



1 Department of Joint Surgery, The Affiliated Traditional Chinese Medicine Hospital of Southwest Medical University, 646000 Luzhou, Sichuan, China
2 Department of Orthopedics, The Affiliated Traditional Chinese Medicine Hospital of Southwest Medical University, 646000 Luzhou, Sichuan, China
3 Center for Orthopedic Diseases Research, The Affiliated Traditional Chinese Medicine Hospital of Southwest Medical University, 646000 Luzhou, Sichuan, China
Abstract
Tendon–bone healing has always been a difficult point in clinical orthopedics, tissue engineering, and sports medicine. The most important structure for stress transmission is the tendon–bone junction, which is the transition from soft tissue to hard tissue. Biological effects can be produced by a variety of cytokines in different cells. During the remodelling and repair of the tendon–bone junction, the key factor is the inflammatory microenvironment regulated by macrophages through various physiological processes such as autophagy, differentiation, and polarization, which mediate cytokine release and influence other cellular functions. This provides a theoretical basis for the development of new mechanisms for tendon–bone junction repair. This article aims to review the potential role of macrophage autophagy, differentiation, and polarization in the repair of tendon–bone injury. In addition, we propose that future research should integrate multidisciplinary approaches such as molecular biology and bioinformatics to conduct in-depth analyses of the dynamic networks of autophagy and polarization in macrophages, thereby guiding future research directions on the specific roles of macrophage autophagy in tendon–bone junction repair.
Keywords
- macrophage
- tendon-bone junction
- autophagy
- cytokines
- mechanism
- osteoclasts
- review
The tendon-bone junction—where tendon inserts into bone—is prone to rupture from stress concentration and represents a common sports medicine injury [1]. High retear rates and suboptimal outcomes after repair impose substantial socioeconomic burdens; 32 million US musculoskeletal injuries annually involve tendons/ligaments in 45% of cases [2]. Structurally categorized as direct/indirect attachments, repaired junctions exhibit significantly reduced biomechanical strength, increasing rerupture risk [3]. This weakness stems from early inflammatory cell influx causing excessive scar tissue that impedes fibrocartilage formation, or insufficient stem cell proliferation/chondrogenic differentiation to regenerate native structure [4].
During initial inflammation, peripheral blood/bone marrow/synovial-derived macrophages recruit to the junction, primarily polarizing to M1 phenotype. These macrophages phagocytose debris, eliminate pathogens, and secrete inflammatory factors to promote healing. M2 macrophages dominate subsequent remodeling, facilitating repair. Macrophages demonstrate activation states spanning pro-inflammatory to reparative, regulated by extracellular signals and metabolic programs. Macrophage autophagy, an immunologically regulated lysosomal pathway, clears damaged organelles/protein aggregates/pathogens. Activated macrophages initiate light chain 3 (LC3)-lipidated autophagosome formation, identify substrates, and phagocytose debris. Through secreted factors, they further regulate osteoblast/osteoclast differentiation to promote tendon-bone healing. This review examines macrophage autophagy, differentiation, and polarization in tendon-bone repair, exploring future therapeutic strategies.
The human Achilles tendon-bone interface is narrow (
The healing process following tendon-bone junction injury comprises three distinct phases. The first phase is the inflammatory phase, characterized primarily by the infiltration of inflammatory cells and the phagocytosis and digestion of necrotic material by macrophages [13, 14]. The second phase is the repair phase, during which macrophages release growth factors to recruit and activate tendon fibroblasts. These fibroblasts collaborate to synthesize fibronectin and type III collagen, forming an initial amorphous extracellular matrix. Subsequently, this type III collagen is replaced by the mechanically superior type I collagen [15]. The third phase is the remodeling phase, wherein elongated spindle-shaped tenocytes and collagen fibers become aligned along the direction of stress, thereby restoring tendon stiffness and tensile strength. Concurrently, type III collagen is progressively replaced by type I collagen, and tendon fibroblasts gradually differentiate into myofibroblasts [16].
