



 , Liqi Yang 1,*
, Liqi Yang 1,*
1 Department of Pediatrics, The Second Affiliated Hospital of Anhui Medical University, 230601 Hefei, Anhui, China
2 Department of Pediatrics, Fuyang People’s Hospital, 236000 Fuyang, Anhui, China
Abstract
Spinal muscular atrophy (SMA) is a severe neuromuscular disorder caused by mutations in the survival motor neuron 1 (SMN1) gene, resulting in progressive motor neuron loss and muscle atrophy. The urotensin 2 (UTS2) gene, located on chromosome 9q34.2, plays a significant role in cellular activities such as proliferation, apoptosis, and inflammatory responses. Notably, elevated expression levels of UTS2 have been observed in SMA patients. However, its precise contribution to disease pathogenesis remains unclear. This study aimed to investigate the effects of UTS2, which is overexpressed in SMA patients, in SMA cell models using a UTS2 inhibitor.
We conducted genomic sequencing and bioinformatics analysis on clinical samples to identify proteins highly expressed in association with SMA. Using RNA interference technology, we suppressed SMN1 gene expression in bone marrow mesenchymal stem cells (MSCs) to establish an in vitro cellular model of SMA. To assess the biological consequences of SMN1 gene knockdown, we employed molecular biological techniques such as immunofluorescence, reverse transcription quantitative polymerase chain reaction (RT-qPCR), and western blotting. Furthermore, we treated the SMA cellular model with the urantide UTS2 receptor inhibitor and examined its effects on cell proliferation, apoptosis, and the expression of relevant proteins.
UTS2 was successfully identified as a highly expressed protein associated with SMA. A stable MSC model with SMN1 gene knockdown was established. RNA interference (RNAi) technology effectively suppressed SMN1 gene expression, leading to changes in cellular morphology and neuron-specific marker expression. Urantide intervention significantly affected both proliferation and apoptosis in the SMA cell model in a dose-dependent manner. Techniques such as the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay, TUNEL fluorescence staining, and flow cytometry analysis revealed that uride decreased cell viability while increasing the proportion of apoptotic cells. Following urantide intervention, there was a notable increase in caspase-3 messenger ribonucleic acid (mRNA) levels, as well as an increase in caspase-3 protein expression, as demonstrated by immunofluorescence analysis.
We elucidated the role of the UTS2 gene in an SMA cell model, emphasizing its dysregulation and identifying potential therapeutic targets. Urantide, a UTS2 inhibitor, had significant biological effects on the SMA cell model, indicating that it is a promising therapeutic strategy for SMA. These findings provide valuable insights for advancing drug development and clinical treatment of SMA.
Keywords
- spinal muscular atrophy
- RNAi
- UTS2
- urantide
- Caspase-3
Spinal muscular atrophy (SMA) is a prevalent genetic neurodegenerative disorder that primarily affects pediatric populations and is characterized by the progressive degeneration of motor neurons, leading to muscle weakness and atrophy. The incidence rate of SMA is approximately 1 in 10,000 individuals [1, 2, 3], with a carrier frequency of 1 in 250 individuals [2]. The genetic basis of SMA primarily involves the deletion or mutation of exon 7 in the SMN1 gene. Although the survival motor neuron 2 (SMN2) gene shares a similar sequence with SMN1, it predominantly produces an incompletely functional SMN protein that cannot fully compensate for the loss of SMN1 [4, 5]. A reduction in SMN protein levels plays a pivotal role in the survival and functionality of motor neurons, and its depletion leads to alpha motor neuron degeneration, muscle atrophy, and activation of the Caspase3-mediated cell apoptosis pathway [5]. Moreover, the SMN protein performs critical regulatory functions within the nucleus by participating in small nuclear ribonucleoprotein (snRNP) biosynthesis and messenger ribonucleic acid (mRNA) splicing processes that are vital for neuronal viability. Insufficient levels of SMN protein result in RNA metabolism disorders and subsequent cellular death, ultimately manifesting as clinical symptoms observed in SMA [6, 7].
The impact of SMAs on patients’ quality of life is significant, and current treatment options are limited. Despite the approval of certain gene therapies and small-molecule drugs, a definitive cure for SMA has yet to be achieved [8, 9]. Furthermore, these drugs, such as nusinersen and risdiplam, which function by promoting the inclusion of exon 7 in the SMN pre-mRNA, primarily target SMN itself [9, 10]. Research focusing on downstream genes affected by SMN and drug development in this area is scarce, highlighting the urgent need to address specific gene expression regulation and identify potential therapeutic targets [3, 9].
Located on chromosome 9q34.2, urotensin 2 (UTS2) is a member of the urotensin II-related peptide (UII) family, which is involved in various physiological processes, including promoting cell proliferation [11], inhibiting apoptosis [12], and inhibiting the inflammatory response [13]. In the context of SMA, UTS2 expression levels are significantly elevated, suggesting a possible contribution to disease progression [13]. The precise mechanisms by which UTS2 influences SMA pathology are not yet fully understood, but several hypotheses have been proposed. One line of thought is that UTS2 may exacerbate motor neuron degeneration by promoting cell death pathways or disrupting neuromuscular junctions, which are critical for motor neuron function [14]. Additionally, UTS2 could interact with other cellular components affected by SMN deficiency, thereby modulating the cellular response to SMN loss. Despite the growing interest in UTS2, research in this area is still in its infancy. Most studies to date have focused on characterizing UTS2 expression patterns in SMA patients and correlating these findings with disease severity [11, 13, 14]. Few studies have investigated the functional implications of UTS2 upregulation in SMAs [15, 16], and even fewer have explored the therapeutic potential of targeting UTS2. This gap in knowledge underscores the need for more in-depth investigations into the role of UTS2 in SMAs, particularly in terms of its downstream effects on cellular processes and its potential as a therapeutic intervention.
