenesis stimulants, which bind to specific receptors to stimulate signal transduction and cause endothelial cell proliferation | [61] |
| Trabectedin | Tetrahydroisoquinoline alkaloid | Manifested as nausea, constipation, fatigue, vomiting, and headaches, yet in some cases, they can escalate to severe complications like cardiomyopathy, anaphylaxis, neutropenia progressing to sepsis, rhabdomyolysis, or tissue necrosis due to extravasation | Inhibits pro-inflammatory mediators produced by monocytes, macrophages and TAMs, such as CXCL8 and IL-6. In addition, trabectedin directly targets endothelial cells to upregulate metalloproteinase-tissue inhibitor-1, metalloproteinase-tissue inhibitor-2, and thrombin sensitive protein-1, and exerts antiangiogenic activity in myxoid lipoma coma | [62] |
| LPS | TLR agonists | No toxic, non-specific immunogen | Interacting with TLR4 to form LPS signaling complexes leads to the synthesis of pro-inflammatory cytokines, including IL-1 |
[63] |
| Corosolic acid (CA) and oleanolic acid (OA) | Triterpenoid compounds | No cytotoxicity | Inhibited macrophage polarization to M2 phenotype by suppressing STAT3 activation | [64] |
| Avastin | Anti-VEGF antibodies | Anemia, pain, abdominal pain, headache, high blood pressure, diarrhea, nausea, vomiting, loss of appetite | Inhibit tumor angiogenesis by specifically binding and blocking VEGF, thereby inhibiting tumor growth and spread | [65] |
| Bevacizumab | Anti-VEGF antibodies | Gastrointestinal perforation, bleeding, arterial thromboembolism | Binds specifically to VEGF-A and prevents its interaction with VEGFR, thereby inhibiting endothelial cell proliferation, activating survival pathways, and the formation of new blood vessels and angiogenesis | [65] |
| Clodronate | Bisphosphonate drugs | Well tolerated orally, and the main side effects are lower digestive tract symptoms such as diarrhea | An effective macrophage removal tool that can remove macrophages from different tissue sites and blood in animals. It uses the macrophage endocytosis mechanism to bring clodronate into the cell, release and accumulate in the cell under the action of macrophage lysosomal phosphatase, and induce apoptosis of macrophages | [66] |
CCL2, C-C motif chemokine ligand 2; TAM, tumor-associated macrophages; CXCL8,
C-X-C motif chemokine ligand 8; IL-6, interleukin-6; LPS, lipopolysaccharide;
TLR, toll-like receptor; TNF-
Triple-negative breast cancer (TNBC) is a subtype of BC characterized by the
absence of estrogen receptor (ER), progesterone receptor (PR), and human
epidermal growth factor receptor 2 (HER2) expression on the cancer cells. This
subtype is the most invasive form of BC with the poorest prognosis [69]. Among
the infiltrating immune cells, TAMs constitute the main fraction of the TME.
