






 , Geng Huang 1,†, Yu Liu 2, Fulin Pan 1, Canling Long 3, Jia Liu 3, Zhigang Ma 1,*
, Geng Huang 1,†, Yu Liu 2, Fulin Pan 1, Canling Long 3, Jia Liu 3, Zhigang Ma 1,*
1 Nephrology and Rheumatology Department, The Second Affiliated Hospital, School of Medicine, The Chinese University of Hong Kong, Shenzhen & Longgang District People’s Hospital of Shenzhen, 518172 Shenzhen, Guangdong, China
2 Nephrology Department, The Chinese University of Hong Kong, Shenzhen Medical Center, 518172 Shenzhen, Guangdong, China
3 Central Laboratory, The Second Affiliated Hospital, School of Medicine, The Chinese University of Hong Kong, Shenzhen & Longgang District People’s Hospital of Shenzhen, 518172 Shenzhen, Guangdong, China
†These authors contributed equally.
Abstract
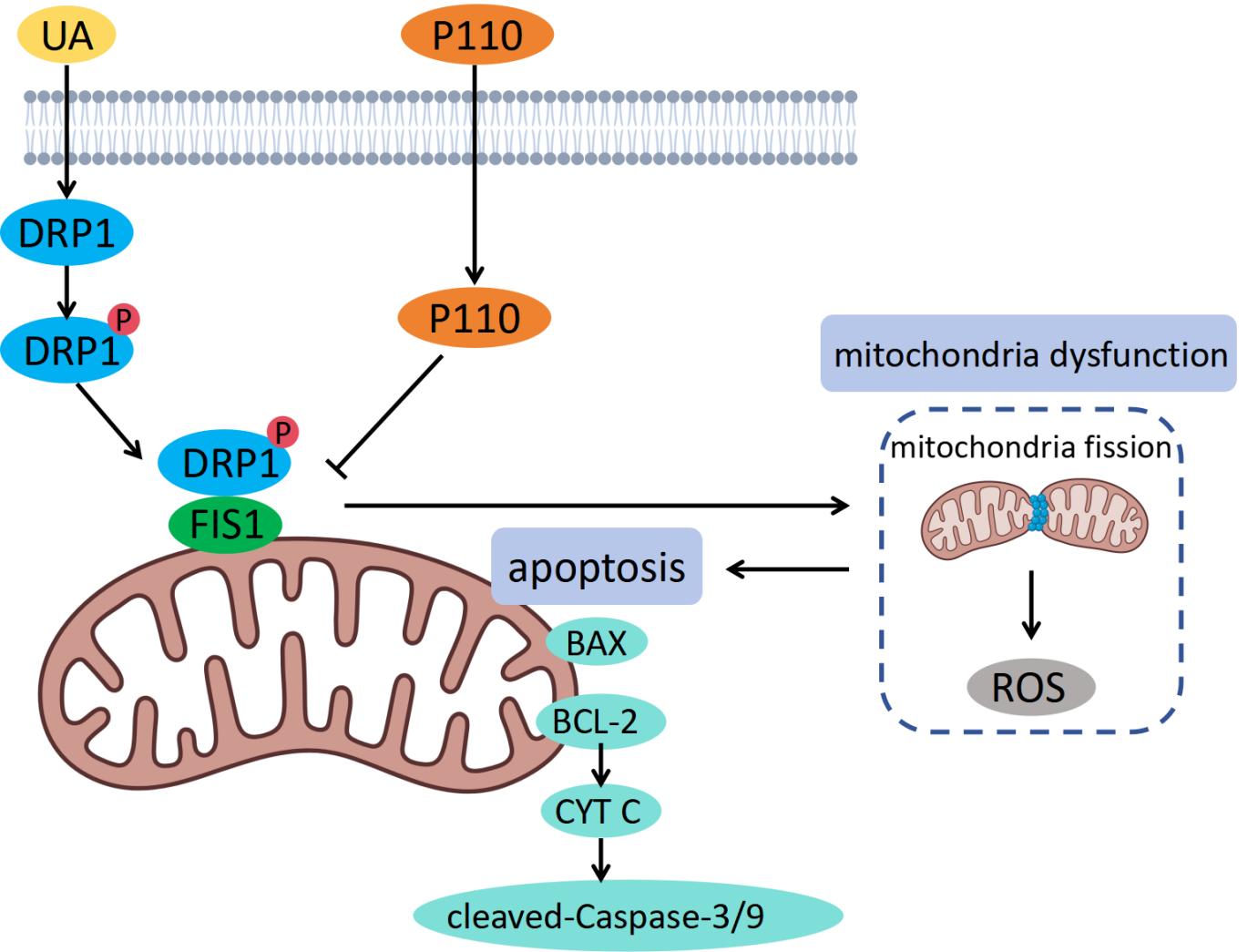
Hyperuricemic nephropathy is associated with mitochondrial dysfunction. Dynamin-related protein 1 (DRP1), a key regulator of mitochondrial fission, is activated under stress and translocates to the mitochondria, where it interacts with adapter proteins such as mitochondrial fission 1 protein (FIS1), thereby promoting excessive mitochondrial fission and apoptosis. Recent research has shown that inhibiting the DRP1/FIS1 interaction can reduce cellular injury in various disease models; however, its role in hyperuricemic nephropathy is unclear.
An in vitro model of hyperuricemic nephropathy was established by treating human renal tubular epithelial cells with uric acid (UA). Reverse transcription quantitative PCR and western blotting, and enzyme-linked immunosorbent assays were used to quantify the mRNA and protein levels of the target molecules. A specific peptide inhibitor, P110, was used to disrupt the binding between DRP1 and FIS1. Co-immunoprecipitation (Co-IP) was performed to confirm the interactions between DRP1 and FIS1. Cell viability was assessed using propidium iodide staining and the Cell Counting Kit-8 assay.
UA significantly upregulated DRP1 expression, activated DRP1, and promoted mitochondrial translocation. P110 inhibited DRP1/FIS1 binding, preventing DRP1 UA-induced mitochondrial translocation. Excessive mitochondrial fission, reactive oxygen species generation, release of inflammatory factors, and apoptosis were significantly alleviated. In addition, inhibition of DRP1 mitochondrial translocation decreased the expression of apoptosis-related markers and apoptosis.
The overactivation of DRP1 is crucial for UA-induced renal tubular epithelial cell injury. P110 exerts a cytoprotective effect by inhibiting the DRP1/FIS1 interaction and modulating the mitochondrial apoptotic pathway. This study proposes a possible target for therapeutic intervention in the treatment of hyperuricemic nephropathy.
Graphical Abstract
Keywords
- uric acid
- renal tubular epithelial cells
- dynamin-related protein 1
- mitochondrial dysfunction
- apoptosis
Hyperuricemia is a chronic metabolic disorder caused by the dysregulation of purine metabolism and/or impaired uric acid excretion, and it shows abnormally high levels of serum uric acid (UA) levels [1]. Persistent hyperuricemia can result in secondary kidney injury, which may progress to uric acid nephropathy [2, 3, 4]. With the increasing consumption of high-purine foods, sugars, and alcohol, the incidence of uric acid nephropathy is also increasing, posing a substantial threat to public health, especially among adult males in China [5, 6]. Therefore, exploring the molecular mechanisms underlying UA-induced kidney injury and devising effective intervention strategies is of great clinical significance.