Autophagy is a conserved mechanism for protein degradation and organelle
recycling in eukaryotic cells [17]. When induced by various cellular stress
conditions, autophagy-associated proteins are recruited to cellular membranes to
form double-membrane phagophores. These phagophores extend and encapsulate
intracellular material, forming distinct autophagosomes [18, 19]. Subsequently,
autophagosomes fuse with lysosomes to form autolysosomes, wherein degradation
occurs under the mediation of acidic hydrolases [20]. The degraded materials
include damaged macromolecules, organelles, and misfolded cytoplasmic proteins,
which are catabolized to provide energy and essential nutrients for intracellular
activities [21]. Autophagy is classified microscopically into xenophagy,
mitophagy, ER-phagy, pexophagy, ribophagy, and chaperone-mediated autophagy, and
macroscopically into selective and non-selective autophagy [18]. Furthermore,
hypoxia or nutrient deprivation serves as a key inducer of non-selective
autophagy, while selective autophagy primarily facilitates the recycling of
misfolded proteins and organelle degradation. Recent studies indicate that
appropriate levels of autophagy also enable osteoblasts and osteoclasts to
survive under extreme conditions [22]. Macrophage autophagy comprises five
distinct phases: initiation, nucleation, elongation, fusion/degradation, and
termination [23]. This process requires multiple regulatory factors. Autophagy
initiation commences with the activation of the Unc-51 like kinase 1 gene (ULK1)
kinase complex, while autophagy related 5 gene (ATG5) plays a critical role in
autophagic vesicle formation among autophagy-related genes [24]. During the
elongation phase, ATG5 and ATG12 are activated by ATG7 and ATG10, forming a
stable complex with ATG16L1 [25]. Microtubule-associated protein 1 LC3 is located
on phagophores and autophagosome membranes. Upon autophagy induction, LC3 is
activated by the complex formed by ATG5-ATG12-ATG16L1 [26]. LC3-I conjugates with
phosphatidylethanolamine (PE) to form LC3-II, which both promotes continuous
extension of the phagophore bilayer membrane structure and facilitates
autophagosome maturation. Consequently, the LC3-II/LC3-I ratio serves as a key
marker for monitoring autophagy occurrence [27]. Autophagy effector protein-1
(Arg1) is a crucial regulator of autophagosome formation. It binds Beclin-1,
which dissociates during stress-induced autophagy to form a complex with class
III phosphatidylinositol 3-kinase (PI3KC3). This complex regulates phagophore
localization and upregulates transcription of cytokine genes, including
IL-1
| Process phase | Key molecules/complexes | Molecular functions | Regulatory mechanisms |
| Initiation signaling | NF- |
Transcriptional activation of autophagy-related genes | I |
| Nucleation of phagocytic vesicles | VPS34-Beclin1-PI3KC3 | Generates PI3P lipid membrane and Recruitment of ATG protein to phagosomes | Beclin1 dissociates from BCL-2 |
| Membrane extension | ATG5-ATG12-ATG16L1Complex | Mediates LC3 lipidation | ATG7/ATG10 activates ATG5-ATG12 |
| Autophagosome labeling | LC3-I |
Autophagosome membrane localization marker | LC3-I cleaved by ATG4 |
| Substrate recognition | p62/SQSTM1 + Parkin p62/SQSTM 1 + Parkin | Recruitment of ubiquitinated substrates | p62 aggregates ubiquitinated proteins |
| Lysosomal fusion | STX17-SNAP29-VAMP8 | Mediates membrane fusion | Rab7-GTP hydrolysis |
| References | [29, 32, 35, 36, 37, 38, 41] | [24, 25, 26, 27, 28, 34, 39, 42] | [24, 25, 26, 27, 34, 40, 43] |
NF-
Apoptosis serves as a critical host defense mechanism, activated through
pathways including p38, NF-
Macrophage necrosis comprises non-programmed and programmed forms. Non-programmed necrosis—typically induced by intense external stimuli—activates receptor-interacting protein kinase (RIPK) and provokes severe inflammation with irreversible damage [48, 49]. Key inducers include high cytokine concentrations, oxidative/ER stress, and fas cell surface death receptor (FAS) ligand activation. Pathogen invasion (e.g., Staphylococcus aureus, Escherichia coli, Streptococcus pneumoniae) generates bacterial toxins causing membrane rupture, mitochondrial depolarization, Adenosinetriphosphate (ATP) depletion, ROS surge, and ultimately programmed necrosis (necroptosis) [50].