Thus, treatments that target TAMs hold great promise as therapeutic strategies
for TNBC. Not only does blocking the macrophage colony-stimulating factor
(M-CSF)/CSF-1R signaling pathway reduce tumor growth in BC xenograft models [70],
but CSF-1R blockade augments the movement and
penetration of CD8+ T cells into tumors [71, 72, 73, 74]. Furthermore, when in combination
with anti-PD-1 treatment, it further promotes the buildup of CD8+ T cells around
cancer cells and reducing tumor growth [53]. Another study indicated that a
combination of CSF-1R inhibitor and an activated anti-CD40 antibody increases M2
to M1, inhibits the conversion of the G2/M cell cycle phase in cancer cells,
promotes the production of TNF-
In addition to the above treatment methods, several other effective methods exist. Phosphoglycerate mutase 1 (PGAM1) plays a pivotal role in the metabolic pathways of cancer, exhibiting highly abundant expression in TNBC and is correlated with an unfavorable prognosis. Inhibition of PGAM1 works synergistically with anti-PD-1 immunotherapy, markedly reshaping the TME. This results in an elevation of immune effector subsets like CD8+ T cell and M1 and a downsize in the infiltration of suppressive immune cells, such as myeloid-derived suppressor cells, M2 macrophages, and Tregs [80]. Moreover, high-mobility group box 1 (HMGB1) is a dynamic redox-actuated protein involved in an array of intracellular activities (e.g., chromosomal architecture reorganization, DNA transcription, and autophagic processes) and extracellular processes (e.g., autoimmune inflammation and immune dysregulation). HMGB1 inhibition leads to a drastic reduction in myeloid-derived suppressor cell (MDSC) and Tregs, an elevated proportion of M1 relative to M2, along with heightened stimulation of DCs and pDCs and an improvement in the success rate of monotherapies using anti-PD-1 in cancer care [81]. Elevated levels of RNA binding motif single stranded interacting protein 1 (RBMS1) in BC are directly linked to higher PD-L1 expression. The absence of RBMS1 leads to the destabilization of beta-1,4-galactosyltransferase 1 (B4GALT1) transcript, preventing the glycosylation of PD-L1, enhancing its ubiquitination, and targeting it for degradation. When combined with CTLA-4 ICIs or chimeric antigen receptor T-cell immunotherapy (CAR-T) therapy, the loss of RBMS1 boosts anti-tumor T cell responses both in vitro and in vivo [82]. High long noncoding RNA NR_109 (lncRNA NR_109) expression is present in M2. NR_109 knockdown inhibits polarization of M2 and their activity in supporting the expansion and invasiveness of malignant cells both in vitro and in vivo [83]. The presence of T cell malignancy 1 (MCT-1) is an innovative predictor of disease progression in individuals with invasive BCs. Oncogenic MCT-1 activation promotes mammary tumor progression and increases M2 TAMs. In contrast, MCT-1 knockdown decreases M2 macrophages and increases tumor-suppressive M1 macrophages [42]. Overall, there are many targets that can regulate TAMs to control tumor progression.
Immunotherapy for BC has its limitations due to low tumor immunogenicity and an immunosuppressive TME. In recent years, researchers have integrated material science, chemistry, and medicine to obtain nanomedicine, which represents an emerging strategy for BC treatment. Nanocarriers transport chemotherapeutic drugs and natural substances, enhancing their lethal effect on BC cells and circumventing the onset of drug resistance [84]. One study introduces a lymphatic tumor homing peptide-1 (LyP-1) and chondroitin sulfate (CS) dual-modified liposome co-loaded with paclitaxel and cryptotanshinone, named CS/LyP-1-PC Lip, designed to bolster chemoimmunotherapy effects in TNBC through triggering immunogenic cell demise and suppressing signal transducer and activator of transcription 3 (STAT3) signaling [85]. This study offers a feasible combination regimen for the development of TNBC chemoimmunotherapy. Another study showed that Rhodiola rosea polysaccharide-based nanoparticles loaded with doxorubicin boost chemoimmunotherapy for TNBC by re-educating TAMs [86]. Combined with immunomodulatory drugs, these nanoparticles activate immune cells to achieve better therapeutic results. Recently, a study reported that these PD-L1-loading microparticles alleviated the suppressed state of the immune microenvironment, eventually impairing the TNBC progression due to boosting the activation and function of CD8+ T cells [87]. Others mediate the transformation of macrophages toward M1 through the interruption of the TANK-binding kinase 1/signal transducer and activator of transcription 6 (TBK1/STAT6) pathway and activity of the protein kinase B/ mammalian target of rapamycin (AKT/mTOR) pathway. Chen et al. [88] designed FA-CD@PP-CpG (FA-CuS/DTX@PEI-PpIXCpG) for synergistic phototherapy (including photodynamic and photothermal therapy) and docetaxel-enhanced immunotherapy. Moreover, loaded docetaxel in FA-CD@PP-CpG promotes the infiltration of cytotoxic T-lymphocytes (CTLs), improves the efficacy of anti-PD-L1 antibody, suppresses MDSCs, and competently polarizes them toward the M1 state, thereby reducing tumor burden and further enhancing the anti-tumor efficacy. In addition, the combination of chemotherapy with atezolizumab efficiently abrogates the state of immunosuppression, providing new insights into clinical TNBC immunotherapy [87].