The objective of this study was to elucidate the pathogenic mechanism underlying SMA through clinical sample analysis and in vitro experiments. Gene chip analysis and bioinformatics methods revealed that the highly expressed protein UTS2 was associated with SMA. RNA interference technology can be employed to inhibit the expression of the SMN1 gene in mesenchymal stem cells (MSCs), thereby establishing an SMA cell model. The ultimate aim of this study was to investigate whether inhibitors targeting UTS2 proteins can effectively reverse the disease phenotype in SMA cells, thus providing novel therapeutic strategies for the clinical management of SMA.
The study was approved by the Fuyang People’s Hospital ethics committee (approval number: [2024]64), and all participants or their families/legal guardians provided informed consent in accordance with the ethical standards of the Helsinki declaration.
In this study, we recruited a cohort of patients diagnosed with SMA (n = 5) and matched healthy controls (n = 5) from Fuyang People’s Hospital between January 2024 and June 2024. The inclusion criteria for participant selection were as follows: (1) met the clinical diagnostic criteria for SMA [17] and (2) had a homozygous deletion of the SMN1 gene detected [4]. The inclusion criterion for the normal control group was a copy number of 2 for the SMN1 gene. The exclusion criteria for participant selection were as follows: (1) the presence of blood disorders; (2) the presence of bone tumors; (3) treatment that could influence SMA progression or symptoms, such as ongoing gene therapy or pharmacological interventions; (4) a history of other neuromuscular disorders that could confound the results; and (5) a family history of genetic disorders that might affect the interpretation of genetic data related to the SMA.
(1) Motor function assessments: Motor function was evaluated via standardized scales to measure muscle strength, coordination, and motor milestones in SMA patients. (2) Neurophysiological measurements: Electromyography (EMG) and nerve conduction studies were performed to assess the functional integrity of motor neurons and neuromuscular junctions. (3) Biochemical markers: The levels of UTS2 and other relevant proteins in the blood and spinal fluid were quantified to correlate with disease severity and response to treatment. (4) Imaging studies: Neuroimaging techniques, such as magnetic resonance imaging (MRI), have been employed to monitor changes in muscle and neuronal structure, providing insights into disease progression. (5) Histopathological examination: Tissue biopsies, when available, were examined for signs of muscle degeneration, neuronal loss, and other pathological features associated with the SMA. (6) Molecular and Cellular Analysis: The expression levels of UTS2 and related genes in patient-derived cells were analyzed to understand the underlying molecular mechanisms.
To obtain differential expression gene information, total RNA was extracted from peripheral blood samples collected from the antecubital vein of the participants. The collection process involved the use of sterile vacutainer tubes containing an appropriate anticoagulant, such as EDTA, to prevent blood clotting. Once collected, the samples were gently inverted several times to ensure thorough mixing with the anticoagulant and then stored on ice.
The total peripheral blood RNA was then extracted via TRIzol Reagent, a monophasic solution of phenol and guanidine isothiocyanate that effectively lyses cells and inactivates RNases, thus preserving the integrity of the RNA. Following extraction, the purity, concentration, and integrity of the RNA were assessed via a combination of NanoDrop (Thermo Fisher Scientific, Waltham, MA, USA) and Agilent 2100/4200 systems (Agilent Technologies, Santa Clara, CA, USA). The NanoDrop provides a rapid and accurate measurement of RNA concentration and purity by determining the absorbance at 260 nm and 280 nm, whereas the Agilent 2100/4200 systems offer a detailed electropherogram that assesses RNA integrity through the analysis of ribosomal RNA peaks.
After assessment, the RNA samples were aliquoted into RNase-free tubes and stored at –80 °C until further use. This low-temperature storage ensures the long-term preservation of RNA integrity, preventing degradation that could occur due to RNase activity or other environmental factors.
Subsequently, 3 µg of RNA was subjected to mRNA enrichment, fragmentation, and complementary DNA (cDNA) synthesis. The cDNA was processed through end-repair, A-tailing, and purification via Hieff NGS® DNA Selection Beads (Yeasen, Shanghai, China). The purified cDNA was amplified to form single-stranded circular DNA (ss-cDNA), which was then transformed into DNA nanoballs (DNBs) through rolling circle amplification (RCA). The final library was quantified with a Qubit (Thermo Fisher Scientific, Waltham, MA, USA) to confirm adequate sequencing input.
The RNA-seq library was sequenced on the DNBSEQ-T7 platform with a PE150 model, leveraging the high-throughput capabilities of next-generation sequencing. This service was provided by Bioyi Biotechnology Co., Ltd. (Wuhan, Hubei, China), ensuring the generation of high-quality paired-end reads for comprehensive transcriptome analysis.