Macrophage pyroptosis exhibits molecular and morphological features intermediate
to apoptosis and necrosis. Pathogen exposure triggers caspase-1-mediated
pyroptosis [51, 52]. Under oxidative stress, Gasdermin D oxidation and NLRP3
inflammasome activation induce mitochondrial membrane potential collapse and ROS
generation [53, 54]. Caspase-1 cleaves Gasdermin D (GSDMD), releasing its
N-terminal domain (NT-GSDMD). This forms membrane pores that facilitate
IL-1
Ferroptosis, initially identified in cancer cells, is a novel mode of programmed death induced by iron-dependent depletion of polyunsaturated fatty acids and aggregation of toxic lipid reactive oxygen species [56]. Three main molecular pathways are involved: first, inhibition of small molecule peroxidation and glutathione peroxidase 4 degradation prevents lipids from being over-oxidised and further induces ferroptosis [57]. Second, inhibition of cystine glutamate transporter receptor reduces peroxidase activity and cellular antioxidant capacity, which eventually progresses to ferroptosis. Third, inhibition of the cystine glutamate transporter receptor system is mediated at the gene level through p53, which induces the activation of pro-apoptotic proteins and transcriptional repression of anti-apoptotic proteins by inhibiting the anti-apoptotic function of BCL-2 [58, 59].
Macrophage autophagy is modulated by NF-
| Autophagy | Necrotic apoptosis | Necrosis | Pyroptosis | Ferroptosis | |
| Biochemical characteristics | Increased lysosomal activity | Caspase1 activation dependent or independent | ATP levels decreased; RIP1, RIP3 and MLK were activated | Activation of caspases oligosome DNA fragments | GSH and GPX4 inhibition; iron accumulation and lipid peroxidation |
| Morphological features | Formation of autophagic lysosomes with double-membrane structures | Plasma membrane blistering, reduced cell and nuclear volume, nuclear fragmentation | Plasma membrane rupture, organelles swelling, chromatin condensation | Nuclear retraction, cell swelling and cell membrane hole, cell collapse and rupture | Mitochondria become small and cristae of mitochondria decrease or disappear |
| Coregulated gene | LC3, ATG5, ATG-7, Beclin1, Other ATG family proteins | p53, Bax, Bak, BCL-2, BCL-XL, BCL-2 family proteins, Other anti-apoptotic | RIP1, RIP3, MLK L | CASP1, CASP11, GSDMD | VDAC2/3, Ras, NOX, TFR1, p53, CARS, GPX4, SLC7A11, HSPB1, NRF2 |
| Regulatory path | PI3K-AKT-mTOR, MAPK-ERK1/2-mTOR | Csapase, P53, BCL-2 | TNF |
Caspase-1, NLRP3 | Gpx4, MVA, HSF1-HSPB1, p62-Keap1-Nrf2, LSH |
| Release of damage associated molecules | HMGB-1 | HMGB1, ATP | DNA, IL-6 | HMGB1, ATP, IL-1 |
HMGB1 |
| Immunological characteristics | anti-inflammatory | anti-inflammatory | Proinflammatory | Proinflammatory | Proinflammatory |
| Detection method | Detection of changes in the levels of autophagy-related proteins such as Atg5, Atg7, BeclinI, LC3, P62, autophagosome fluorescence single/double labelling assay, lysosomal function assays | Mitochondria-free membrane potential assay, An-nexin V/PI, TU-NEL assay, apoptosis-related pathways, apoptosis-related proteins | Immunofluorescence or flowthrough methods by PI or 7-AAD staining | NLRP3, ASC, Procaspase-1, Cleavedcaspase-1, Pro-IL-1 |
Cellular activity assays: CCK-8, intracellular iron levels (PGSK probe), levels of reactive oxygen species, changes in death-associated factors such as COX-2, ACSL4, PTGS2, NOX1, GPX4, and FTH1 |
| References | [15, 16, 17, 18, 19, 20, 21, 63, 64] | [46, 50, 56, 57, 58, 59] | [19, 27, 46, 47, 50, 66] | [20, 31, 61, 62, 65, 66] | [35, 36, 37, 38, 52, 53, 54] |
Stimulated by Receptor Activator of nuclear factor-
| Cell type | Autophagy function | Role in tendon-bone healing | Molecular mechanisms |
