





 , Svetlana Vechkapova 2,3, Dmitriy Lanshakov 1,3,4,*
, Svetlana Vechkapova 2,3, Dmitriy Lanshakov 1,3,4,*1 Postgenomics Neurobiology Laboratory, The Institute of Cytology and Genetics, 630090 Novosibirsk, Russia
2 Federal Research Center for Information and Computational Technologies, 630090 Novosibirsk, Russia
3 Normal Physiology Department, Novosibirsk State Medical University, 630091 Novosibirsk, Russia
4 Natural Science Department, Novosibirsk State University, 630090 Novosibirsk, Russia
Abstract
Pericytes (PCs) are multifunctional mural cells embedded in the basement membrane of microvessels and play essential roles in the development and maintenance of the central nervous system. This review provides a comprehensive synthesis of the current knowledge on PC biology, tracing their trajectory from embryonic origins to specialized functions in the adult brain. During early brain development, PCs are recruited via platelet-derived growth factor B (PDGF-BB)/platelet-derived growth factor receptor beta (PDGFRβ) signaling and contribute to the formation of the blood–brain barrier (BBB), cortical architecture, and vascular stability. Their developmental plasticity is shaped by multiple embryonic origins and dynamic interactions with endothelial and neural precursor cells. In the adult central nervous system, PCs are central to maintaining BBB integrity, regulating cerebral blood flow, and modulating neurovascular coupling. They also participate in immune responses, metabolic waste clearance, and neuroprotection through the secretion of trophic factors and cytokines. Of particular interest is their emerging role in the expression of lipocalin-type prostaglandin D synthase (L-PGDS), which synthesizes prostaglandin D2—a molecule involved in sleep regulation, inflammation, and neurodegeneration. L-PGDS may also act as an amyloid β chaperone, implicating PCs in the pathology of Alzheimer’s disease and other neurodegenerative disorders. The regulatory mechanisms of L-PGDS expression involve nuclear factor kappa B and Notch–Hes signaling, as well as potential modulation via brain-derived neurotrophic factor/tropomyosin receptor kinase B/protein kinase C pathway. By integrating developmental, molecular, and pathophysiological perspectives, this review positions PCs as key cellular regulators of brain function and highlights their potential as therapeutic targets in cerebrovascular and neurodegenerative diseases.
Keywords
- pericytes
- growth & development
- blood–brain barrier
- prostaglandins
The human brain is an extraordinarily complex organ, composed of approximately 3000 distinct cell types [1]. This cellular diversity emerges through intricate developmental processes that generate sophisticated neural circuits responsible for the full spectrum of human behavior, including high-order executive functions. Historically, non-neuronal cells such as astrocytes, microglia, endothelial cells (ECs), and pericytes (PCs) were considered merely supportive elements. However, accumulating evidence increasingly highlights their critical roles in brain function and health. In particular, PCs—mural cells embedded in the vascular basement membrane—have gained attention for their diverse and dynamic contributions to cerebrovascular regulation and neurovascular unit (NVU) integrity. The purpose of this review is to synthesize recent findings on the developmental and functional roles of PCs, emphasizing their emerging importance in maintaining brain homeostasis and supporting neural development.
PCs are specialized mural cells that play indispensable roles in microvascular regulation across all vascularized tissues. Their strategic location abluminal to ECs positions them as key mediators of vascular stability, permeability, and remodeling. This function’s critical role is most apparent in the central nervous system (CNS), given that PC deficiencies are associated with a range of neuropathologies.
PCs are heterogeneous, elongated mural cells embedded within the vascular basement membrane. These multibranched cells exhibit three characteristic morphological features: (1) finger-like cytoplasmic processes containing contractile myofilaments (actin, myosin, tropomyosin); (2) cell bodies typically positioned at capillary branch points; and (3) primary processes extending along vascular branches with secondary processes forming circumferential connections around endothelial tubes. Their processes may either encircle microvessels or extend longitudinally along precapillary arterioles and postcapillary venules [2].
Present in all vascularized tissues, PCs show remarkable density variation across organs. The CNS contains the highest PC coverage, with a 1:1 endothelial-to-PC ratio on approximately 70–80% of microvessels. Other tissues demonstrate progressively lower ratios: 2.5:1 in kidneys and heart, 8:1 in lungs, 10:1 in liver and skin, and 100:1 in skeletal muscle [2]. Localized PC loss induces microvascular dysfunction. For example, proliferative retinopathy is consistently associated with a reduced density of PCs in microvessels of the retina [2].
PC morphology varies according to vascular bed specialization. CNS PCs typically appear as flattened stellate cells, while renal PCs often assume a more rounded conformation. This structural adaptation reflects their tissue-specific functions in vascular regulation and maintenance [2].