To gain insight into the biological functions and pathways associated with these
differentially expressed genes (DEGs), Gene Ontology (GO) and Kyoto Encyclopedia
of Genes and Genomes (KEGG) enrichment analyses were performed. In the quality
control process, the raw sequencing reads were first processed via fastp software
0.21.0 (SBGrid Consortium, Cambridge, MA, USA) to remove low-quality data,
including 3′ end sequences containing adapters and reads with an average
quality score below Q20, ensuring that only high-quality reads were used for
subsequent analysis. These high-quality reads were subsequently aligned to a
reference genome via HISAT2 software 2.1.0 (Center for Computational Biology at
Johns Hopkins University, Baltimore, MD, USA). For gene expression analysis,
String Tie software 2.1.5 (Center for Computational Biology at Johns Hopkins
University, Baltimore, MD, USA) was used to quantify gene expression levels,
which were normalized to fragments per kilobase of transcript per million mapped
reads (FPKM). Furthermore, DESeq2 software 1.30.1 (Bioconductor Project,
Dortmund, Germany) was used to identify DEGs on the basis of the criteria of
Finally, to investigate the enrichment of differentially expressed genes in
biological functions and pathways, Cluster Profiler software 3.18.1 (Bioconductor
Project, Dortmund, Germany) was used for Gene Ontology (GO) and Kyoto
Encyclopedia of Genes and Genomes (KEGG) enrichment analyses. Significance was
defined as p
Human bone marrow mesenchymal stem cells (hBM-MSCs) were isolated from healthy
donors via a sterile procedure, which typically involves the following steps:
bone marrow aspiration under aseptic conditions, density gradient centrifugation
to enrich the MSC population, initial plastic adherence to select for MSCs, and
subsequent culture expansion in a sterile incubator while monitoring for cell
confluence and viability, ensuring that the entire process adheres to good
manufacturing practice (GMP) standards to maintain sterility and cell purity. The
bone marrow was flushed with L-DMEM (Gibco, #11885084, Thermo Fisher Scientific,
Waltham, MA, USA) supplemented with 10% FBS (Gibco, #10082147, Thermo Fisher
Scientific, Waltham, MA, USA) to create a cell suspension, which was then
centrifuged at 1500 rpm for 10 minutes to remove the adipose tissue and
supernatant. The resulting cell pellet was resuspended in fresh L-DMEM and
homogenized by gentle pipetting. Mononuclear cells were separated via lymphocyte
separation medium (density of 1.077 g/mL), washed twice, counted, and plated at a
density of 1
hBM-MSCs were cultured in low-glucose complete DMEM (BL301A, Pricella, Shanghai, China) and maintained at 37 °C in a humidified atmosphere containing 5% CO2, and the medium was subsequently changed every 3–4 days. All cell lines were validated by short tandem repeat (STR) profiling and tested negative for mycoplasma.
The experimental design included several groups to evaluate the impact of short hairpin RNA (shRNA)-mediated SMN1 knockdown and the subsequent effects of UTS2 inhibition with urantide on human bone marrow mesenchymal stem cells (hBM-MSCs). The groups included a time point group transfected with SMN1-targeting shRNA at the beginning (0 h), a group assessed 48 hours posttransfection, a nontargeting shRNA control group (pshRNA-0), and an SMN1 shRNA treatment group (pshRNA-SMN1). Additionally, a baseline control group of untransfected cells was included, alongside an SMA cell model group (SMA), and two groups were treated with low (10 nM, L-Urantide+SMA) and high (20 nM, H-Urantide+SMA) concentrations of uranide.
For transfection, hBM-MSCs were plated in 24-well plates at a density of 50,000 cells per well and allowed to adhere overnight under cell culture conditions at 37 °C and 5% CO2. The following day, the cells were transfected with the recombinant pLKO.1-shRNA plasmids via Lipofectamine 3000 (#L3000008, Thermo Fisher Scientific, Waltham, MA, USA) in Opti-MEM (#31985062, Thermo Fisher Scientific, Waltham, MA, USA) reduced serum medium following the manufacturer’s instructions. The DNA-Lipofectamine complexes were added to the cells, which were subsequently incubated for 6 hours to facilitate complex formation and uptake, after which the medium was replaced with fresh complete growth medium. After a 48-hour incubation to allow for gene silencing, the cells were harvested for analysis, such as reverse transcription quantitative polymerase chain reaction (RT-qPCR). Western blot analysis, immunofluorescence staining and cell viability and apoptosis assays.
The efficiency of SMN1 knockdown was determined by quantifying the expression of SMN1 mRNA via reverse transcription quantitative polymerase chain reaction (RT-qPCR), and the results were normalized to those of the housekeeping gene GAPDH to determine relative expression levels. The shRNA sequences used were as follows: SMN-shRNA-1F-ATAGTATACGAAAATAGTCACTTCAAGAGAGTGACTATTTTCGTATACTAT, SMN-shRNA-1R-GACTATTTTCGTATACTATTTAAGTTCTCTAAATAGTATACGAAAATAGTC, SMN-shRNA-2F-AAATAGTATACGAAAATAGTCTTCAAGAGAGACTATTTTCGTATACTATTT, SMN-shRNA-2R-CTATTTTCGTATACTATTTCGAAGTTCTCTCGAAATAGTATACGAAAATAG, SMN-shRNA-3F-TGTAAACAGGGGTTGAAAGGTTTCAAGAGAACCTTTCAACCCCTGTTTACA, SMN-shRNA-3R-CTTTCAACCCCTGTTTACAAGAAGTTCTCTCTTGTAAACAGGGGTTGAAAG, sh-NC-F-CACCGTTCTCCGAACGTGTCACGTTTCAAGAGAACGTGACACGTTCGGAGAATTTTTTG, sh-NC-F-GATCCAAAAAATTCTCCGAACGTGTCACGTTCTCTTGAAACGTGACACGTTCGGAGAAC.