| Macrophages | Remove damaged mitochondria | Inhibition of NLRP3 inflammasome |
PINK1/Parkin mediates mitochondrial autophagy |
| Osteoclasts | Promotes crease margin formation | Enhance bone resorption |
ATG5-dependent Rab7 localisation |
| Osteoblasts | Maintains cell survival | Resist stress-induced apoptosis |
Autophagy degrades misfolded proteins |
| Chondrocytes | Inhibits hypertrophic apoptosis | Protects fibrocartilage layer |
Autophagy removes damaged ER |
| Tendon stem cells | Maintain stemness | Promote differentiation to tendon cells |
Activation of p62/KEAP1-NRF2 axis |
| References | [15, 16, 17, 18, 19, 20, 21, 63, 64] | [46, 50, 56, 57, 58, 59] | [19, 27, 46, 47, 50, 66] |
During early inflammation, macrophages (M
M1 macrophage polarization induces chondrocyte apoptosis and hypertrophy,
extracellular matrix (ECM) degradation, and ultimately exacerbates cartilage
damage and OA progression [77]. Selective macrophage depletion significantly
reduces the M1/M2 ratio, downregulates IL-1
M1 macrophages secrete cytokines to induce osteoclastogenesis from osteoclast
precursor cells (OCPs). Studies demonstrate that M1 macrophages enhance
osteoclast precursor proliferation or directly induce their differentiation into
osteoclasts via cytokine secretion, while increased pro-inflammatory factor
release indirectly upregulates RANKL secretion by osteoblasts and stromal cells,
thereby augmenting osteoclast formation [80, 81]. In contrast, M2 macrophages
polarize into the M2a subtype upon IL-4/IL-13 stimulation, secreting insulin-like
growth factor-1 (IGF-1) and arginase-1, releasing chemokines such as CCL13 and
CCL17, and recruiting basophils, eosinophils, and Th2 cells [82]. Notably, high
surface expression of CD86 and CD163 facilitates neovascularization and collagen
deposition. Glucocorticoids, IL-10, and TGF-
Osteoclasts, regulated by autophagy or pathway-associated upstream/downstream cytokines, also feedback by releasing bioactive signals that inversely modulate macrophages and autophagic complexes, further regulating tendon-bone junction healing (relevant cytokines are detailed below) (Table 4, Ref. [30, 63, 64, 72, 73, 74, 75, 76, 84, 85, 86, 87, 88]).
| Regulatory module | Key components | Biological significance | References |
| M1 polarisation pathway | TLR4/NF- |
Pro-inflammatory signals inhibit autophagy initiation, but ROS can activate stress-induced autophagy | [72, 73, 74, 75, 76, 84, 85] |
| M2 Polarisation Pathway | IL-4/STAT6 |
Inhibits autophagy by activating Akt-mTOR, promoting a reparative phenotype | [72, 73, 74, 75, 76, 86, 87] |
| Autophagy regulates polarisation | TG5 degradation STAT1 |
Resist stress-induced apoptosis |
[63, 64, 73, 76] |
| Metabolic switch | Hypoxia |
Removes damaged mitochondria, reducing M1-related inflammation | [30, 88] |
TNF-
IL-1
IL-4 is an anti-inflammatory factor secreted by M2 and inhibits osteoclast
formation by suppressing the expression of RANKL and TNF-
IL-6, a downstream product of the NF-
IL-10 is negatively associated with the development of osteoporosis and inhibits osteoclast formation by decreasing the secretion of pro-inflammatory factors and inhibiting the expression of RANKL. IL-10 levels are significantly lower in osteoporotic patients compared to healthy individuals [97].
M1 macrophages release chemokines (e.g., CCL2, CXCL8, SDF-1, etc.) to recruit MSCs to the injury site to participate in tendon bone healing [98]. In the early stage of tendon-bone junction healing, a large number of M1 macrophages infiltrate to phagocytose cellular debris and foreign pathogens, and at the same time recruit MSCs to the tendon-bone junction by secreting chemokines [99].