Recent studies have revealed that a hyperuricemic environment can trigger mitochondrial dysfunction, which manifests as a decrease in the mitochondrial membrane potential, abnormal mitochondrial morphology, and increased apoptosis [7, 8, 9, 10]. As an organ with high oxygen consumption, the kidney’s tubular epithelial cells are rich in mitochondria, which are highly susceptible to oxidative stress. UA can induce the overproduction of reactive oxygen species (ROS), thereby disrupting mitochondrial function and establishing a vicious cycle of “ROS-mitochondrial damage”, which significantly exacerbates renal injury [11]. Hence, maintaining mitochondrial homeostasis is essential to alleviate UA-mediated renal pathology.
Mitochondria are highly dynamic organelles whose morphology and function are precisely regulated by the balance between fusion and fission [12]. Dynamin-related protein 1 (DRP1) is a key molecule involved in regulating mitochondrial fission [13]. The activity of DRP1 is modulated by various post-translational modifications. Phosphorylation at Ser616 activates DRP1 and facilitates its binding to mitochondrial outer membrane receptors, such as mitochondrial fission 1 protein (FIS1) and mitochondrial fission factor (MFF), mediating DRP1 translocation to the mitochondria and initiating fission [14, 15]. Overactivation of DRP1 can lead to mitochondrial fragmentation, loss of membrane potential, ROS burst, impaired adenosine triphosphate (ATP) synthesis, and further induce mitochondrial outer membrane permeabilization and Cytochrome C (CYT C) release via B-cell lymphoma 2 (BCL-2) family proteins (e.g., BCL2-associated X protein (BAX)), thus activating downstream apoptotic pathways [16, 17].
P110 is a specific peptide inhibitor that targets activated DRP1. It blocks the interaction between DRP1 and FIS1, effectively inhibiting DRP1 mitochondrial translocation and overactivation [18]. This inhibitor is composed of a seven-amino acid functional peptide fused with the cell-penetrating transactivator of transcription (TAT, 47–57) peptide, endowing it with high cell membrane permeability, effective in vivo delivery, and no toxic side effects, providing a solid basis for its therapeutic potential [18, 19]. Previous studies have shown that P110 has protective effects in various disease models, including neurological disorders, heart diseases, and cancer [20, 21, 22, 23]. Additionally, downregulation of DRP1 alleviates UA-induced renal injury, suggesting that DRP1 may be a potential therapeutic target in kidney diseases [24]. However, the specific role of DRP1 overactivation in the pathogenesis of hyperuricemic nephropathy and the corresponding interventional strategies, especially the protective mechanism of P110-mediated inhibition of the DRP1/FIS1 interaction in this model, remain to be clarified.
This study identifies the critical role of DRP1 overactivation in the pathogenesis of hyperuricemic nephropathy. Further investigation confirmed that P110, by specifically inhibiting the DRP1/FIS1 interaction, effectively blocked DRP1 overactivation and attenuated UA-induced renal tubular epithelial cell apoptosis via the mitochondrial pathway, demonstrating significant protective effects. This study identified a potential molecular target for the treatment of hyperuricemic nephropathy.
Human renal proximal tubular epithelial cells (HK-2) were procured from Procell
Life Science & Technology Co., Ltd. (Wuhan, Hubei, China; Product No. CL-0109)
with accompanying official certificates for STR profiling and mycoplasma-free
status. To ensure ongoing quality, we perform routine in-house testing using the
BeyoDirect™ Mycoplasma qPCR Detection Kit (Beyotime, Shanghai,
China; Product No. C0303S); all results have been negative, consistent with the
kit’s criteria (sample Ct
The viability of cells was evaluated using the Cell Counting Kit-8 (CCK-8; Beyotime, Shanghai, China; Product No. C0037). Cells were plated in 96-well plates (2000 cells/well), subjected to compounds at different concentrations for specific time periods, and then each well was incubated with 10 µL of CCK-8 reagent. After incubation, absorbance was measured at 450 nm using a microplate reader (BioTek, Winooski, VT, USA).
Intracellular ROS levels were detected using 2′,7′-dichlorodihydrofluorescein diacetate (DCFH-DA; Beyotime, Shanghai, China; Product No. S1105M). Cells were incubated with DCFH-DA in serum-free medium for 30 minutes at 37 °C in the dark. After washing with PBS, the fluorescence intensity was quantified using a microplate reader (BioTek, Winooski, VT, USA) with excitation and emission wavelengths set at 495/529 nm.
Cell viability was evaluated by propidium iodide (PI; Servicebio, Wuhan, Hubei, China; Product No. G1021) staining. After treatments, cells were gently washed twice with PBS and stained with PI solution (10 µg/mL in PBS) for 10 minutes at room temperature in the dark. The nuclei were stained again with Hoechst 33342 (10 µg/mL; Thermo Fisher Scientific, Waltham, MA, USA; Product No. R37605). Fluorescent images were acquired using a Leica STELLARIS 5 confocal laser-scanning microscope (Leica Microsystems, Wetzlar, Germany).
According to the manufacturer’s guidelines, total RNA was extracted from cells using AG RNAex Pro Reagent (Accurate Biotechnology, Changsha, Hunan, China; Product No. AG21102). Complementary DNA (cDNA) was synthesized from 1 µg total RNA with the Evo M-MLV RT Premix (Accurate Biotechnology, Changsha, Hunan, China; Product No. AG11728). Quantitative PCR (qPCR) was carried out on a QuantStudio 3 Real-Time PCR System (Thermo Fisher Scientific, Waltham, MA, USA) using the SYBR® Green Premix Pro Taq HS qPCR Kit (Accurate Biotechnology, Changsha, Hunan, China; Product No. AG11718) and specific primers. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) served as an internal reference. Gene expression levels were calculated by the 2-ΔΔCT method. The sequences of the primers used were as follows: 5′-CCAGTGAAGCCGGTCAATCT-3′ (F, DRP1); 5′-GCTGCCGTGACAGAAAACAC-3′ (R, DRP1); 5′-CTGAACGAGCTGGTGTCTGT-3′ (F, FIS1); 5′-GAACAGGGAAAGGACAGCGA-3′ (R, FIS1); 5′-AAAGATGGTGTGGCCGATGT-3′ (F, SOD2); 5′-CAAGCCAAACGACTTCCAGC-3′ (R, SOD2); 5′-GTGCCACCAAGTTCAAGCAG-3′ (F, HO-1); 5′-CACGCATGGCTCAAAAACCA-3′ (R, HO-1); 5′-TGCTTACACTTACGCTGCCAT-3′ (F, NQO-1); 5′-CCAGTGGTGATGGAAAGCAC-3′ (R, NQO-1); 5′-CGGGAAGGTGAAGCGCAATG-3′ (F, MFN); 5′-CTGTCCAGCTCTGTGGTGAC-3′ (R, MFN); 5′-CTGTGGCCTGGATAGCAGAA-3′ (F, OPA1); 5′-GCGAGGCTGGTAGCCATATT-3′ (R, OPA1); 5′-AATGGGCAGCCGTTAGGAAA-3′ (F, GAPDH); 5′-TCGTGTTGCACTGGTTAAAGC-3′ (R, GAPDH).