The hyperexpression of HER2, which occurs in about 15–20% of BC instances, is linked to an unfavorable prognosis and reduced PFS and overall survival (OS). Over the past few decades, HER2-targeting monoclonal antibodies and 3 inhibitors with CAR-T cells. The elimination of (TKI) have been part of the diverse treatment strategies for HER2+ BC. Not to mention, patients who undergo disease progression following multiple HER2-directed therapies frequently face a limited array of additional treatment choices [82, 89]. Considering TAM’s role, the selective targeting of TAMs could be a strategy to regulate the TME.
Tucatinib significantly inhibits tumor growth and increases the number of
CD8+/PD-1+ and CD8+/TIM3+ T cells, CD49+ NK cells, monocytes, and DCs and
macrophages, while decreases myeloid-derived suppressor cells. Additionally,
tucatinib effectively manages the TME, which is a significant aspect of its
therapeutic impact on BC. Multitherapy with anti-PD-L1 or anti-PD-1 showed
strengthened effectiveness in HER2+ patients [90]. Additionally, VEGF and
placental growth factors are abundantly expressed in BC cells and M2 in the
tissues, mediating tumor progression and immunosuppression. The study reported
the application of VEGF siRNA and placental growth factor siRNA to both M2 and BC
cells for successful anti-tumor immunotherapy [91]. Given that elevated
sphingosine 1-phosphate receptor 3 (S1PR3) expression is linked to resistance to
PD-1-based immunotherapy and higher levels of T cell exhaustion, employing an
S1PR3 antagonist can boost the oncostatic efficacy of CAR-T
cell therapy. This is achieved by curbing T cell exhaustion and reshaping the
TME, which involves attracting pro-inflammatory macrophages [92]. The case
further justifies the combination of S1PR3 inhibitors with CAR-T cells. The
elimination of proline-rich tyrosine kinase 2 ch1 signaling and hinders CCL2
production by BC cells but co (PYK2) decreases the penetration of TAMs and
simultaneously prevents angiogenesis and tumor growth. Specifically, PYK2
ablation not only hampers Notch1 signaling and hinders CCL2 production by BC
cells but concurrently curbs the generation of chemokine receptor 2 (CCR2),
chemokine receptor 4 (CXCR4), and interleukin-4 receptor subunit alpha
(IL-4R
Many treatments, including radiotherapy, chemotherapy, and targeted therapy, are not effective in a majority of cancer patients [97, 98, 99]. Immunosuppression is the primary barrier to fully realizing the therapeutic potential of immunotherapies [9, 100]. Moreover, a higher degree of M2-like macrophage infiltration is related to immune suppression. Thus, seven combined treatment methods are summarized below to provide a reference for clinical treatments.
The aim of vaccines designed for anti-tumor therapy is to provoke a highly specific cellular immune response against the cancer. Moreover, prompt T-cell reactions preempt the recurrence of tumors by establishing a long-term immune memory [101]. A DNA vaccine targeting the cysteine protease legumain, which is highly expressed within TAMs, has the ability to slump TAM density and inhibits cancerous growth, neovascularization, and metastasis [102]. A combination therapy of a DNA vaccination encoding the HER2 protein and trastuzumab demonstrates positive outcomes in the treatment of patients with HER2+ mBC [103]. The combination with GM-CSF is performed to increase DC function and to hamper Tregs. GM-CSF also promotes the polarization of macrophages toward the M1 phenotype and activates their anti-tumor functions. For example, the GVAX vaccine, an anti-tumor vaccine genetically modified with GM-CSF gene, combined with cyclophosphamide and trastuzumab, is utilized to treat HER2-negative mBC [104]. According to Garcia’s findings [105], an anti-CD47 antibody along with PD-L1 blockade increases the frequency of innate and adaptive immune regulatory reactions and strengthens the vaccinal effect of treatments. In addition, immunotherapeutic vaccination triggers immune identification and elimination of BC by embedding malignant antigens such as HER2. HER2/neu-based vaccination combined with GMCSF vaccine amplifier and trastuzumab is an effective treatment for TNBC [106].