The accurate identification of PCs has represented a significant challenge in vascular biology due to their morphological overlap with other perivascular cells. For decades, the lack of specific markers hindered precise characterization until advances in fluorescence microscopy, confocal imaging, and genetic labeling techniques revealed over two dozen molecular signatures associated with these cells [3, 4].
Platelet-derived growth factor receptor beta (PDGFR
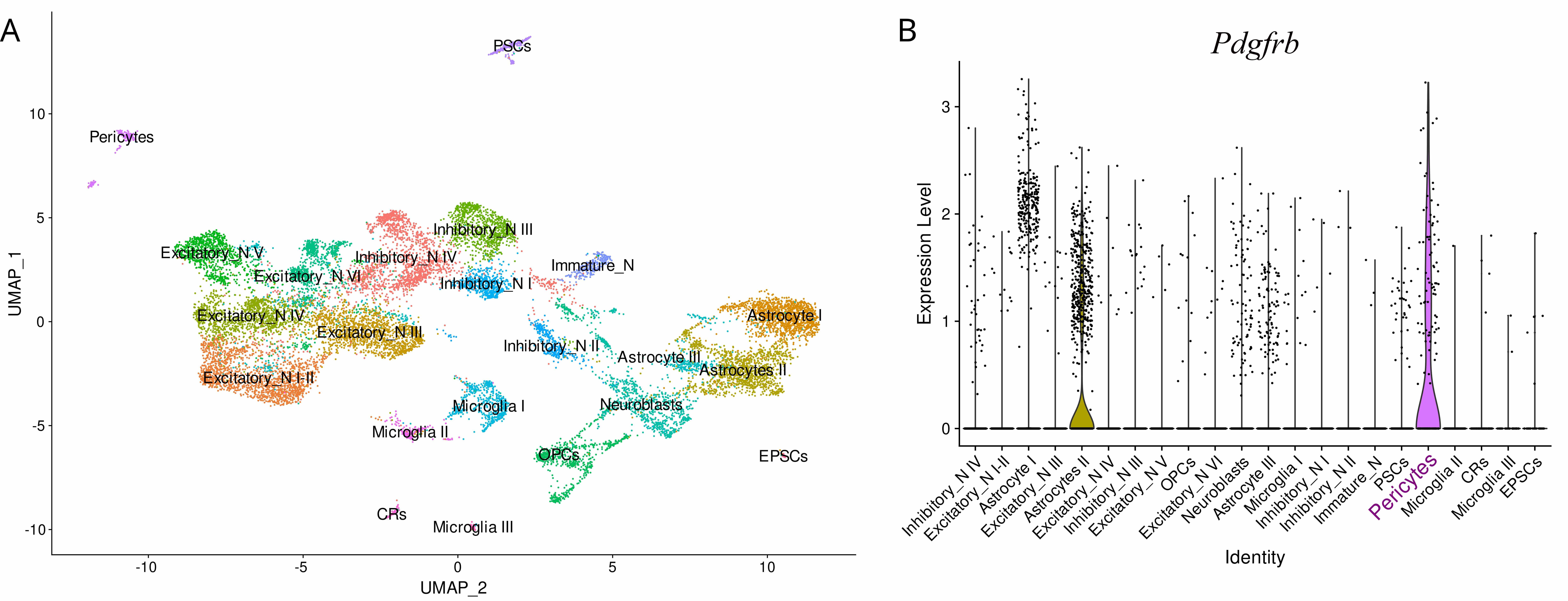 Fig. 1.
Fig. 1.
Schematics showing diversity of cell types in the rat neonatal cortex and expression of molecular marker genes in them including pericytes. (A) Cell clustering in the rat neonatal cortex based on the single-nucleus RNA sequencing data by Chen et al. [5]. The cells are represented by dots. The close proximity of the dots indicates a similar state of the transcriptome in the cells. The names of the clusters are written above the dots. CRs, Cajal–Retzius cells; EPSC, ependymal stem cells; PSCs, perivascular stromal cells; OPCs, oligodendrocyte progenitor cells. (B) Violin plots showing stable expression of platelet-derived growth factor receptor beta (Pdgfrb) in pericytes and astrocyte-2. Dots represent cells located on the y-axis, according to expression of the gene of interest in that cell. Fig. 1 was built based on Chen et al. [5] data NCBI GEO (accession number: GSE185538) using Seurat package ver. 5.3.1. (https://cran.rproject.org/web/packages/Seurat/index.html) in R.
Melanoma cell adhesion molecule cluster of differentiation 146 (CD146) demonstrates particularly interesting dynamics during vascular maturation. In the mouse brain, Cd146 is initially expressed exclusively on immature ECs of nascent capillaries. As vessels mature and acquire PC coverage, Cd146 expression undergoes a complete switch, becoming restricted to PCs while disappearing from ECs [9]. This transition makes Cd146 particularly useful for studying PC–endothelial interactions during vascular development.