Cytosolic and mitochondrial fractions were isolated using a Mitochondria
Isolation Kit (Thermo Fisher Scientific, Waltham, MA, USA; Product No. 89874)
following the manufacturer’s protocol. Briefly, the cells were washed with
ice-cold PBS, resuspended in ice-cold mitochondrial isolation buffer, and
homogenized. The homogenate was centrifuged at 800
Total cellular protein was isolated by employing RIPA lysis buffer (Thermo
Fisher Scientific, Waltham, MA, USA; Product No. 89901) that had been
supplemented with protease and phosphatase inhibitors (Sigma-Aldrich, St. Louis,
MO, USA; Product No. 78440). The concentration of the extracted protein was
measured via the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham,
MA, USA; Product No. 23227). Subsequently, equivalent quantities of the protein
samples were subjected to separation through sodium dodecyl
sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) and then transferred onto
polyvinylidene difluoride (PVDF) membranes. Membranes were blocked and probed
with primary antibodies overnight at 4 °C in TBST (Tris-buffered saline
with 0.1% Tween-20). The blocking agent and dilution buffer was 5% skim milk
for all antibodies, except for the anti-p-DRP1 (Ser616) antibody, for which 5%
BSA was used to minimize background. The primary antibodies used were as follows:
anti-GAPDH (1:5000, Proteintech, Wuhan, Hubei, China; Product No. 60004-1-Ig),
anti-DRP1 (1:2000, Proteintech, Wuhan, Hubei, China; Product No. 12957-1-AP),
anti-p-DRP1 (Ser616; 1:1000, Abcam, Cambridge, UK; Product No. Ab314755),
anti-COX IV (1:2000, Abcam, Cambridge, UK; Product No. Ab202554), anti-FIS1
(1:2000, Proteintech, Wuhan, Hubei, China; Product No. 10956-1-AP),
anti-IL-1
Protein-protein interactions between DRP1 and FIS1 were assessed using the Pierce™ Classic IP Kit (Thermo Fisher Scientific, Waltham, MA, USA; Product No. 88804). Briefly, the magnetic beads were washed and incubated with anti-FIS1 antibody or control IgG for 60 min. Cell lysates were introduced to antibody-bound beads and subjected to an overnight incubation at 4 °C under continuous rotation. Once the incubation period was completed, the beads were washed, and the immunoprecipitated complexes were then eluted. The existence of DRP1 within the FIS1 immunoprecipitates was examined through SDS-PAGE followed by Western blotting analysis.
Cells from each group were harvested by trypsinization, pelleted by centrifugation, and fixed in 2.5% glutaraldehyde (Servicebio, Wuhan, Hubei, China; Product No. G1102-100ML) for 3 h at 4 °C. Samples were then post-fixed in 1% osmium tetroxide in 0.1 M PBS (pH 7.4) for 2 h. After dehydration using a graded ethanol series, the pellets were embedded in epoxy resin. Ultrathin sections (50 nm) were prepared, stained with uranyl acetate and lead citrate and examined under a transmission electron microscope (Hitachi High Technologies, Tokyo, Japan).
Mitochondrial membrane potential was assessed using the potentiometric dye JC-1
(MCE, Monmouth Junction, NJ, USA; Product No. HY-15534). After treatments, cells
were incubated with 20 µM JC-1 in complete medium for 20 minutes at 37
°C in a 5% CO₂ atmosphere. Cells were washed with PBS, and JC-1
fluorescence (monomeric form: green, Ex
Total DNA was extracted from HK-2 cells using Qiagen DNeasy Blood and Tissue
Kits (Product No. 69504; Hilden, Germany) according to standard methods. To
determine mitochondrial DNA (mtDNA) copy number, mtDNA was quantified by
normalizing the mitochondrial-encoded gene ND1 to the nuclear-encoded gene B2M
using RT-PCR and the
The mtDNA copy number was normalized to that of B2M to calculate its relative value.
Concentrations of interleukin-1
Data are presented as mean
To establish a hyperuricemic cell injury model, HK-2 cells were subjected to different concentrations of UA (0, 1, 2, 3, and 4 mM) for a duration of 36 h. The CCK-8 assay was employed to evaluate the viability of the cells. The data clearly demonstrated a dose-dependent decline in cell viability as UA concentration increased (Supplementary Fig. 1). Among the tested concentrations, treatment with 3 mM UA led to a reduction of cell viability by approximately 50%, which corresponded to the half-maximal inhibitory concentration. Therefore, this concentration was selected for subsequent experiments, as it effectively represents a condition where significant cell injury is induced while maintaining a measurable cell population for further analysis.
Next, to evaluate the effect of UA on cell membrane integrity, we compared the UA-treated group with the control group. Propidium iodide staining was used because PI can only enter cells with compromised membranes. The results showed a marked increase in the number of PI-positive cells in UA-treated groups (Fig. 1A,B). This substantial increase in PI-positive cells clearly indicated that the cell membranes in the UA-treated group were damaged, resulting in the loss of their normal barrier function.
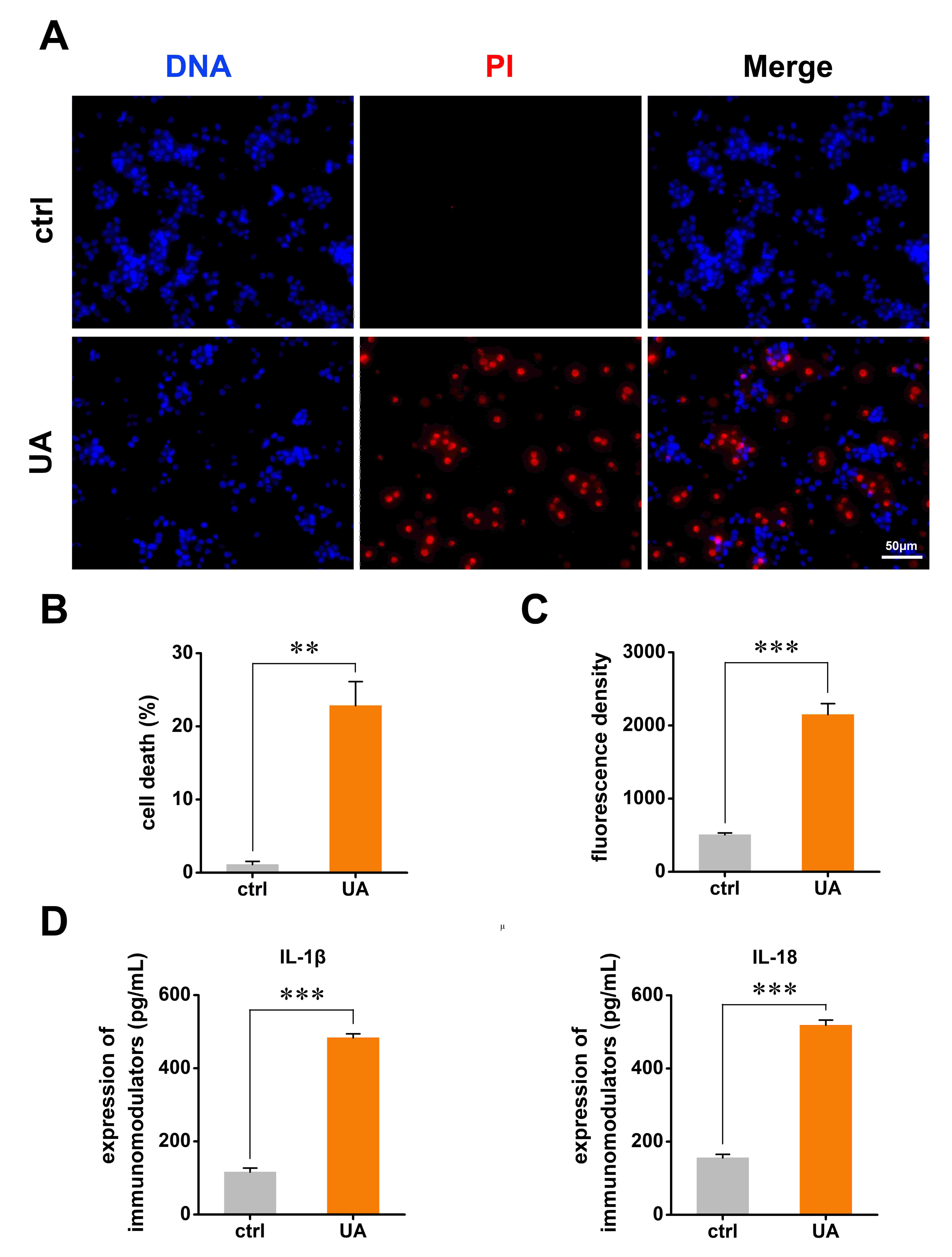 Fig. 1.