Thus far, numerous immunological checkpoint inhibitors have been documented, yet
the predominant ones utilized in clinical settings are those targeting PD-1 and
PD-L1. The PD-1/PD-L1 axis and CTLA-4 both act as suppressive signals that dampen
the immune response of T cells. Preventing the PD-1/PD-L1 axis to boost the
cytotoxic capabilities of T cells has shown efficacy in resolving malignancies
[107]. Administering pembrolizumab alongside standard chemotherapy enhances PFS
in individuals with TNBC when compared to those undergoing chemotherapy alone
[14]. Moreover, pre-treatment with cisplatin and doxorubicin followed by
nivolumab leads to a more effective therapeutic outcome, a result linked to the
chemotherapeutic drugs’ ability to modulate the immune system and create a
supportive TME for PD-1 inhibitors. Tumor
cells stimulate macrophages to upregulate the expression of IL-15 receptor alpha
subunit (IL-15R
Remodeling the TME with nanocarriers represents a promising approach to boost
the efficacy of immunotherapy. CCL2 attracts macrophages and monocytes, which
then evolve into M2 and MDSCs. Lipid-protamine nanostructures are designed to
deliver a plasmid that traps CCL2 and inhibits its ability to recruit M2 and
MDSCs, thereby enhancing anti-tumor immunity and impeding cancer advancement
[113]. Cationic nanoparticles encapsulating siRNA-CCR2
(CNP-siCCR2) target the CCL2-CCR2 signaling pathway, which leads to the
downregulation of CCR2 in monocytes [114], these nanoparticles can alter the TME,
thereby inhibiting both tumor expansion and spreading. Jung et al. [115]
developed 7C1 nanoparticles that are packed with CX3CL1. These nanoparticles have
been successful in reducing the level of CX3CL1 and preventing the gathering of
macrophages in the TME. In addition, liposomal formulations of zoledronic acid
have been shown to deplete TAMs, reduce the presentation of the M2 marker CD206,
inhibit the expression of CD31, and consequently decreasing neovascularization
and breast tumor growth in TNBC. Like zoledronic acid, liposomal
nanoparticle-delivered guano-sine monophosphate–adenosine monophosphate (GAMP)
also suppresses TNBC development by the reprogramming of the M2 phenotype back to
the M1 state [116, 117]. Although signal regulatory protein alpha
(SIRP
Inhibitors, such as poly ADP ribose polymerase (PARP), HER2, PI3K, AKT, mTOR,
EGFRs, and VEGF, could serve as treatments to prevent the advancement of BC due
to their roles in the cell cycle, angiogenesis, and metastasis [121, 122].
Rapamycin, a mTOR inhibitor, has a positive therapeutic effect on
hormone-resistant metastatic ER+ BC. A synergy regimen of everolimus with
exemestane or trastuzumab yields favorable therapeutic outcomes
in both HER2+/– BC [123]. Early-phase research suggests that PARP inhibitors
increase cytoplasmic DNA and activate stimulator of interferon genes (STING)
protein, which increases INF-
The use of paclitaxel in BC leads to an increase in CSF-1, which in turn boosts
the migration of TAMs [3]. When CSF-1 inhibitors are administered with
paclitaxel, it not only raises the number of T cells in the tumor and improves
the treatment’s effectiveness but also curbs metastasis [4].
The CSF-1R inhibitor BLZ-945 and the chemotherapeutic drugs
doxorubicin and epirubicin specifically target and eliminate TAMs [128, 129].