A recent single-cell study identified more cell diversity in the NVU recapitulating a gradual endothelial and punctuated mural cell continuum at the transcriptome level [10, 11]. PCs are clustered into two subtypes based on function rather than morphology: one subtype is rich in proteins involved in small molecule transmembrane transport (solute carrier family 6 member 1 [Slc6a1], Slc1a3, Slc6a12, Slc38a11), while the other is characterized by proteins involved in organizing the extracellular matrix (ECM) (collagen type IV alpha 1 chain [Col4a1], Col4a2, laminin subunit alpha 2 [Lama2], glypican 5). Beyond these core markers, PCs express other functionally significant proteins including regulator of G protein signaling 5 (activated during vessel remodeling and tumor development) [12], cluster of differentiation 13 (brain-enriched zinc-dependent metalloprotease), and contractile elements (alpha smooth muscle actin and desmin). The Hedgehog pathway effector glioma-associated oncogene homolog 1 [13] and transcription factor T-box transcription factor 18 [14] further illustrate the molecular diversity of PC subpopulations. Other molecular markers enriched in brain PCs, including phospholipase A1 member A and cytochrome C oxidase subunit 4I2, help distinguish PC subpopulations from vascular smooth muscle cells [15]. While expressed in some neurons and glia, these markers significantly expand our toolkit for PC identification when combined with conventional approaches.
Breakthrough work with the fluorescent Nissl dye, NeuroTrace 500/525, demonstrated specific labeling of live brain PCs without inadvertently staining other vascular components after topical administration to the brain’s surface. This advancement has enabled unprecedented intravital imaging of PC dynamics and confirmed their status as a distinct cellular population with unique molecular and functional properties [4, 16].
Nevertheless, current understanding emphasizes that no single marker can
universally identify all PCs across tissues and developmental stages. The
reliability of identification depends critically on contextual factors including
the cell’s differentiation status, local microenvironmental cues, and
pathological conditions [17]. Consequently, contemporary best practices require
combinatorial approaches using at least two complementary markers—typically
PDGFR
Formation of the blood–brain barrier (BBB) is a complex process in which PCs play a central role by regulating the maturation and stability of cerebral vessels. To understand how these cells contribute to barrier function, it is essential to trace their development from the earliest stages.
PC development in mice begins on about embryonic day 9.5 (E9.5) [18], coinciding
with two critical events: the onset of neurogenesis (E9–E10) [19] and the
initiation of brain vascularization [18, 20]. During this period, the foundation
of the future vascular network is established, and the first PC precursors
emerge. Interestingly, these cells originate from multiple embryonic sources,
contributing to their heterogeneity. Most PCs develop from neural crest and
mesenchymal cells [21, 22], while a subset originates from yolk sac-derived
macrophages [23]. By E10, these macrophages (CD31+F4/80+) migrate into the
developing midbrain and subsequently express classical PC markers such as
PDGFR
Between E9.5 and E11.5, neural crest-derived cells actively migrate into the
telencephalon, accompanied by the growth of endothelial tubes, which is driven by
vascular endothelial growth factor (VEGF) [18, 20]. Notably, many already express
PC markers (PDGFR
One of the central mechanisms ensuring PC integration into the vascular network
is the PDGF-BB/PDGFR
Beyond PDGF-BB/PDGFR
The BBB becomes functional by E15.5 in mice (E17 in rats) [31], yet its development continues postnatally [32]. This indicates that even after the basic barrier structure is established, PCs remain involved in fine-tuning its properties, such as selective permeability, resistance to oxidative stress, and microcirculatory stability.
In summary, PC development is a multistage process governed by the coordinated activity of several signaling pathways. Understanding these mechanisms not only advances fundamental knowledge but also opens avenues for researching neurodegenerative diseases linked to BBB dysfunction. Future studies could explore how disruptions in PC differentiation influence the risk of cerebrovascular disorders postnatally, bridging developmental biology with clinical neuroscience.
PCs, however, are not confined to the microvasculature alone. Their presence is equally critical in the leptomeninges — the delicate inner membranes enveloping the CNS. The leptomeninges are the two innermost layers of the meninges — the protective coverings that surround the brain and spinal cord. They consist of pia and arachnoid. Pia is a thin layer that is directly attached to the surface of the brain and spinal cord, following all of their contours and grooves. Arachnoid is a web-like, transparent membrane that lies just above the pia mater and beneath the outer dura mater. These two layers together form the leptomeninges, which differ from the outermost layer, the dura mater (a tough, fibrous membrane). The leptomeningeal layer consists of mesenchymal stem cells, PCs, and ECs (Fig. 2). ECs in the leptomeninges are essential for forming the cortical vascular plexus that supports neuronal growth [33].