Fig. 1.
UA triggers HK-2 cell injury, oxidative stress, and inflammatory
response. (A) PI/Hoechst double-staining fluorescence images showing cell
membrane damage in the UA-treated group (3 mM) (PI red, nuclei blue). Scale bar:
50 µm. (B) Quantitative analysis of PI-positive cells/signals. (C)
Quantitative results of intracellular ROS levels detected by DCFH-DA fluorescence
probe. (D) Protein expression levels of inflammatory factors IL-1
In addition to cell membrane integrity, we investigated oxidative stress and
inflammatory responses. ROS levels were measured, and the results showed a
significant increase in the UA-treated group compared to that in the control
group (Fig. 1C). Moreover, the expression of inflammatory factors such as
IL-1
To verify the pivotal role of oxidative stress in UA-mediated injury, we used the antioxidant, NACA. As shown in Supplementary Fig. 2A, UA treatment significantly reduced HK-2 cell viability, whereas NACA pretreatment (UA + NACA) markedly restored cell viability. NACA alone did not exhibit cytotoxicity. Consistent with the viability assay results, UA stimulation led to a substantial increase in intracellular ROS levels (Supplementary Fig. 2B). This oxidative burst was effectively suppressed by NACA pretreatment. These results demonstrate that UA-induced oxidative stress is the primary cause of cytotoxicity.
To further explore the mechanisms underlying UA-induced cell injury, we focused on mitochondrial dynamics, specifically the role of DRP1, a key regulator of mitochondrial fission. RT-qPCR was performed to examine the mRNA expression levels of DRP1 and FIS1. The results revealed that UA treatment significantly upregulated the mRNA expression of both DRP1 and FIS1 (Fig. 2A), suggesting that UA initiates mitochondrial fission at the transcriptional level.
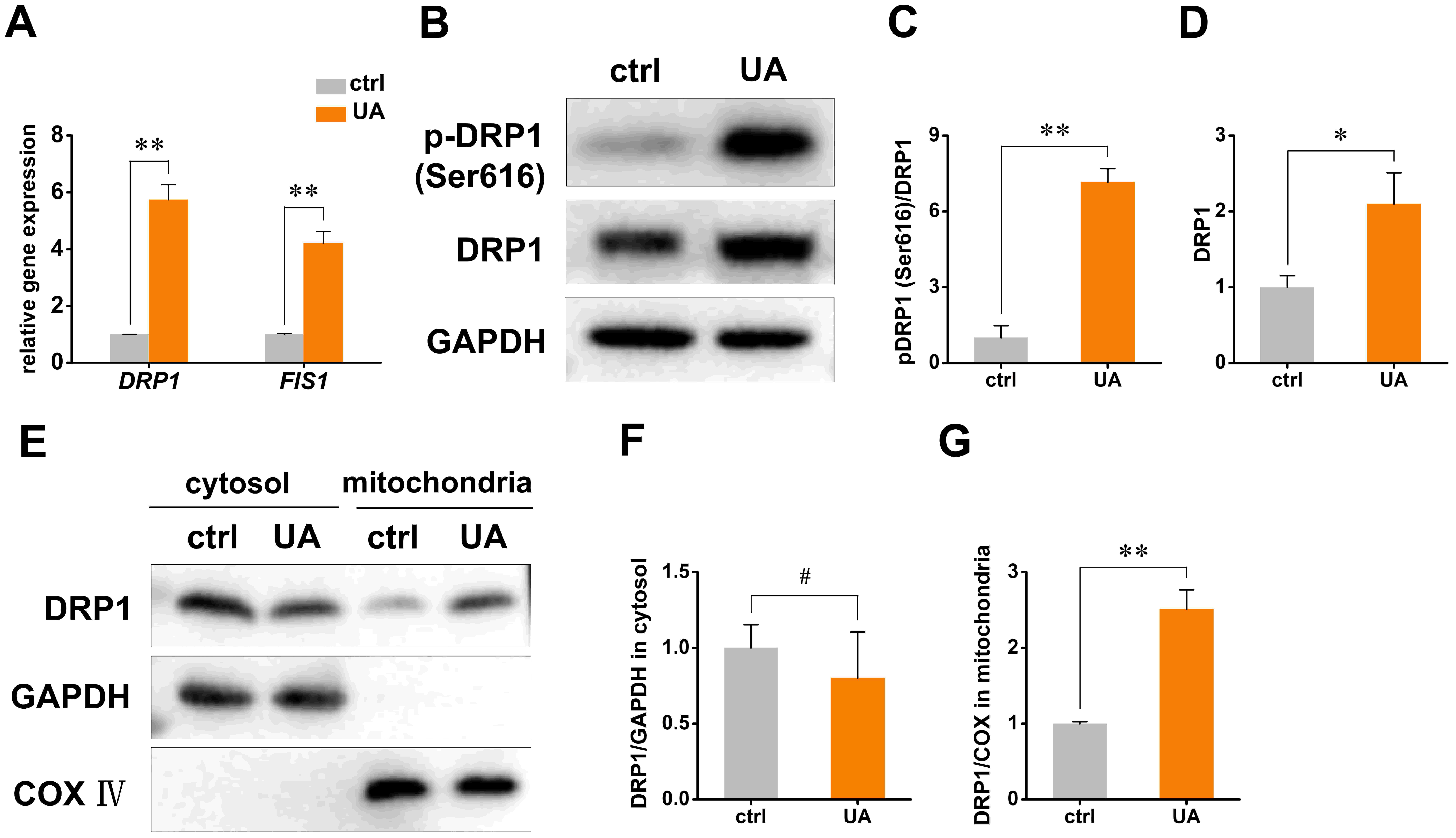 Fig. 2.
Fig. 2.
UA promotes DRP1 activation and mitochondrial translocation.
(A) RT-qPCR detection of DRP1 and FIS1 mRNA expression levels.
(B) Western blot detection of total DRP1 and phosphorylated DRP1 (p-DRP1 Ser616)
protein expression. (C,D) Quantitative analysis of DRP1 and p-DRP1 protein
expression in (B). (E) Western blot detection of DRP1 enrichment in the
mitochondrial fraction. (F,G) Quantitative analysis of DRP1 distribution in
cytosolic and mitochondrial fractions. UA, Uric acid; DRP1, Dynamin-related
protein 1; RT-qPCR, Reverse transcription quantitative polymerase chain reaction;
FIS1, Mitochondrial fission 1 protein; p-DRP1, Phosphorylated DRP1. The data are
presented as the mean
At the protein level, Western blot analysis was carried out to assess the expression of total DRP1 and its activated form, phosphorylated DRP1 at Ser616 (p-DRP1 Ser616). These results indicated that UA treatment significantly increased the expression of both total DRP1 and p-DRP1 Ser616. Notably, the increase in p-DRP1 Ser616 was more pronounced than that in total DRP1 (Fig. 2B–D). This differential increase suggests that UA not only enhances the overall expression of DRP1, but also promotes its activation, which is crucial for its function in mitochondrial fission.