TGF-
M2 macrophage polarization enhances endogenous vascular endothelial growth factor (VEGF) expression, promoting vascular neovascularization and fat infiltration in ischemic conditions. This creates a favorable microenvironment for tendon-bone healing [88]. BMSC-derived exosomes induce M2 polarization, elevate VEGF expression, and stimulate peripheral vascular neovascularization around injured tendon-bone interfaces. This improves ligament healing strength without increasing adhesion [105]. Bone morphogenetic proteins (BMPs), abundant in bone matrix, promote chondrogenic and osteogenic differentiation of bone marrow stromal stem cells. BMP-2—among the most active isoforms—drives trilineage differentiation (adipogenesis, chondrogenesis, osteogenesis), enhances vascular density, and upregulates alkaline phosphatase and osteocalcin [106, 107] (Table 5, Ref. [32, 40, 41, 65, 84, 85, 86, 87, 89, 90, 91, 92, 93, 94, 95, 100, 101, 102, 103, 104]).
| Cytokine | Regulatory role of autophagy | Effects on tendon-bone healing | Molecular pathways | References |
| TNF- |
Negative: Induces tendon stem cell apoptosis + inhibits type I collagen synthesis | NF- |
[32, 65, 84, 85, 87, 89, 94, 95] | |
| Positive: Low concentrations promote M1 differentiation into OC precursors | Inhibits AMPK |
|||
| IL-1 |
Negative: Promotes MMP expression |
NLRP3-Caspase-1/GSDMD pathway activation | [40, 41, 85, 89, 90, 91, 92, 93] | |
| Positive: Enhances cellular stress survival | PI3K-AKT-FOXO3a feedback loop | |||
| IL-4 | Positive: Inhibits RANKL expression |
STAT6 phosphorylation |
[86, 87] | |
| Negative: Delays damage clearance | Activates Akt |
|||
| TGF- |
Positive: Promotes fibrocartilage regeneration + inhibits scar formation | Smad2/3 phosphorylation |
[41, 100, 101, 102, 103, 104] | |
| Positive: Clears damaged ECM |
Inhibits PI3K-Akt |
|||
| IL-10 | Positive: Inhibits pro-inflammatory cytokine storm + promotes M2 polarisation | Activates JAK1-STAT3 |
[89, 93] | |
| Positive: Protects macrophage anti-inflammatory phenotype | PINK1/Parkin ubiquitinates damaged mitochondria |
These growth factors, when highly expressed, modulate both the biological and mechanical microenvironments of the tendon-bone junction through the following mechanisms: First, distinct growth factor types are upregulated at different stages—those recruiting inflammatory cells and promoting neoangiogenesis are predominant during the inflammatory phase, while those driving stem cell differentiation into specific lineages and collagen synthesis dominate during the reparative and remodeling phases [108]. Second, acting as signaling molecules, these growth factors regulate cellular behaviors via paracrine or autocrine mechanisms to facilitate tendon-bone healing [109]. Third, growth factors exhibit interactive effects [103]. However, due to their short half-lives, sustained local delivery via appropriate carriers is necessary to maintain their temporal efficacy.
Shear stress, distraction stress, mechanical stimulation, and noose stress can affect collagen and extracellular matrix synthesis, and under limited conditions can promote the differentiation of cells at the tendon-bone junction into cartilage [110]. Existing studies have found higher expression of type I collagen and glycosaminoglycans in the cartilage matrix of the loaded stress group, with a higher resistance to distraction and healing closer to normal tissue morphology [111].
The Other tendon-bone junction repair method include biomechanical materials, cellular applications (e.g., osteoinductive materials, biodegradable scaffolds, bionic patch gene therapy and cellular therapy, etc.), and postoperative rehabilitation strategies [105, 109].