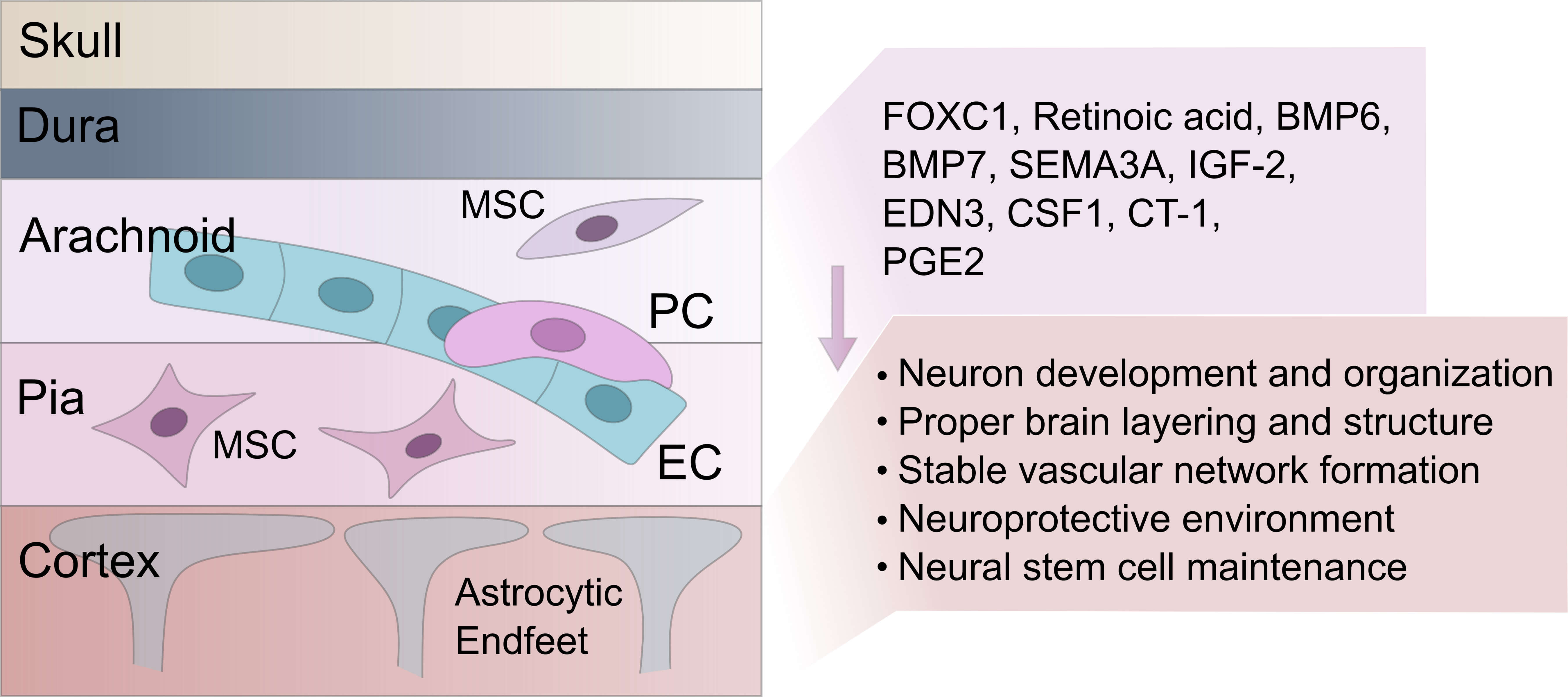 Fig. 2.
Fig. 2.
Schematic illustration of the skull, meningeal layers, and the adjacent cerebral cortex. The diagram shows the anatomical organization from the outer skull through the dura, arachnoid, and pia mater, down to the underlying cortical tissue. Within the cortex, astrocytic end-feet are shown extending toward the pial surface. Within the arachnoid, key cellular components — endothelial cells (ECs), pericytes (PCs), and mesenchymal stem-like cells (MSCs) — are depicted. The leptomeninges secrete molecular cues such as forkhead box C1 (FOXC1), retinoic acid, insulin-like growth factor 2 (IGF-2), bone morphogenetic protein 6 (BMP6), BMP7, semaphorin 3A (SEMA3A), endothelin 3 (EDN3), colony stimulating factor 1 (CSF1), cardiotrophin-1 (CT-1), and prostaglandin E2 (PGE2), which influence cortical development and survival. Fig. 2 was drawn in the Inkscape software ver. 1.2.1 (https://inkscape.org).
During brain development, the leptomeninges actively interact with neurons and
neural stem cells, playing crucial roles beyond structural support. They harbor
stem/progenitor cells that can differentiate into neurons. These cells express
markers such as nestin and can form neurospheres in vitro, showing
similar properties to neural stem cells from traditional brain niches [34].