Since the activation of DRP1 is known to mediate its translocation to the mitochondria, we investigated the subcellular distribution of DRP1. Cellular fractionation, a technique that separates cellular compartments based on their physical properties, was combined with Western blot analysis. This approach allowed quantification of DRP1 in the mitochondrial fraction. The outcomes indicated that UA treatment notably enhanced the enrichment of DRP1 in mitochondria (Fig. 2E–G). This finding provides direct evidence that UA treatment promotes the mitochondrial translocation of DRP1, which is likely to be of crucial importance in the mitochondrial fission event and contribute to UA-induced cell injury.
To explore the significance of the DRP1/FIS1 interaction in UA-induced cell injury, we used the specific peptide inhibitor P110. Co-IP assays were carried out, and the results clearly demonstrated that P110 treatment effectively inhibited the UA-induced protein interaction between DRP1 and FIS1 (Fig. 3A,B). This finding serves as an initial indication that P110 can disrupt relevant molecular interactions associated with cell injury.
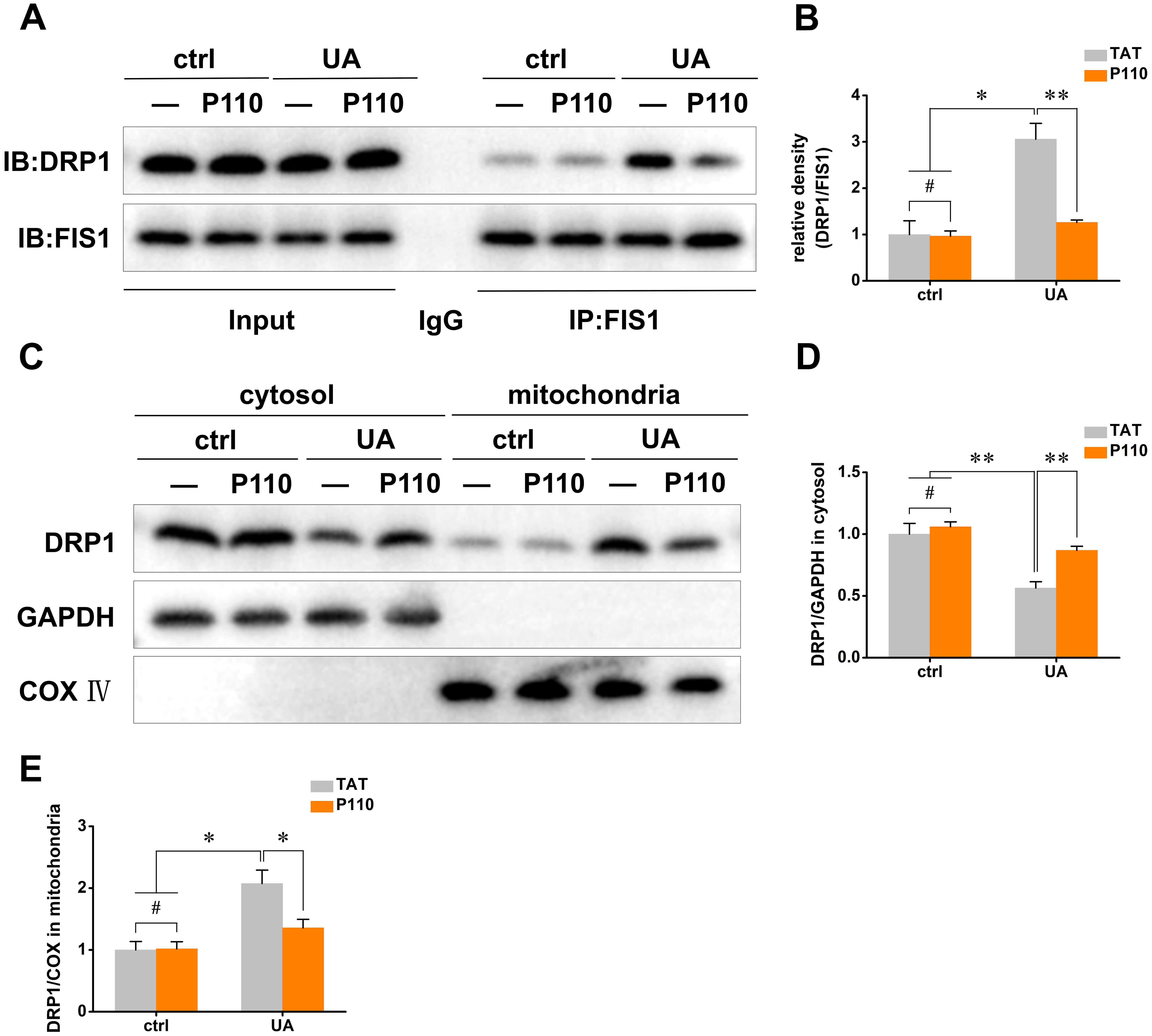 Fig. 3.
Fig. 3.
P110 inhibits DRP1/FIS1 interaction and DRP1 mitochondrial
translocation. (A) Co-IP analysis of the interaction between DRP1 and FIS1. (B)
Quantitative analysis of DRP1 and FIS1 binding levels. (C) Western blot detection
of DRP1 distribution in mitochondria. (D,E) Quantitative analysis of DRP1 protein
levels in mitochondria and cytoplasm. Input: 10% total lysate; IgG, negative
control; IP, anti-FIS1 immunoprecipitated; DRP1, Dynamin-related protein 1; FIS1,
Mitochondrial fission 1 protein; Co-IP, Co-immunoprecipitation. The data are
presented as the mean
Subsequently, we investigated the impact of P110 on the intracellular distribution of DRP1. Western blot analysis revealed that P110 did not affect the total expression of DRP1. However, it significantly reduced the distribution of DRP1 in the mitochondrial fraction (Fig. 3C–E). This reduction in mitochondrial DRP1 levels indicates that P110 effectively blocks the mitochondrial translocation of DRP1, which is likely to be an important step in alleviating UA-induced cell injury.
In terms of cellular function, the P110 treatment had a significant positive effect on cell viability. As shown in Fig. 4A, P110 significantly improved the UA-induced reduction in cell viability, suggesting a cytoprotective effect. In the context of oxidative stress, P110 effectively counteracted UA-induced excessive ROS induced by UA stimulation (Fig. 4B). Moreover, it significantly upregulated the expression of antioxidant enzymes including SOD2, HO-1, and NQO1 (Fig. 4C). These changes in antioxidant enzyme expression imply enhanced cellular antioxidant defense capacity, which is crucial for cell survival under oxidative stress conditions.