Total RNA was extracted from cells via the Cell/Tissue Total RNA Isolation Kit V2 (Vazyme, Nanjing, Jiansu, China). The HiScript lll 1st Strand cDNA Synthesis Kit (Vazyme, China) was used to synthesize cDNA, followed by RT-qPCR using Taq Pro Universal SYBR qPCR Master Mix (Vazyme, China). The relative expression levels of fl-SMN mRNA were calculated via the 2-ΔΔCt method, with GAPDH serving as the endogenous control. The primers used were as follows: SMN-F-CCACTTACTATCATGCTGGCTG, SMN-R-CCACTTACTATCATGCTGGCTG, Caspase-3-F-TGCTATTGTGAGGCGGTTGT, Caspase-3-R-TCACGGCCTGGGATTTCAAG, GAPDH-F-ACCACAGTCCATGCCATCAC, and GAPDH-R-TCCACCACCCTGTTGCTGTA.
Protein extraction was performed via RIPA lysis buffer (Beyotime, Shanghai,
China) supplemented with PMSF (Beyotime, China). Protein concentrations were
determined via a BCA protein assay kit (NCM Biotech, Nanjing, Jiangsu, China).
Equal amounts of protein were separated by SDS‒PAGE and transferred onto PVDF
membranes (Sigma‒Aldrich, St. Louis, MO, USA). The membranes were probed with
primary antibodies against SMN/Gemin 1 (1:1000, ab108531, Abcam, Cambridge, UK)
and
The cells were fixed with 4% paraformaldehyde and permeabilized with 0.25% Triton X-100. After being blocked with goat serum, the cells were incubated with primary antibodies against neuron-specific markers [NSE (1:200, Abcam, UK) and neurofilament (NF) (1:200, Cell Signaling Technology, USA)] overnight at 4 °C, followed by incubation with fluorescently labeled secondary antibodies [goat anti-rabbit IgG H&L/HRP (1:200, Bioss, China) and goat anti-mouse IgG H&L/HRP (Bioss, China)]. Nuclei were counterstained with DAPI, and images were captured via a fluorescence microscope.
To evaluate whether the reduction in cell viability is attributed to an increase in apoptosis, we employed TUNEL fluorescence and Annexin V, which enabled us to compare the apoptotic rates among the different experimental groups.
Cell viability was assessed via the MTT assay (Beyotime, China), and apoptosis was measured via the TUNEL assay (Beyotime, China) and an Annexin V-FITC apoptosis detection kit (Beyotime, China) according to the manufacturers’ instructions.
The data were analyzed via GraphPad Prism 9.4.0 (GraphPad Software, LLC, Boston,
MA, USA) and are presented as the means
The principal component analysis (PCA) plot demonstrated distinct separation
between the SMA and control groups, except for samples C1 and C2, which showed
some overlap with the SMA group, indicating a unique transcriptional profile
associated with SMA (Fig. 1A). The heatmap of differentially expressed genes