Single-cell transcriptome profiling of fibroblast-like embryonic brain meningeal
cells in Col1a1-gfp mice revealed molecular markers for dural (cellular
retinoic acid-binding protein 2 [Crapb2], matrix Gla protein),
arachnoid (Crabp2, aldehyde dehydrogenase 1 family, member A2,
S100a6) and pial cells (nerve growth factor receptor,
Lama2, S100a6) [35]. Leptomeningeal cells produce biologically active
proteins including retinoic acid and insulin-like growth factor 2 (IGF2), which
support neuronal survival and differentiation during development [36, 37]. Recent
transcriptomic studies have revealed that leptomeningeal cells at the brain
surface produce a rich array of growth factors (Bmp6, Bmp7,
Sema3a, Igf2, endothelin 3, colony stimulating factor 1 (CSF1),
cardiotrophin 1) that communicate with neurons, astrocytes, and immune cells in
cortical layer 1, suggesting an ongoing regulatory dialogue [38]. Leptomeningeal
secretions such as prostaglandin E2 (PGE2) promote the expression of
TGF-
Removing the leptomeninges during embryonic stages disrupts proper cortical layering, indicating their essential role in guiding neuron migration and development [37]. Research has shown that leptomeninges and the transcription factor forkhead box C1 (Foxc1) play critical roles in brain development, particularly by influencing cortical development and vascularization. Foxc1 is a crucial regulator of somitic, cardiovascular, calvarial, renal, and ocular development processes [40]. It is expressed in the embryonic mesenchyme that gives rise to the leptomeninges. It is also highly expressed in PCs, and conditional deletion of Foxc1 in these cells leads to vascular overgrowth and microhemorrhages, showing their functional importance in leptomeningeal vasculature [41]. Mutations in Foxc1 lead to defective meningeal differentiation, which in turn causes severe cortical malformations such as dysplasia, heterotopias, and abnormal cortical layering [40]. Foxc1 positively regulates the expression of stromal cell-derived factor 1 in the mesenchyme, which is essential for the survival and migration of cerebellar radial glia and Purkinje cells. Loss of Foxc1 causes rapid glial degeneration and neuronal misplacement [42]. Foxc1 expressed in leptomeninges and PCs regulates brain vascular development. Its loss leads to vascular instability, abnormal PC proliferation, and focal breakdown of the BBB [41].
Thus, appropriate development of the cortical and cerebellar laminae relies on proper PC signaling during brain development. Dysfunction in the development of neuronal precursor cells or in neuronal communication can result in brain abnormalities, impacting the formation of neuronal circuits and the regulation of gene expression. This, in turn, contributes to future cognitive deficits and behavioral abnormalities.
In adulthood, PCs primarily support the integrity of the BBB. As essential components of the NVU, they contribute not only to brain development but also to its continued function throughout life. PCs regulate the expression of tight junction (TJ) proteins, help stabilize microvasculature, and control vessel diameter [43]. These actions collectively sustain BBB function and enable the precise regulation of cerebral blood flow, especially during periods of increased neuronal activity [44, 45, 46].
Traditionally, PCs were considered key mediators of rapid capillary constriction and relaxation during neurovascular coupling. However, recent findings indicate that their effects on capillary diameter and blood flow occur with significantly slower kinetics than previously assumed [46]. Rather than acting through immediate contractile responses, PCs appear to influence cerebral perfusion via long-term mechanisms such as vascular remodeling and the release of paracrine factors. This emerging perspective refines earlier models of PC involvement in NV regulation and highlights their broader modulatory role within the NVU.
Beyond vascular regulation, PCs respond dynamically to environmental stressors such as hypoxia, inflammation, infection, and elevated blood glucose levels [47]. They also contribute to neuroinflammation and facilitate the clearance of metabolic waste. Impaired PC function or PC loss triggers a cascade of dysfunctions, including increased BBB permeability [18, 48], disrupted NV coupling [49, 50], accumulation of neurotoxins, and ultimately neuronal damage or death [51, 52]. PC dysfunction has been implicated in a range of CNS disorders including Alzheimer’s disease, Parkinson’s disease, dementia, stroke, diabetic retinopathy, glaucoma, and intracranial vascular malformations [53, 54].
PCs, located within the basal lamina between ECs and astrocytic end-feet, exert
broad regulatory functions within the NVU, primarily via paracrine signaling.