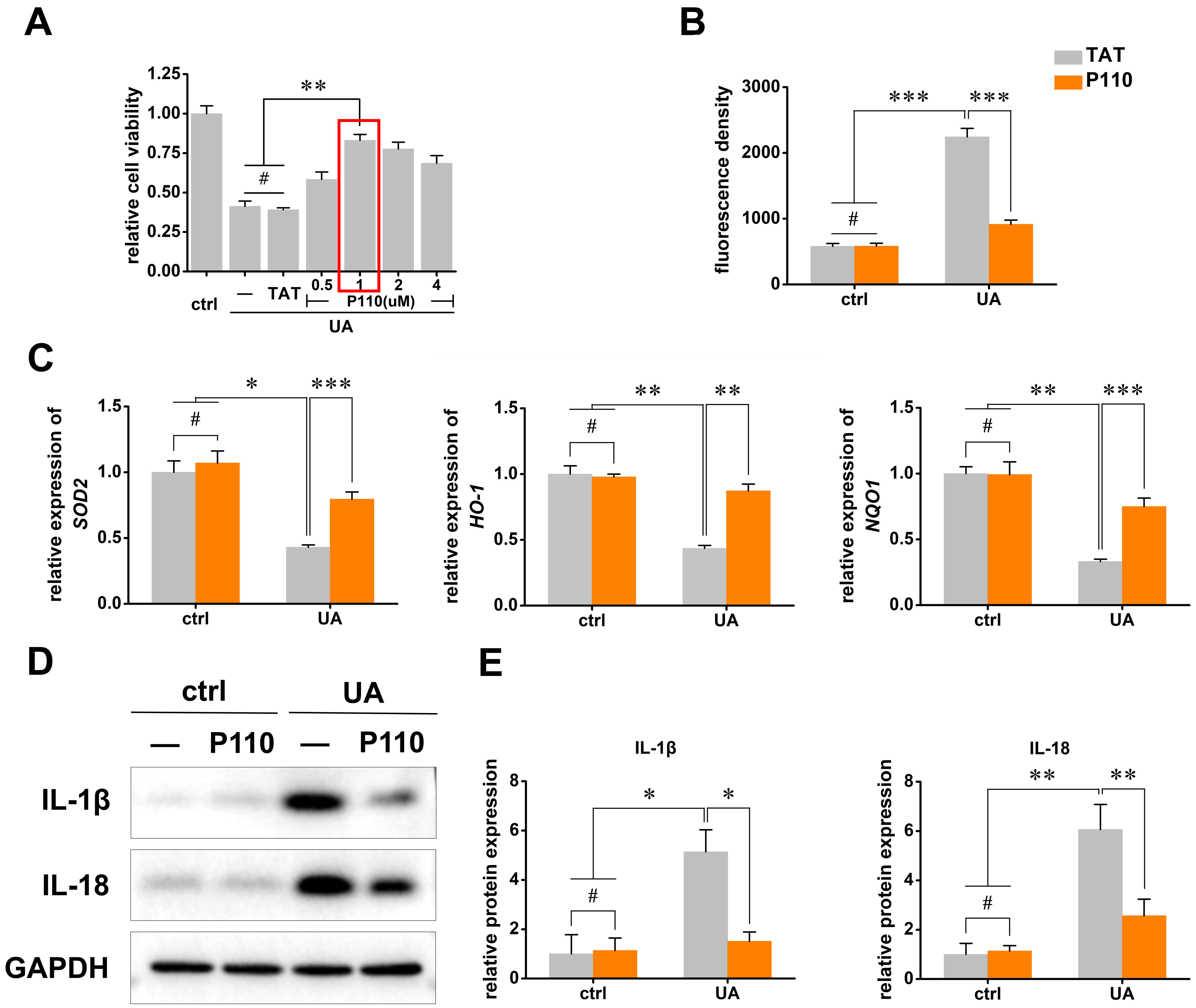 Fig. 4.
Fig. 4.
P110 alleviates UA-induced cell injury. (A) CCK-8 assay
detecting cell viability under P110 or TAT treatment in UA-induced cell injury.
The red box indicates that treatment with 1 µM of P110 significantly
improved cell viability. Additionally, 1 µM of P110 will be used in the
subsequent experiments. (B) Detection of intracellular ROS levels by DCFH-DA
fluorescence probe. (C) RT-qPCR detection of antioxidant enzymes SOD2,
HO-1, and NQO1 expression. (D) Western blot detection of
inflammatory factors IL-1
Additionally, P110 treatment had a notable effect on the inflammatory response.
It markedly suppressed the upregulation of inflammatory factors such as
IL-1
To gain an in-depth understanding of the cytoprotective mechanism of P110, we focused on its regulatory effects on mitochondrial homeostasis. RT-qPCR was performed to examine the expression of mitochondrial fusion proteins. The results showed that P110 treatment significantly reversed UA-induced downregulation of the mitochondrial fusion proteins MFN2 and OPA1 (Fig. 5A). This restoration of fusion protein expression indicates that P110 may play a role in promoting mitochondrial fusion, which is an important aspect of mitochondrial dynamics.
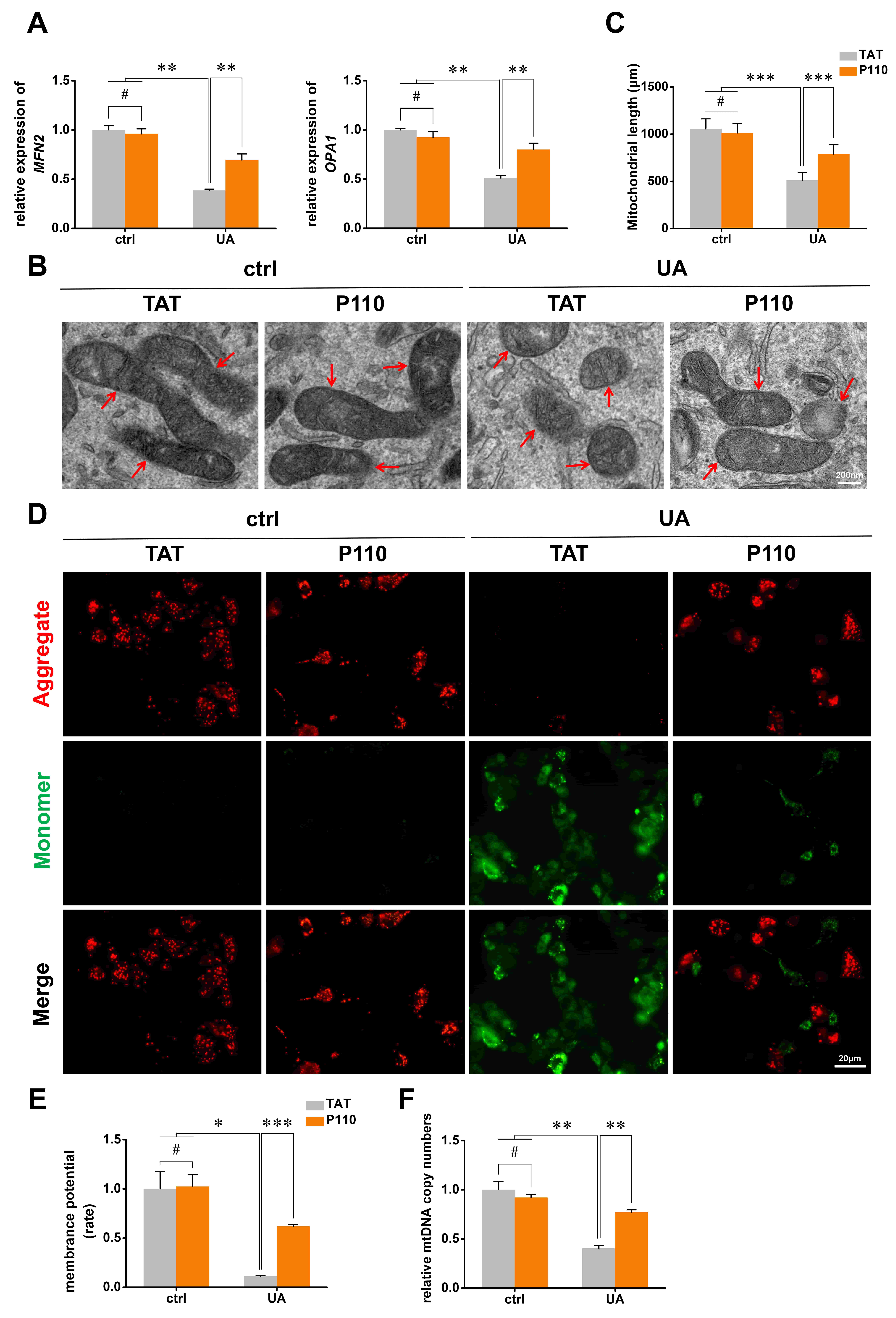 Fig. 5.