(DEGs) further emphasized the difference between the two groups, highlighting
that differential gene expression can serve as a robust classifier (Fig. 1B).
Comparative analysis revealed 196 upregulated genes and 129 downregulated genes
in the SMA group compared with the control group (Fig. 1C), suggesting a more
pronounced effect of upregulated genes on SMA pathogenesis. The results indicated
significant enrichment of upregulated genes in various biological processes and
pathways (Fig. 1D,E), leading us to focus subsequent research on upregulated
genes. The application of stringent criteria for gene selection based on a
log2FoldChange greater than 1.5 and a p value
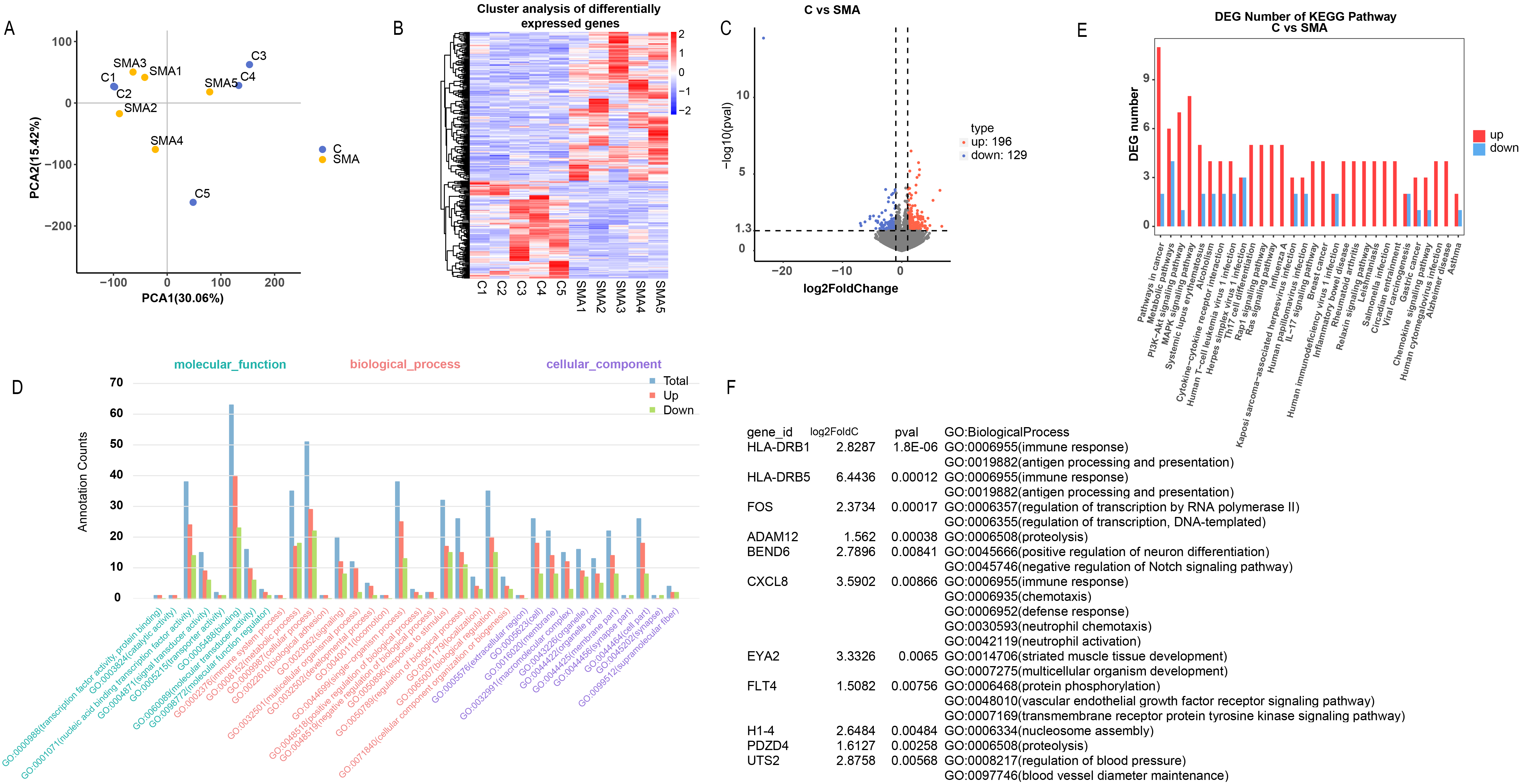 Fig. 1.
Fig. 1.
Bioinformatics analysis of clinical samples. (A) Principal component analysis (PCA) plot showing the separation between the spinal muscular atrophy (SMA) and control groups. (B) Heatmap of differentially expressed genes (DEGs) differentiating the two groups. (C) Volcano plot indicating the number of upregulated and downregulated genes in the SMA group compared with the control group. (D,E) Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) enrichment analyses of the DEGs. (F) Selection of UTS2 among the upregulated genes on the basis of stringent filtering criteria.
After the knockdown of SMN1 in MSCs, we observed significant changes in cellular morphology and marker expression indicative of neuronal differentiation. The absence of CD34 expression and the presence of CD44 expression are associated with MSCs (Fig. 2A). Successful transfection was indicated by the positive signal of GFP (green fluorescent proteins) (Fig. 2B), while both the RNA (Fig. 2C) and protein (Fig. 2D) expression of SMN1 were significantly suppressed by shRNA.
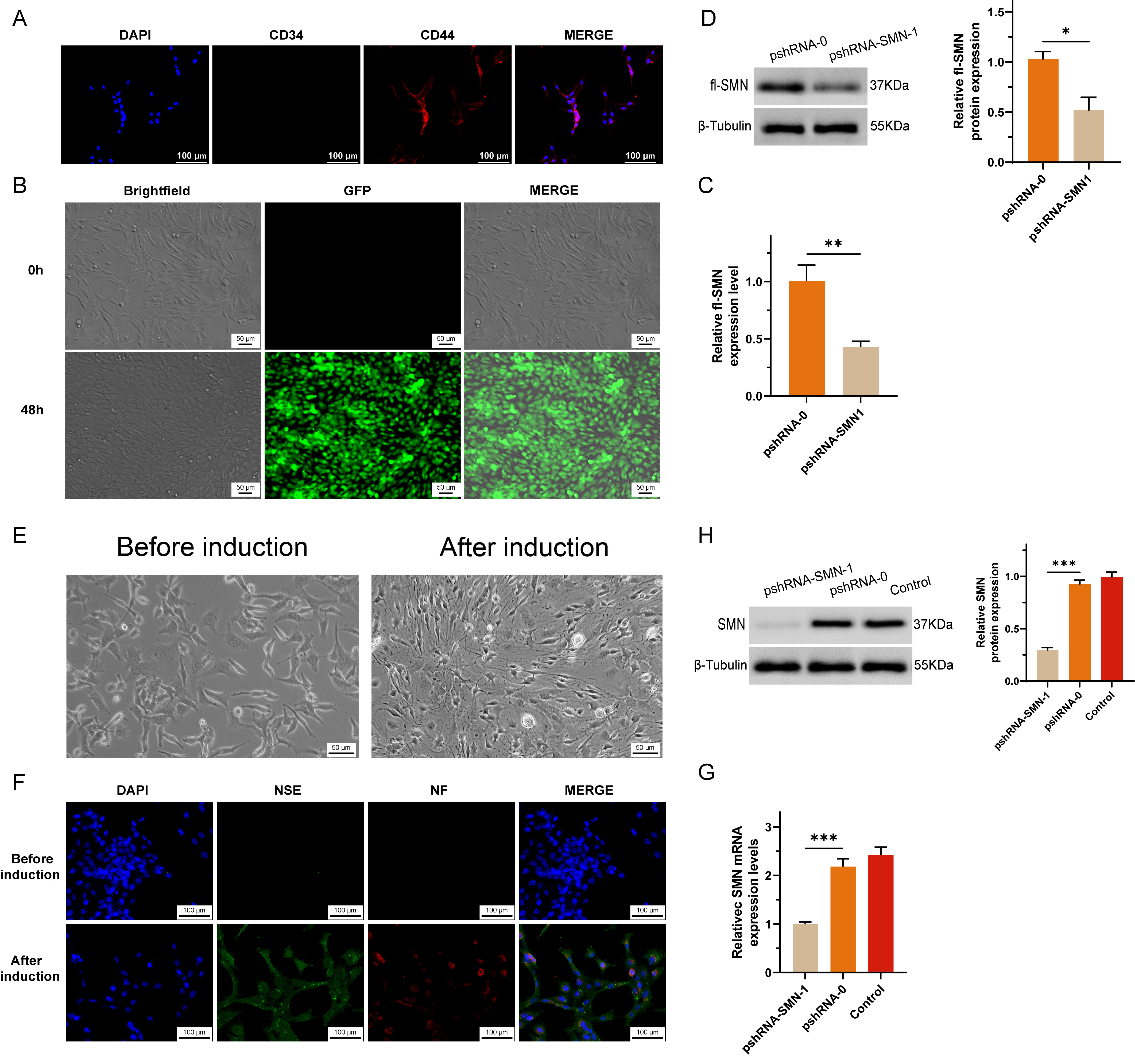 Fig. 2.
Fig. 2.
Establishment and characterization of SMN1 knockdown in
MSCs. (A) Bone marrow was obtained from non-SMA patients, excluding those with
concurrent blood diseases or bone tumors, to isolate and purify MSCs. (B) A short
hairpin RNA (shRNA) construct targeting the survival motor neuron 1
(SMN1) gene was synthesized and cloned and inserted into a vector,
creating a recombinant plasmid designed to express SMN1-shRNA within
cells. (C,D) SMN1 knockdown in MSCs was evaluated by measuring SMN mRNA levels via RT-qPCR (C) and full-length SMN protein levels via Western blotting (D).
A significant reduction in SMN expression
confirmed the efficacy of the shRNA construct in silencing the SMN1
gene. (E) Morphological changes indicative of neuronal differentiation in MSCs
after infection with SMN1-targeting shRNA lentivirus. The images show