Their secretome includes a range of inflammatory molecules, growth factors, and
ECM components such as neurotrophins, IL-9, IL-10, IL-13, tumor necrosis factor
alpha, granulocyte colony stimulating factor 1 (CSF), interferon gamma, ANG1, and
intercellular adhesion molecule 1 [55]. These molecules influence EC development,
survival, and function, including modulation of endothelial barrier permeability
[56, 57]. The interaction between PCs and ECs is reciprocal and tightly regulated
by key signaling molecules including PDGF-BB, TGF-
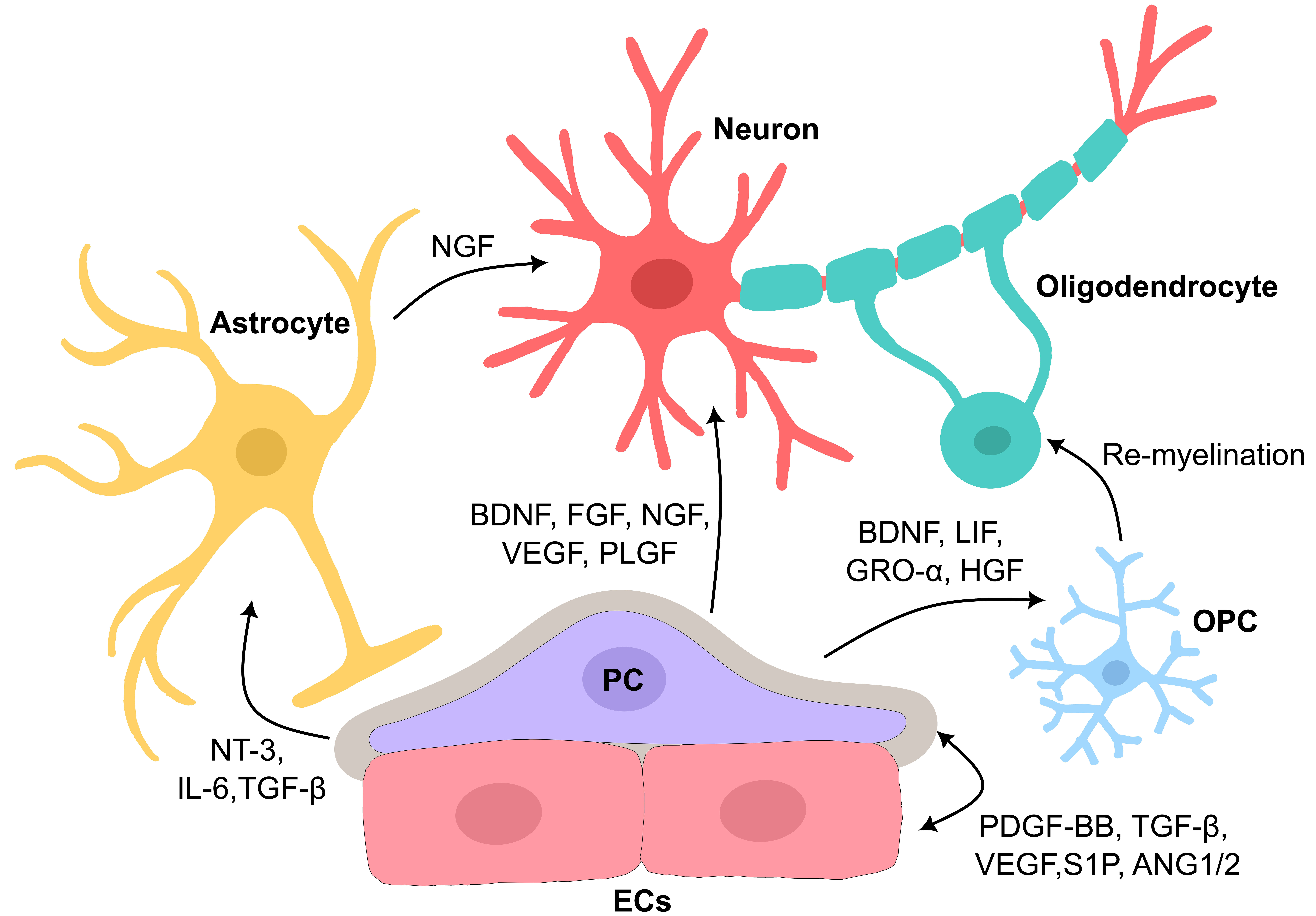 Fig. 3.
Fig. 3.