Pembrolizumab in association with chemotherapy drugs for locally recurrent
unresectable or metastatic TNBC patients. Atezolizumab and nab-paclitaxel prolong
PFS among patients with metastatic TNBC and PD-L1–positive subgroup [16].
Blocking the CCL2 pathway with anti-CCL2 antibodies in BC models has been shown
to reduce both tumor growth and migration. This approach is even more effective
when the monoclonal antibody against CCL2 (such as carlumab) is used
with other chemotherapy drugs for patients [47, 59]. In
addition, PLX3397 with radiotherapy suppresses the variation of monocytes into
TAMs and inhibits STAT6 tyrosine phosphorylation, consequently reducing drug
resistance [130, 131]. The inhibition of the CD47-SIRP
Oncolytic viruses (OVs) represent a promising category of cancer treatments that proliferate within cancerous cells and elicit anti-tumor reactions. OVs trigger immunogenic cell death in cancer cells, stimulating T-cell activation, and fostering an immune response that protects against tumor growth, such as the HSV-derived oncolytic virus T-VEC has successfully passed Phase III clinical trials and has received approval from the FDA (U.S. Food and Drug Administration) for utilization in immunological cancer treatments [133]. Furthermore, studies in the early phases of clinical research, encompassing various solid tumor types such as BC, have indicated that OVs possess therapeutic potential with low toxicity [134, 135, 136, 137]. Given their therapeutic benefits, safety profiles, and minimal side effects, the deployment of these engineered viruses in immunological cancer treatments could mark a significant advancement in oncology. OVs have demonstrated the ability to stimulate the anti-tumor activities of specific immune cells, such as T cells. Research has shown that HSV1716 is capable of reconfiguring TAMs towards a phenotype that is less suppressive to the immune response. Administration of HSV1716 elevated the count of F4/80+ TAMs that displayed pro-inflammatory, M1 markers, like IL-12 and NOS2, compared to the controls without the virus. In addition, treatment with HSV1716 markedly decreased the quantity of F4/80+ MRC1+ TAMs [138]. This transformation of TAMs could potentially shift the balance within the TME, converting M2 into M1, which in turn facilitates the attraction of adaptive immune cells and enhances cytotoxic capabilities. In addition, OVs have been shown to increase sensitivity to immune checkpoint blockers. For instance, treatment with OVs makes typically unresponsive TNBC susceptible to immune checkpoint inhibitors, preventing recurrence in the majority of animals that received the treatment. The combination of OV therapy with ICIs serves as a potential neoadjuvant strategy within the interval between the diagnosis of TNBC and surgical removal [139]. Hedberg et al. [140] demonstrated through single-cell analysis that the presence of TAMs was elevated following HSV-C134 treatment, and these TAMs exhibited higher levels of the pro-inflammatory marker STAT1, a transcription factor that plays a role in the expression of interferons. Denton et al. [141] demonstrated that treatment with liposomal clodronate led to a reduction in TAM infiltration, which enhanced this OVs’ anti-tumor effectiveness without an increase in viral replication. Finally, OVs designed to target specific anti-tumor functions of TAMs hold great promise. Nevertheless, to amplify the interaction between TAMs and OVs for enhanced anti-tumor immunity, it will be essential to leverage the distinctions between TAMs’ anti-tumor and antiviral reactions.
A pair of herbs, Hedyotis diffusa and Scutellaria barbata, have been discovered to curb the differentiation of TAMs towards M2 in vitro. This effect hampers the migration of BC cells, suggesting a potential role in impeding the spread of BC [142].
The application of hyperthermia directly to the breast tumor environment is an additional potential approach for immunotherapy, as it can lead to the direct elimination of cancer cells [143, 144]. It is thought that hyperthermia augments immune cells, such as NK cells and CD8+ T cells, to eliminate tumor cells. Moreover, treatment of macrophages with CD40 agonists such as sotigalimab and selicrelumab has been shown to significantly enhance the generation of inflammatory factors, activate DCs, and promote the polarization of M1 TAMs. This approach is of particular interest in cancer immunotherapy due to its potential to reshape the TME and enhance the immune response against cancer cells.