that neurite outgrowth and changes in the cell body are characteristic of
neuron-like cells. (F) Immunocytochemical staining confirming the expression of
the neuron-specific markers NSE and NF in differentiated neuron-like cells,
indicating successful differentiation. (G) RT-qPCR analysis demonstrating a
sustained reduction in SMN mRNA levels in differentiated neuron-like cells
compared with preinduction levels. (H) Western blot analysis showing decreased
SMN protein expression in differentiated cells, which was consistent with the
mRNA results and confirmed the maintenance of the SMN1 knockdown effect.
* represents p
After induction, the morphology of MSCs transformed into neuronal-like cells, characterized by cytoplasmic flattening and shrinkage toward the nucleus, as well as peripheral extension of cell membrane protrusions (Fig. 2E). Immunocytochemical staining confirmed the expression of neuron-specific markers, including neuron-specific enolase (NSE) and neurofilament (NF) proteins, in the differentiated cells (Fig. 2F). To assess the impact of SMN1 knockdown on the differentiation process, we measured the expression levels of SMN mRNA and protein in neuron-like cells via reverse transcription quantitative polymerase chain reaction (RT-qPCR) and Western blot methods, respectively. The results revealed a reduction in SMN expression postdifferentiation compared with preinduction levels, indicating that the knockdown effect was maintained throughout the differentiation process (Fig. 2G,H).
The cell survival curves revealed a positive proliferation trend in the SMA group, which was attenuated in a dose-dependent manner by urantide treatment, as observed in the L-urrantide group and H-urrantide group (Fig. 3A). MTT assays corroborated these findings, revealing a significant decrease in cell viability with urantide treatment (Fig. 3B). TUNEL assays and flow cytometry via Annexin V staining revealed a significant increase in the number of apoptotic cells in response to urantide treatment, with higher concentrations leading to further increases in apoptosis rates than those in the SMA group without intervention (Fig. 3C,D). RT-qPCR analysis revealed the upregulation of Caspase-3 mRNA in the urantide-treated groups, suggesting the induction of apoptotic pathways in the SMA cell model (Fig. 3F). Immunofluorescence staining revealed an increase in Caspase-3 protein expression in the urantide-treated groups, with higher expression observed in the H-urrantide+SMA group than in the L-urrantid group, and SMN protein expression was decreased in accordance with the mRNA levels (Fig. 3E).