Paracrine interactions between pericytes and other cellular
components of the neurovascular unit. Pericytes (PCs), located between
endothelial cells (ECs) and astrocytic end-feet, secrete a wide range of
signaling molecules that regulate neurovascular function. These include growth
factors e.g., neurotrophin 3 (NT-3), transforming growth factor beta
(TGF-
During pathological states such as stroke, these mechanisms become especially
vital. The restoration of blood flow in the infarcted area is crucial for
neuronal survival [61], and this process depends on PC function. In
Pdgfrb+/- mice lacking PCs, reperfusion and vascular repair are
significantly impaired [62], highlighting the importance of PC survival in
peri-infarct zones. Following stroke, ECs upregulate PDGF-BB expression, which in
turn, increases PDGFR
PC interactions within the NVU extend beyond ECs. In the healthy brain, PCs
influence astrocyte polarization by shaping their end-feet, thus supporting the
coordinated function of both astrocytes and the endothelium [9, 66]. After stroke,
PDGFR
For example, NT-3 secreted by PCs in response to hypoxia acts on astrocytes via the tropomyosin receptor kinase C (TrkC)–ERK1/2 signaling pathway, which triggers astrocytic secretion of nerve growth factor (NGF) [67]. NGF, in turn, binds to TrkA receptors on neurons, promoting their survival by protecting them from apoptosis. Notably, TrkA expression is upregulated on neurons during hypoxia, even in the absence of endogenous NGF production [68] (Fig. 3).
In addition to supporting neurons via astrocytes, PCs contribute to
remyelination in peri-infarct areas. They facilitate the differentiation of
oligodendrocyte progenitor cells (OPCs) by releasing trophic factors such as
brain-derived neurotrophic factor (BDNF), leukemia inhibitory factor (LIF),
growth-regulated oncogene alpha (GRO-
PCs support neuronal survival and functional integrity not only by maintaining the BBB but also through the secretion of neuroprotective and pro-regenerative molecules. Among the diverse set of factors produced by PCs are BDNF, NGF, VEGF, fibroblast growth factor (FGF), and placental growth factor (PLGF) (Fig. 3) [71]. BDNF and NGF promote neuronal survival, growth, and differentiation [72, 73]. FGF has neuroprotective and restorative effects on rodent models of stroke and Parkinson’s disease [74, 75]. PLGF, an angiogenic factor from the VEGF family, enhances the survival of primary cortical neurons under conditions of oxygen–glucose deprivation [76]. Following stroke, PLGF expression increases in blood vessels, neurons, and astrocytes, contributing to neuroprotection during cerebral ischemia [77, 78]. Notably, these neurotrophic factors are primarily secreted by PCs via exosomes rather than through classical secretion pathways [71]. However, the direct impact of PC-derived growth factors on neurons remains to be fully elucidated.
Beyond their interactions within the NVU, PCs also participate in immune regulation. While brain PCs do not initiate inflammatory responses, they act as key sensors of neuroinflammation and modulate multiple signaling pathways involved in immune–neural communication [79]. In response to inflammatory threats, PCs secrete a range of cytokines and chemokines. Notably, PDGF-BB stimulation leads to increased expression of growth factors and anti-inflammatory cytokines in PCs. By contrast, lipopolysaccharide (LPS) exposure activates pro-inflammatory responses, promoting the secretion of cytokines involved in immune cell proliferation, maturation, and activation, including T cells, B cells, natural killer cells, and neutrophils.
This dichotomous response is mediated by distinct intracellular signaling
pathways: PDGF-BB promotes reparative signaling, whereas LPS activates the
Toll-like receptor 4–nuclear factor kappa B (NF-
Among a wide range of inflammatory molecules secreted by PCs, lipocalin-type
prostaglandin D synthase (L-PGDS) has recently been identified as a significant
factor (Fig. 4A, Ref. [5]) [80]. Previous research has shown that L-PGDS is
primarily located in the CNS, specifically within leptomeningeal cells, choroid
plexus epithelial cells, and oligodendrocytes [81, 82]. However, some researchers
have suggested that brain PCs, which are resistant to ischemia and can survive in
ischemic regions even after a permanent ischemic stroke [83, 84, 85], may serve as
L-PGDS-producing cells in these areas following stroke [80]. L-PGDS is
responsible for synthesizing PGD2 by catalyzing the isomerization of PGH2 [86].