Fig. 5.
P110 improves mitochondrial fusion and restores mitochondrial
homeostasis. (A) RT-qPCR detection of the mRNA expression levels of
mitochondrial fusion proteins MFN2 and OPA1. (B) Transmission
electron microscopy images showing mitochondrial ultrastructure. Red arrows
indicate mitochondria. Scale bar: 200 nm. (C) Quantitative analysis of mitochondrial length. (D)
JC-1 staining fluorescence images showing mitochondrial membrane potential (red:
aggregates, green: monomers). Scale bar: 20 µm. (E) Quantitative
analysis of JC-1 red/green fluorescence ratio. (F) RT-qPCR detection of mtDNA
expression. P110, Mitochondrial division inhibitor 1; RT-qPCR, Reverse
transcription quantitative polymerase chain reaction; MFN2, Mitofusin 2; OPA1,
Optic atrophy 1; JC-1,
5,5′,6,6′-Tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine
iodide; mtDNA, Mitochondrial DNA. The data are presented as the mean
Transmission electron microscopy was used to directly observe morphological changes in the mitochondria. The images revealed that P110 intervention markedly alleviated UA-induced mitochondrial fragmentation and effectively increased mitochondrial length (Fig. 5B,C). These morphological changes suggest that P110 can improve the mitochondrial structure, which is closely related to its function.
Furthermore, we evaluated the effects of P110 on
Mitochondrial dysfunction is a key event in intrinsic apoptotic pathway. To determine whether P110 exerts its protective effects via this pathway, we examined mitochondria-mediated apoptosis-related indicators. Western blotting was performed to assess the expression of apoptosis-related proteins. The results showed that UA treatment significantly upregulated the expression of pro-apoptotic proteins including BAX, CYT C, cleaved Caspase-9, and cleaved Caspase-3, and concurrently downregulated the level of the anti-apoptotic protein BCL-2. However, P110 treatment reversed these abnormal expression patterns (Fig. 6A,B).
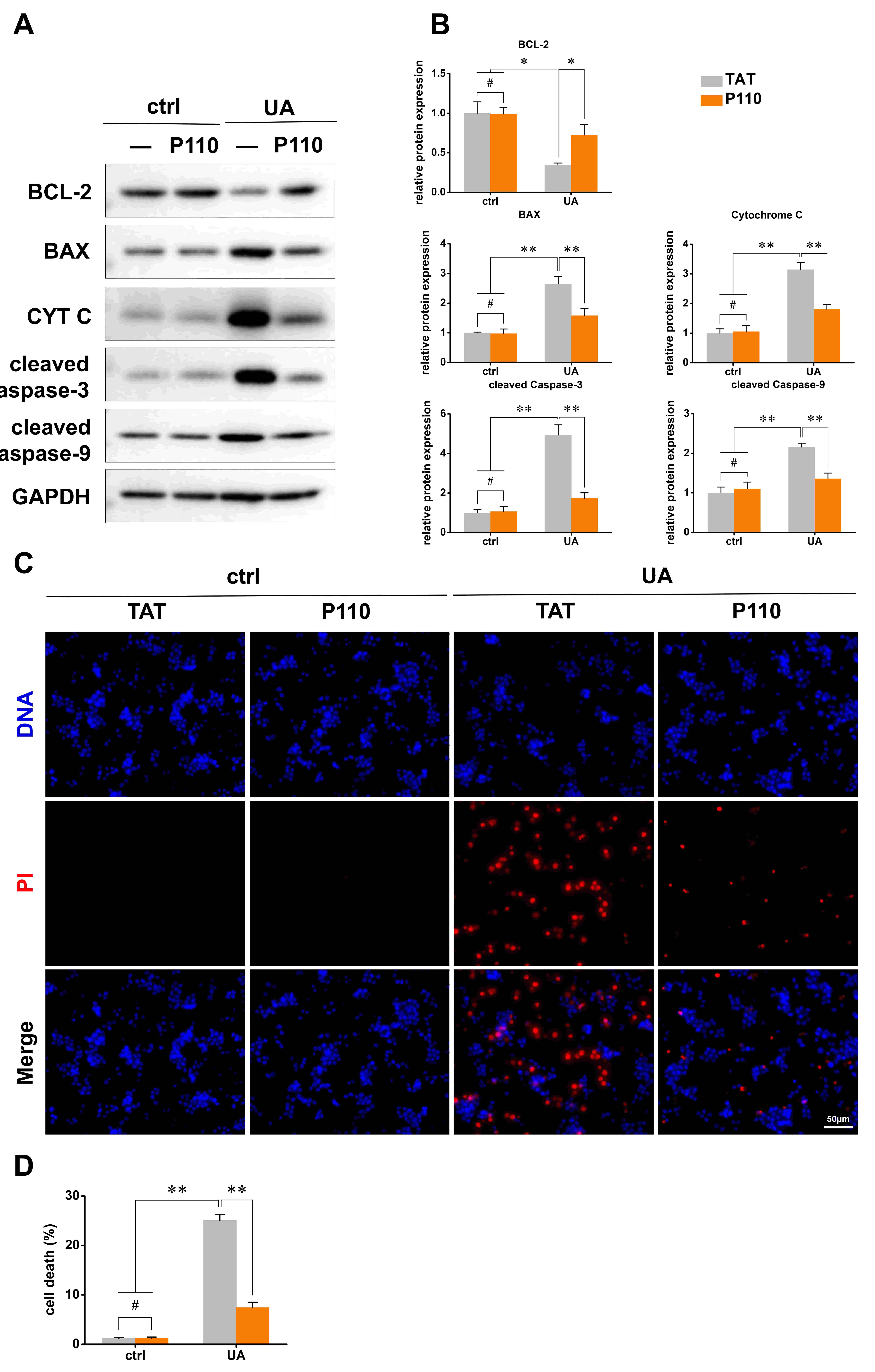 Fig. 6.
Fig. 6.
P110 suppresses cell apoptosis through the mitochondrial
pathway. (A) Western blot detection of apoptosis-related proteins BAX, BCL-2,
CYT C, cleaved Caspase-9, and cleaved Caspase-3 expression. (B) Quantitative
analysis of protein expression in (A). (C) Cell PI fluorescence staining images
showing apoptosis. Scale bar: 50 µm. (D) Quantitative statistics of
apoptosis rate. BAX, BCL2-associated X protein; BCL-2, B-cell lymphoma 2; CYT C,
Cytochrome C; PI, Propidium iodide. The data are presented as the mean
To further confirm the anti-apoptotic effect of P110, apoptosis was detected using PI staining. The results showed that P110 treatment significantly reduced UA-induced apoptosis (Fig. 6C,D). These findings suggest that P110 attenuates mitochondria-mediated apoptosis by inhibiting DRP1/FIS1 interaction, thereby protecting against UA-induced renal tubular epithelial cell injury.
In summary, this study comprehensively demonstrated that P110 can specifically inhibit the DRP1/FIS1 interaction, which in turn leads to a series of beneficial effects, including the alleviation of oxidative stress, restoration of mitochondrial homeostasis, and inhibition of apoptosis, ultimately protecting cells from UA-induced injury.