In addition, CAR-Ps or chimeric antigen receptors for phagocytosis, is an innovative approach that engineers macrophages to directly engulf tumor cells or facilitate the degradation of the ECM components, thereby inhibiting the growth and progression of solid tumors. Zhang and colleagues [145] developed a HER2-specific CAR macrophage therapy, which contains an adjustable domain that binds to HER2 to elevate MMP production for the degradation of the ECM and another intracellular region consisting of CD147, which amplifies T cell infiltration within the TME, subsequently slowing down tumor growth. Overall, these approaches represent a promising new direction in cancer immunotherapy, providing a potential strategy to enhance the immune system’s ability to target and eliminate tumors.
In summary, we have compiled a list of several drugs and the efficacy of combination therapies in Table 2, while treatment strategies are outlined in Table 3.
| Drugs | Combinations | Functions |
| GM-CSF immunoadjuvant and trastuzumab | DNA vaccine against the cysteine protea selegumain; HER2/neu-based vaccination; Sipuleuce l-T vaccine | Decreases M2 and blocks tumor growth, angiogenesis and metastasis; triggers immune recognition and destruction; prolongs the survival of patients |
| IL-2, GM-CSF, and trastuzumab treatment | HER2-plasmid DNA vaccination | Induce M1 macrophage polarization; enhances DC functions and limits Treg regulation |
| Cyclophosphamide and trastuzumab | A granulocyte-macrophage colony-stimulating factor gene-transfected tumor vaccine | / |
| Anti-CD47 antibody | PD-L1 blockade | Enhances the anti-tumor effect |
| Pembrolizumab | Standard chemotherapy | Improves PFS in patients with metastatic TNBC |
| Nivolumab treatment | Cisplatin and doxorubicin | boost the gathering of CD8+ T cells |
| PD-1 antibody | IL-15Rc blocking peptide | Suppresses tumor growth and prevents oncogenic escape from treatment |
| Anti-VEGF-antibody | Avastin or Bevacizumab | Inhibits macrophage infiltration; prevents TAMs from releasing pro-angiogenic factors |
| Neoadjuvant trastuzumab | Anti-HER2 antibody, PD-L1 antagonist and IDO | Enhances anti-tumor immunity |
| Blocking SIRP |
Lipid-protamine nanostructures for the delivery of plasmids that trap CCL2 | Prevents its action in recruiting M2 and MDSCs |
| CSF-1R blocking | siRNA-CCR2 encapsulated cationic nanoparticles | Prevents the recruitment of macrophages to the tumor region |
| PD-1 antibody | Nanoparticle delivering small interfering RNA YTHDF1 | Boosts CD8+ T cell infiltration and M1-type TAMs, and reduces Tregs and M2-type TAMs in the TME |
| Everolimus | Exemestane or trastuzumab | / |
| PARP inhibitors | PD-1 antibody | Increases INF- |
| Estradiol | Inhibition of CCL2 and CCL5 | Increases macrophage influx and angiogenesis |
| Tucatinib | Immunotherapy or chemotherapeutic agents | Clinical benefits in patients with trastuzumab resistance |
| Paclitaxel | CSF-1 inhibitors | Increases CCL8, IL-34, and CSF-1 |
| Chemotherapy drug | Anti-CCL2 antibodies | Decreases both tumor growth and migration |
| BLZ-945 (a CSF-1R inhibitor), CD47 antibodies | Chemotherapeutic drug | Strengthen phagocytosis of TAMs and heighten immunotherapy, chemotherapy, and combination strategy |
| Pembrolizumab | Chemotherapy | Prolongs PFS among patients with metastatic TNBC and PD-L1–positive subgroup |
| Atezolizumab | Nab-paclitaxel |
GMCSF, granulocyte-macrophage colony-stimulating factor; HER2, human epidermal
growth factor receptor 2; TNBC, triple-negative breast cancer; IDO, indoleamine
2,3-dioxygenase; SIRP
| Current field of therapeutic direction | Combined to inhibit angiogenesis |
| Combined to inhibit immunosuppressive cell recruitment | |
| Combined to inhibit the production of tumor promoting factors | |
| Combined anticancer vaccine | |
| Combined immune checkpoint inhibitors | |
| Combination with Oncolytic viruses | |
| Combined nanomaterial drugs | |
| Joint signaling pathway target inhibitor | |
| Combination chemotherapy drug | |
| Combined with herbal medicine treatment | |
| Potential therapeutic directions | Modified CAR-T to secrete chemokines and cytokines to enhance T cell killing and intratumoral infiltration |
| Regulate TAM lipid metabolism and promote M1 polarization | |
| Screening old drugs for new ones |
CAR-T, chimeric antigen receptor T-cell.