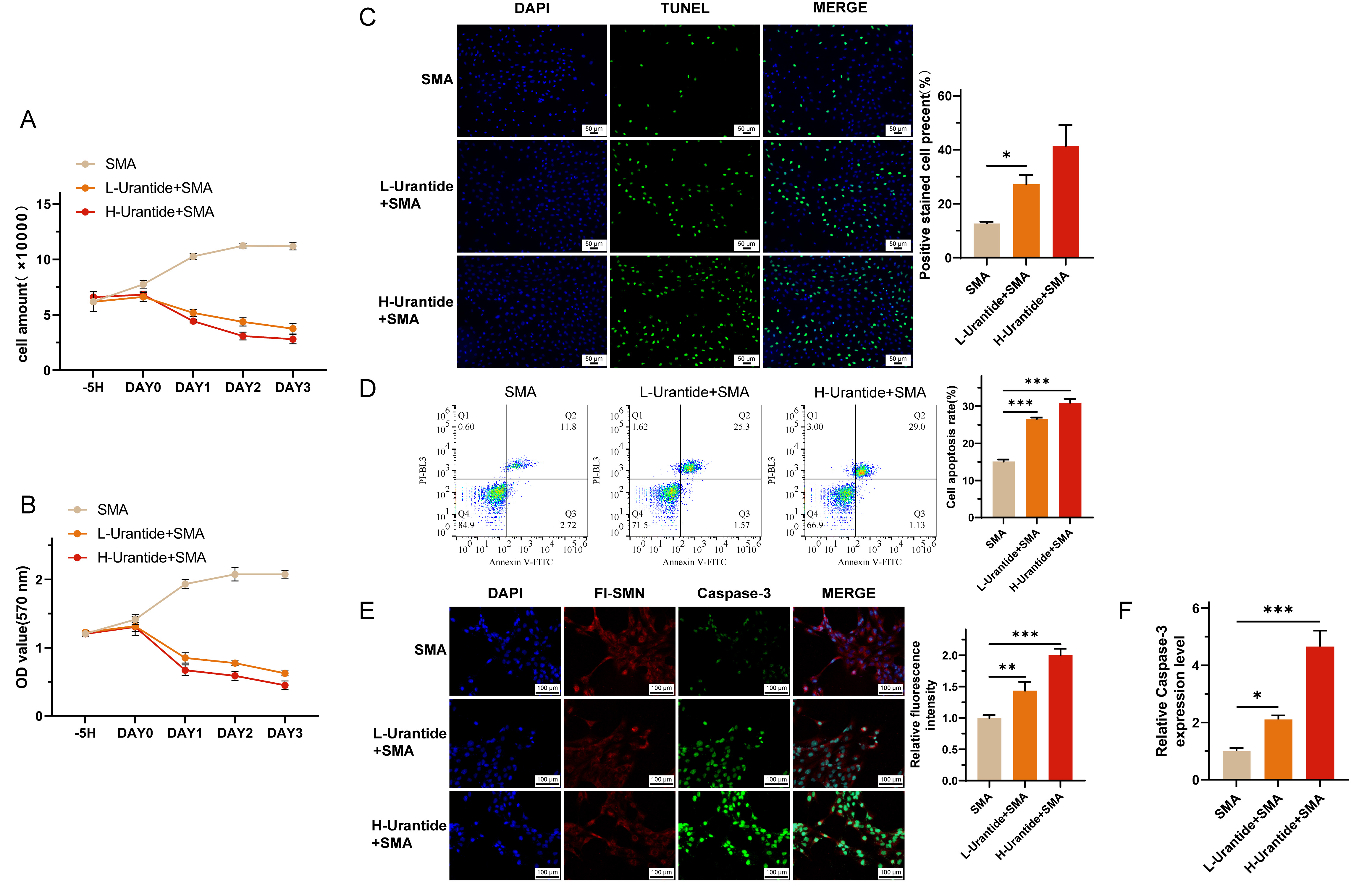 Fig. 3.
Fig. 3.
Urantide-induced apoptosis in SMA cell models. (A) Cell
survival curves depicting a dose-dependent attenuation of proliferation in SMA
cells treated with urantide, as observed in the L-urrantide and H-urrantide
groups. (B) MTT assays confirmed a significant decrease in cell viability with
urantide treatment, indicating cytotoxic effects. (C) TUNEL fluorescence staining
revealed a significant increase in apoptotic cells in response to urantide
treatment, with higher concentrations leading to further increases in apoptosis
rates. (D) Flow cytometry via Annexin V staining further demonstrated the
proapoptotic effect of uride in a dose-dependent manner. (E) Immunofluorescence
staining showing increased Caspase-3 protein expression in the urantide-treated
groups, with the H-urrantide+SMA group exhibiting higher expression than the
L-urrantide group and decreased SMN protein expression aligning with the mRNA
levels. (F) RT-qPCR analysis confirming the upregulation of Caspase-3 mRNA in the
urantide-treated groups, suggesting the activation of apoptotic pathways. *
represents p
Our study presents significant findings that enhance the current understanding of SMA pathology. The identification of UTS2 as a highly expressed protein in SMA patients suggests its potential involvement in disease pathogenesis. The successful establishment of a stable MSC model with SMN1 gene knockdown provides a valuable tool for investigating the effects of UTS2 inhibition. The observed effects of urantide on cell proliferation and apoptosis indicate a complex regulatory role of UTS2 in SMA-related cellular processes.