This study comprehensively explored the pivotal role and molecular mechanisms of DRP1-mediated imbalance in mitochondrial dynamics during UA-induced injury of renal tubular epithelial cells. The key findings were as follows: (1) UA treatment notably enhanced DRP1 expression, activation, and mitochondrial translocation; (2) the interaction between DRP1 and FIS1 mediated UA-induced excessive mitochondrial fission, ROS production, and the inflammatory response; and (3) blocking this interaction with the specific inhibitor P110 effectively preserved mitochondrial homeostasis and suppressed cell apoptosis, thus exerting renal protective effects.
Numerous studies have established a strong association between mitochondrial dysfunction and development and progression of hyperuricemic nephropathy [7, 25, 26]. This study further linked this pathological process to DRP1-mediated excessive mitochondrial fission. After UA treatment, we observed that DRP1 was phosphorylated and activated at Ser616, translocated to the mitochondria, and bound to the receptor FIS1, resulting in mitochondrial fragmentation (Figs. 2,5). This finding is consistent with reports on neurodegenerative diseases and cancer, suggesting that abnormal DRP1 activation is a common pathway in diverse cellular stress responses [20, 23].
Notably, a recent study by Wu demonstrated that the traditional Chinese medicine compound shizhifang ameliorated hyperuricemia-induced renal tubular injury and maintained mitochondrial homeostasis by inhibiting DRP1-mediated mitochondrial fission [24]. This result is consistent with the conclusions of our study and highlights the significance of DRP1 as a crucial therapeutic target for hyperuricemic nephropathy.
Excessive mitochondrial fission not only impairs energy metabolism, but also promotes substantial ROS production, disrupts the oxidative/antioxidant balance, forms a vicious cycle, and exacerbates cellular damage. In this study, we confirmed that P110 treatment significantly upregulated the antioxidant enzymes expression, for instance, SOD2, HO-1, and NQO1 (Fig. 3C). This suggests that by maintaining mitochondrial structural integrity, P110 indirectly enhances cellular antioxidant defense capacity, thereby interrupting the positive feedback loop of UA-induced oxidative stress.
More importantly, this study revealed the complete signaling axis from DRP1 activation to apoptosis. Mitochondria serve not only as energy-producing organelles, but also as integration centers for apoptotic signals. Our results showed that by inhibiting the DRP1/FIS1 interaction, P110 stabilized the mitochondrial membrane potential, decreased the release of CYT C, and consequently suppressed the downstream activation of Caspase-9 and Caspase-3, and modulated the BCL-2/BAX balance (Fig. 6). This clearly demonstrates that the protective effect of P110 was achieved by interfering with the mitochondrial apoptotic signal transduction pathway.
Wu et al. [24] also reported improved expression of apoptosis-related proteins after shizhifang treatment in an animal model, further supporting the generalizability of targeting the DRP1-mitochondria-apoptosis axis to alleviate hyperuricemic renal injury. Additionally, recent research has reported a direct molecular interaction between DRP1 and the pro-apoptotic protein BAX [27]. Activated DRP1 can directly bind to BAX and induce its conformational activation and oligomerization, leading to mitochondrial outer membrane permeabilization and CYT C release, ultimately triggering apoptosis. Therefore, the protective effect of P110 may not be limited to inhibition of mitochondrial fragmentation. By preventing DRP1 recruitment to the mitochondria, P110 likely blocks the direct activation of BAX by DRP1, a key apoptosis-inducing pathway, thereby suppressing the initiation of mitochondrial apoptosis at its origin. This provided a novel mechanistic explanation for the potent cytoprotective effects of P110.
The primary innovation of this study is the first-time validation of the therapeutic potential of P110 in a model of hyperuricemic nephropathy. As a peptide inhibitor targeting the DRP1/FIS1 interaction, P110 has advantages, such as high specificity, efficient cell membrane penetration, and effective in vivo delivery. The results of this study are in line with those of previous reports, indicating that the downregulation of DRP1 expression protects against hyperuricemic renal injury. Moreover, this study further refines the intervention strategy at the “protein interaction” level, providing a more precise target for the development of novel therapeutic drugs.
Our study found that UA concurrently upregulated DRP1 and FIS1, suggesting a coordinated cellular response to fission stress, as evidenced by their reported functional coupling [28]. We focused on DRP1 because it is a central fission effector and its phosphorylation at Ser616 is a key regulatory event that directly governs mitochondrial translocation. This focus aligned with our aim to evaluate P110, a specific inhibitor of the DRP1/FIS1 interaction. Our results confirmed that disrupting this interaction with P110 was sufficient to mitigate fission and apoptosis. Although genetic knockdown of DRP1 or FIS1 offers deeper mechanistic insights, as supported by studies in which DRP1 knockdown alleviated UA-induced injury [24, 29] and FIS1 modulation was protective in other models [29], our work demonstrates that pharmacologically targeting the DRP1/FIS1 complex with P110 is a viable and effective therapeutic strategy for hyperuricemic nephropathy.
However, this study had certain limitations. First, the study was restricted to HK-2 cells. The therapeutic potential and pharmacokinetic profile of P110 requires further investigation using in vivo preclinical models and future clinical studies. Second, DRP1 activation involves various upstream regulatory kinases (e.g., CDK1 and ERK), and the specific signaling pathway through which UA triggers DRP1 phosphorylation requires further investigation.
In this study, we successfully established a renal tubular epithelial cell injury in uric acid nephropathy by treating HK-2 cells with UA. Exposure to UA led to the upregulation and phosphorylation (at Ser616) of DRP1, promoting its mitochondrial translocation, which subsequently induced excessive mitochondrial fission, ROS burst, inflammatory response, and cell apoptosis. In contrast, treatment with the specific peptide inhibitor P110 effectively inhibited the interaction between DRP1 and FIS1, blocking DRP1 mitochondrial translocation. This action alleviates mitochondrial fragmentation, restores mitochondrial membrane potential, and enhances antioxidant capacity. P110 significantly improves UA-induced cell injury by inhibiting mitochondria-mediated apoptosis. These results indicate that DRP1 overactivation is a key molecular event in the process of UA-induced injury of renal tubular epithelial cells and that targeted inhibition of the DRP1/FIS1 interaction provides a potential strategy for the treatment of hyperuricemic nephropathy.
UA, uric acid; Co-IP, co-immunoprecipitation; ROS, reactive oxygen species; DRP1, dynamin-related protein 1; CYT C, cytochrome C; BAX, BCL-2-associated X protein; TAT, transactivator of transcription; NACA, N-acetylcysteine; HK-2, human renal proximal tubular epithelial cells; MCE, MedChemExpress; CCK-8, Cell Counting Kit-8; DCFH-DA, 2′,7′-dichlorodihydrofluorescein diacetate; PI, propidium iodide; cDNA, complementary DNA; qPCR, quantitative polymerase chain reaction; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; TEM, transmission electron microscopy; ELISA, enzyme-linked immunosorbent assay.
All data and materials are available from the corresponding author upon reasonable request.
YS, GH, and CL designed the experiments. YL and CL performed the experiments. YS, GH, and YL analyzed the data. FP, JL, and ZM contributed to data interpretation and manuscript revising. YS and ZM wrote the paper. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
We thank Mrs. Lei Zhang from Longgang District People’s Hospital of Shenzhen for her administrative assistance.
This work was supported by the Longgang District Science and Technology Innovation Special Fund of Shenzhen (LGWJ2022-31). This work was supported by the Longgang Medical Discipline Construction Fund.
The authors declare no conflicts of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/FBL46700.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.