LIPUS delivers non-invasive mechanical stimuli that activate the PI3K-Akt
pathway via mechanotransduction, polarizing macrophages toward the M2 phenotype
[112]. This polarization upregulates IL-10 while downregulating pro-inflammatory
cytokines (TNF-
Osteoclast (OC) differentiation and maturation are regulated by multiple signaling pathways, including mTORC1 which mediates autophagy and influences OC differentiation [97]. mTORC1 is a serine/threonine kinase regulating protein synthesis. Under nutrient-rich conditions, mTORC1 binds and inhibits the ULK1 complex, suppressing autophagy; during malnutrition, their dissociation activates ULK1 through dephosphorylation, initiating autophagy [66, 117]. The mTOR pathway is modulated by upstream regulators including AMPK, Akt, and PI3K pathways [118, 119]. mTORC1-mediated autophagy may bidirectionally regulate OC. AMPK maintains cellular homeostasis by activating during energy depletion. High glucose decreases p-AMPK and p-ULK1 while increasing p-mTOR [120]. Concomitantly, reduced LC3-II and Beclin-1 with elevated P62 correlate with decreased OC production and bone resorption, indicating mTORC1-mediated autophagy promotes osteoclastogenesis [121].
Osteoclasts (OCs) migrate to and adhere to bone surfaces during differentiation
and maturation to execute bone resorption. Their migratory capacity depends on
adaptive morphological changes and the continuous formation and degradation of
pseudopodia. Integrin
Osteoclasts (OCs) attach to bone surfaces forming ruffled borders, secreting acid and proteolytic enzymes to resorb bone. ATG4B, ATG5, ATG7, and LC3 critically regulate ruffled border formation and lysosomal secretion [24, 39]. ATG5/ATG7 knockout impairs ruffled border formation and reduces bone resorption without affecting OC differentiation [42]. Autophagy and resorption functions interconnect via lysosomes: Fusion of lysosomal vesicles forms ruffled borders, while vesicular transport of resorptive factors relies on GTP hydrolases. Rab7 localizes to ruffled borders in an ATG5-dependent manner; ATG5 deficiency disrupts Rab7/LC3 expression and reduces cathepsin K secretion [43, 124]. ATG7 and ATG4B/LC3 further recruit Rab7 to enhance resorption.
The dynamic interplay between macrophage autophagy and polarization offers
actionable targets for refining therapeutic strategies in tendon-bone junction
(TBJ) repair. For graft integration, targeted modulation of macrophage phenotypes
can optimize the foreign body response. Biomimetic scaffolds (e.g.,
gradient-mineralized hydrogels [7, 109]) engineered to sustainably release
autophagy inducers or M2-polarizing cytokines could promote early M2 dominance,
suppress chronic inflammation, and enhance fibrocartilage regeneration at the
graft-host interface. Crucially, scaffold surface topography and biochemical cues
can direct macrophage polarization toward regenerative phenotypes, reducing
fibrotic encapsulation and improving biomechanical integration [105]. In scaffold
design, incorporating “smart” nanocarriers (e.g., exosomes) enables
spatiotemporally controlled delivery of autophagy modulators (e.g., ATG5/7
agonists) or anti-inflammatory miRNAs specifically to TBJ-resident macrophages
[105, 125]. For postoperative recovery, LIPUS exemplifies a non-invasive
mechanobiological intervention. By activating the PI3K-Akt pathway [112, 126],
LIPUS polarizes macrophages toward M2 phenotypes, enhances autophagy flux, and
upregulates BMP-2/TGF-
Macrophage polarization is a key regulatory factor in tissue repair processes,
but its regulatory mechanisms exhibit significant context-dependent discrepancies
across different parts of the musculoskeletal system [127]. This difference is
particularly evident in tendon-bone interface (TBJ) versus skin/muscle healing,
where M2 macrophage polarization may even exert diametrically opposite effects.