The buildup of TAMs is also linked to poor clinical outcomes in a range of solid
malignancies [146, 147, 148]. Furthermore, the abundance of TAMs shows a positive link
with malignant inflammation, estrogen receptor, and E‑selectin expression [149].
This indicates the importance of TAMs for tumorigenesis and is also key to
whether tumor treatment is effective. Studies revealed that TAMs have evolved to
the M2 and activate
Targeting TAMs offers hope for successful disease control, but their therapeutic
response is not optimal, and a personalized treatment regimen may be required due
to the heterogeneity of TAMs. Moreover, M2-type TAMs can inhibit the activity of
T cells and NK cells by secreting immunosuppressive factors, thus promoting the
immune escape of tumors and reducing the clinical therapeutic effect. For
instance, TAMs express a variety of cytokines to reduce the cytotoxic immune
response of T cells and NK cells, including the ligands of EGFR family [153],
TNF, MMPs, TGF-
BC is the most common cancer worldwide and the leading cause of cancer-related mortality among women. Present therapeutic selections are limited to surgery, adjuvant chemotherapy, and radiotherapy. However, some patients do not qualify for surgical measures at initial diagnosis. Immunotherapy represents a promising treatment option by targeting TAMs, or the inhibitors of signaling pathways [154, 155] and agonists [156, 157]. This review summarized the immunosuppressive and immune escape role of macrophages in the TME. The proposed targeting of TAMs presents a promising therapeutic strategy. The changes in the TME following the targeting TAMs, with a focus on TNBC and HER2+ BC, are also described. The review concludes with an overview of several combination therapies, aiming to provide a breakthrough in clinical treatments. Immunotherapy has made significant advances in the treatment of BC, with agents such as atezolizumab and pembrolizumab showing partial benefits. However, these therapies are not yet sufficient to eradicate the disease in all patients. In summary, combination immunotherapy may offer a more effective approach to enhance the treatment outcomes for BC.
Although many treatments for TAMs have been reported, new treatments are
urgently needed. For example, actively screening existing drugs for new
therapeutic uses is a promising strategy. As the discovery of new drugs is
challenging, it is beneficial to actively explore new approaches for screening
targeted drugs with FDA approval. In addition, Treg cells deter the secretion of
INF-
YH: conceptualization, methodology, software, investigation, data curation, visualization, resources, writing—original draft, writing—review and editing. QL: supervision, visualization, writing—review, and editing. ZHL: conceptualization, methodology, software. QH & LW: conceptualization, methodology. ZFG: (i) made substantial contributions to conception and design, or acquisition of data, or analysis and interpretation of data; (ii) been involved in drafting the manuscript or reviewing it critically for important intellectual content, and (iii) given final approval of the version to be published. All authors have read and agreed to the published version of the manuscript. All authors have participated sufficiently in the work to take public responsibility for appropriate portions of the content and agreed to be accountable for all aspects of the work in ensuring that questions related to its accuracy or integrity. All authors contributed to editorial changes in the manuscript.
Not applicable.
We would like to thank the staff at each participating institution for their support with data collection.
Guangdong Natural Science Foundation Committee - Youth Promotion Project (2023A1515030182).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.