The development of a stable model for the knockdown of the SMN1 gene in MSCs represents a significant advancement and provides a valuable resource for research on SMA. This model, which is specifically designed to reduce SMN1 gene expression on the basis of existing SMA-derived induced pluripotent stem cell (iPSC) models [19], enables meticulous examination of the intricate interactions between UTS2 regulation and SMA pathophysiology. The observed modulation of cell proliferation and apoptosis by the Urantide UTS2 receptor antagonist further highlights the complex regulatory functions of UTS2 in the SMA, demonstrating its multifaceted influence. By simulating key aspects of SMA pathology, this model serves as an important platform for exploring the therapeutic potential of UTS2 inhibition and advancing our understanding of the cellular dynamics in the SMA.
Our findings build upon the existing body of research that supports the neuroprotective role of UTS2 in neuronal survival and synaptic plasticity [15, 16, 17, 18, 20]. With respect to spinal muscular atrophy (SMA), our study elucidates the intricate functions of UTS2 in diverse contexts. Traditionally, UTS2 is believed to contribute to cell survival; however, our findings indicate that the aberrant expression of UTS2 in SMA patients’ nerve cells is deleterious to human health. This observation does not contradict the established neuroprotective role of UTS2 but rather enhances our comprehension of its involvement under distinct pathological conditions [21, 22]
The observation that urantide treatment significantly influences cell proliferation in SMA cell models is particularly intriguing. This finding is consistent with previous studies that have implicated UTS2 in promoting cell division and vascularization in cancerous tissues [11, 17, 23, 24, 25, 26]. The dual role of UTS2 in both neurodegenerative diseases and cancer highlights the complexity of its biological functions and suggests that targeted therapies must be carefully designed to address the specific pathological context [27, 28, 29]. In conclusion, our study not only broadens the understanding of the role of UTS2 in the SMA but also underscores the need for a more comprehensive evaluation of its functions under different physiological and pathological conditions. The contrasting effects of UTS2 in SMAs versus its neuroprotective role in other contexts necessitate further investigation into the molecular mechanisms underlying these divergent outcomes. This knowledge is crucial for the development of targeted therapeutics that can effectively modulate UTS2 activity in a disease-specific manner.
The observed increase in Caspase-3 expression following urantide treatment suggested that UTS2 may regulate apoptotic pathways in the SMA. This finding is consistent with the known role of UTS2 in neuronal cell death [14] and suggests a potential mechanism by which UTS2 contributes to SMA progression.
The significant biological effects of urrantide on the SMA cell model, including reduced cell viability and increased apoptosis, indicate its potential as a therapeutic agent. These results support the need for further investigations into UTS2 inhibitors as novel treatment strategies for SMA, which may lead to improved clinical management. While our study provides valuable insights into the role of UTS2 in SMAs, it is not without limitations. First, the in vitro nature of the experiments limits the extrapolation of these findings to the in vivo context, and the relatively small sample size consisting of only five SMA patients and five controls for bioinformatics analysis may limit the generalizability of our findings and introduce potential sampling bias. Second, while our study emphasized Caspase-3 upregulation posturrantide treatment as a marker of apoptosis, we recognize the importance of exploring alternative apoptosis pathways and potential off-target effects. Third, concerning the dose-dependent effects of urantide, we acknowledge the absence of toxicity studies and the impact of urantide on non-SMA cells, which currently limits the therapeutic applicability of our findings. Future research should focus on in vivo models and need to be validated in larger cohorts to confirm the therapeutic potential of uride and explore its long-term effects on SMA progression; investigate additional apoptotic pathways beyond Caspase-3, including those involving Caspase-9 and Caspase-8, to comprehensively understand the cell death mechanisms influenced by uride; assess the toxicity and off-target effects of urantide, especially in non-SMA cells, to fully evaluate its safety profile and therapeutic index; and expand the sample size to increase the statistical robustness and generalizability of the findings.
Our study elucidates the role of UTS2 in SMA and identifies UTS2 as a potential therapeutic target. The use of urantide to modulate the SMA cellular phenotype presents a promising avenue for future research and clinical development. Further studies are warranted to fully understand the mechanisms by which UTS2 influences the SMA and to harness this knowledge for the benefit of patients.
The data used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Conceptualization: XZ; Data curation: XZ; Formal analysis: LQY; Investigation: LQY; Writing—original draft: XZ. Both authors contributed to editorial changes in the manuscript. Both authors read and approved the final manuscript. Both authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
The study was approved by the Fuyang People’s Hospital ethics committee (approval number: [2024]64), and all patients or their families/legal guardians provided informed consent in accordance with the ethical standards of the Helsinki declaration.
The authors express their appreciation to staff in the second affiliated hospital of Anhui Medical University for their technical assistance.
This research was funded by the health research project of Fuyang Health Commission (grant number: FY2023-141).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.