The divergence in fibrotic outcomes is a prominent manifestation of this
discrepancy. In skin wound healing, excessive M2 polarization drives pathological
fibrosis formation by overactivating the TGF-
Recent studies reveal that dysregulation of macrophage polarization disrupts the
intrinsic healing program of tendons, thereby promoting the occurrence of ectopic
ossification [134]. This phenomenon stands in sharp contrast to the healing
process of bone defects: in bone repair, polarization of M2 macrophages typically
favors osteogenesis; whereas in tendon injury, persistent activation of M1
macrophages drives pathological chondro-osteogenic differentiation through unique
mechanisms. The shift in the cytokine microenvironment is a key mechanism
therein. Activated M1 macrophages significantly increase the secretion of
oncostatin M; this factor synergizes with BMP2 signaling to potently activate the
Runx2 transcription factor in tendon-derived stem cells [125, 135]. This signaling
axis overrides the canonical TGF-
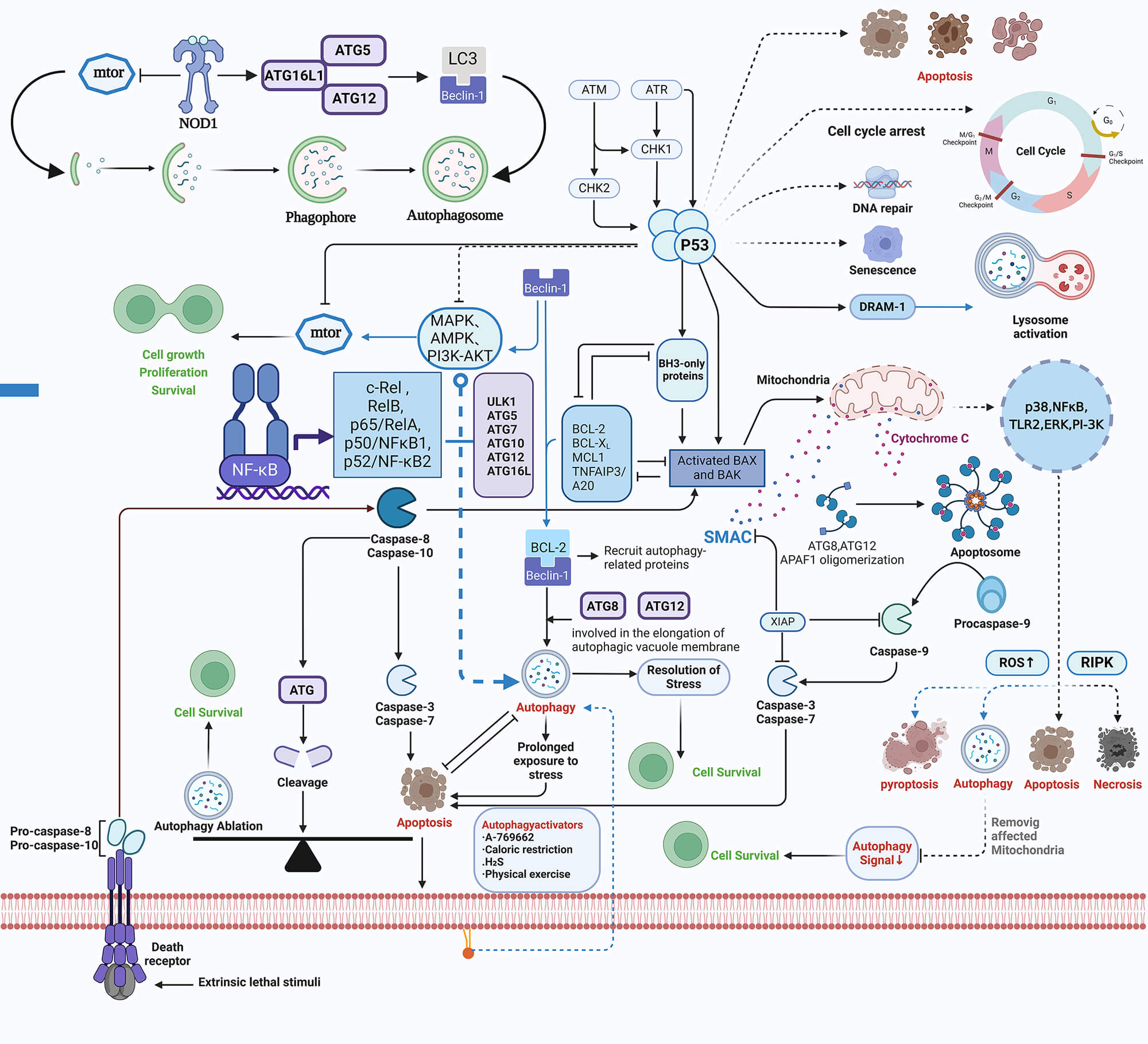 Fig. 1.
Fig. 1.
Signaling pathways and the sequential process of autophagy. This figure illustrates: (1) the roles of cytokines and various molecules in autophagy through different pathways; (2) the morphological differences between autophagy and other forms of apoptosis; (3) the complete process of autophagy, from initiation, vesicle formation, membrane extension, substrate recognition, to lysosomal fusion.
Emerging evidence confirms cytokine-mediated autophagy as pivotal for macrophage
function at tendon-bone interfaces. However, physical stimuli represent an
underutilized therapeutic lever. Coman et al. [115] demonstrated that
mechanical strain activates PI3K-Akt via integrin
Interaction mechanisms between autophagy and polarization: Future studies should clarify whether autophagy-related proteins (e.g., ATG5, Beclin-1, LC3) modulate macrophage polarization direction (M1/M2) by degrading key polarization proteins or activating specific transcription factors [79]. Spatiotemporal-specific regulation of autophagy and polarization: Autophagy levels may differ between the inflammatory phase (M1-dominated) and the reparative phase (M2-dominated) [73, 74, 75, 76].
Optimization of drug delivery systems: Current autophagy modulators
(inducers/inhibitors) lack tissue specificity and elicit systemic side effects
[18, 21, 22, 23, 24]. Future investigations should explore nanocarrier-based systems (e.g.,
exosomes, liposomes) encapsulating autophagy regulators to achieve spatiotargeted
modulation of local TBJ macrophages. Synergistic effects of biomaterials and
autophagy: Biomimetic scaffolds (e.g., gradient mineralized hydrogels) can
replicate the four-layer architecture of the tendon-bone junction (TBJ) while
co-loading bioactive factors (e.g., IL-4, TGF-
Bioinformatics and high-throughput sequencing: transcriptome characterization of macrophage subsets during TBJ repair by single-cell RNA sequencing, combining bioinformatics to predict key targets [42].
Large animal experiments with clinical trials: testing intervention strategies targeting autophagy in large animal models of rotator cuff injury or anterior cruciate ligament reconstruction, and a gradual transition to clinical trials [31, 56].
Conformal LIPUS transducers such as 3D-printed devices to match TBJ stiffness
gradients to optimize integrin
Combining autophagy-modulating nanoparticles with LIPUS stimulation could provide a synergistic approach to resolving chronic inflammation at tendon-bone junctions and enhancing matrix regeneration at the repair site [126]. Combine LIPUS with thermosensitive liposomes co-loaded with autophagy inducer. Nanoscale LIPUS materials could be developed in future to achieve this goal.
Despite advances in understanding macrophage autophagy in tendon-bone junction
repair, critical limitations persist: Current models (e.g., rodent rotator cuff
injuries) fail to fully recapitulate human TBJ complexity, particularly regarding
mechanotransduction and immune cell crosstalk. Biomaterial scaffolds show promise
in vitro but lack clinical validation for sustained immunomodulation. We
also put forward some testable hypotheses. Hypothesis 1: Precision timing of
autophagy induction will accelerate M1-to-M2 transition, reducing scar formation
(Testable via ATG7-knockout murine models). Hypothesis 2: Scaffold-driven
autophagy (e.g., activated integrin
M1, Macrophage-1; M2, Macrophage-2; OC, Osteoclasts; OB, Osteoblasts; BMP, Bone Morphogenetic Protein-2; BMSCs, Bone marrow mesenchymal stem cells; MMPs, Matrix MetalloProteinases; mTOR, Mammalian target of rapamycin; IL-, Interleukin-; AMPK, AMP-activated protein kinase; GSH, Glutathione; GSSG, Oxidized Glutathione; GPX4, Glutathione PeroXidase4; ATG, Autophagy-related gene; NF-
DJY: Conceptualization, Methodology, Writing—Original Draft Preparation, Supervision. GWG, JFS: Collecting References. JJC: Collecting References, Resources, Writing—Review & Editing. GYW, JCL: Collecting References, Project Administration, Funding Acquisition, Supervision. All authors contributed to editorial changes in the manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
The Affiliated Traditional Chinese Medicine Hospital of Southwest Medical University, Biomechanical research on cartilage transplantation based on the theory of “equal emphasis on muscle and bone”, 2023ZYQJ02.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.