PGD2 is the most abundant prostaglandin in the healthy brain and plays a crucial
role in various pathological processes. The physiological functions of PGD2 are
mediated through G protein-coupled receptors DP1 and DP2. The DP1 receptor is
coupled to G
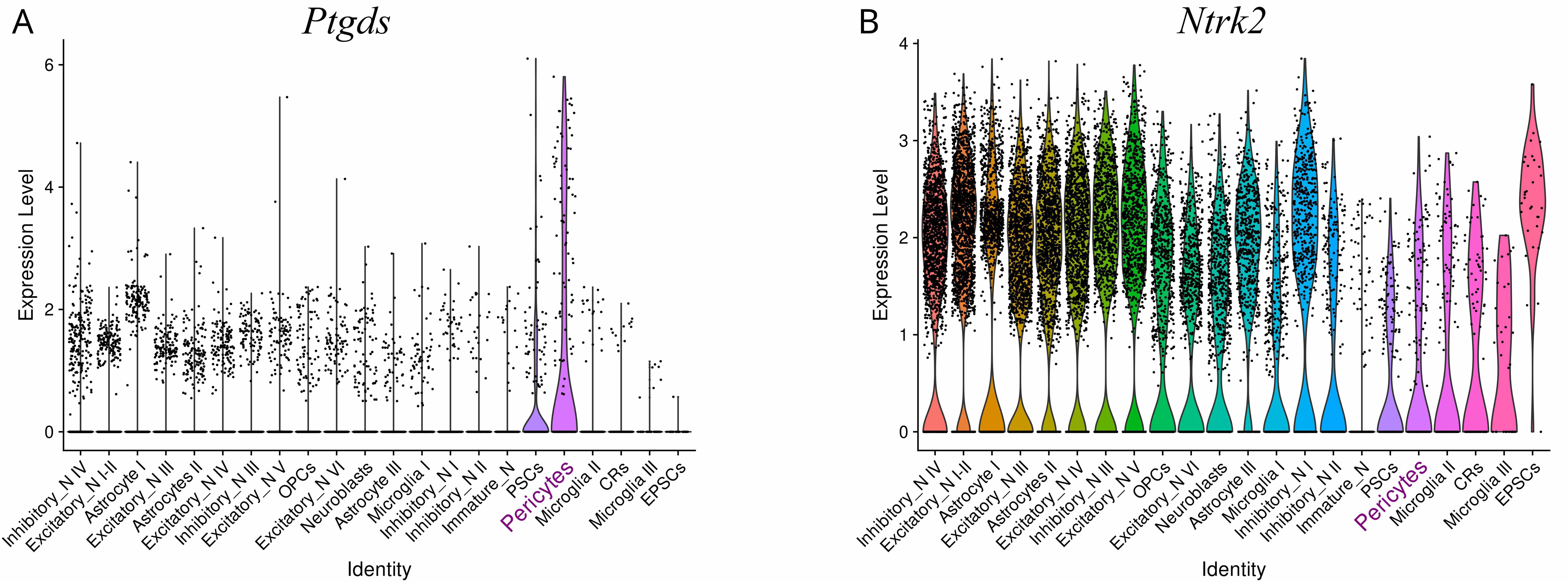 Fig. 4.
Fig. 4.
Expression of selected gene markers in the neonatal rat cortex. (A) Violin plots showing marked expression of prostaglandin D2 synthase (Ptgds) in pericytes. (B) Violin plots showing tropomyosin receptor kinase B (TrkB, Ntrk2) expression in cell types of the rat neonatal cortex. Fig. 4 presents data by Chen et al. [5] (NCBI GEO Accession No. GSE185538), analyzed using Seurat package ver. 5.3.1. (https://cran.rproject.org/web/packages/Seurat/index.html) in R.
In addition to the isomerization of PGH2 into PGD2 and its transportation,
L-PGDS has the capacity to bind to a broad array of molecules. This includes
lipophilic hormones such as retinoids and thyroid hormones, heme and
heme-degradation products, lipids, gangliosides, lipophilic drugs, and
nicotinamide coenzymes [86]. It is imperative to note that L-PGDS binds amyloid
The mechanisms regulating L-PGDS gene (Ptgds) expression in PCs are
still not well understood. However, it is known that the NF-
Beyond inflammatory cues, PCs are exquisitely sensitive to systemic metabolic disturbances, particularly in diabetic conditions. Chronic hyperglycemia induces mitochondrial overload in brain PCs, driving excessive reactive oxygen species (ROS) production through accelerated oxidative metabolism of glucose. This oxidative stress triggers progressive PC loss via apoptosis through mitochondrial pathways involving cytochrome c and apoptosis-inducing factor [117]. Pharmacological inhibition of mitochondrial carbonic anhydrase, particularly isoform VA, reduces ROS generation and protects PCs from glucotoxicity [118].
To counteract metabolic stress, PCs initially engage adaptive responses such as
upregulation of glycolytic enzymes and mitochondrial antioxidant defenses (e.g.,
superoxide dismutase 2), enabling temporary maintenance of energy homeostasis
under hyperglycemic conditions [119]. However, these mechanisms appear
insufficient for long-term compensation. Accumulation of advanced glycation
end-products (AGEs) promotes TGF-
Insulin resistance may exacerbate PC dysfunction by impairing insulin receptor signaling, thereby reducing glucose uptake [121], altering survival pathways, and intensifying oxidative vulnerability in the context of diabetes [122]. Recent models suggest that brain PCs express insulin receptors [123] and rely on insulin-mediated signaling for metabolic regulation [121], implicating insulin resistance as a contributor to maladaptive stress responses [122].
The resulting vascular instability manifests through coordinated molecular
changes, namely, downregulation of TJ proteins (notably claudin-5 and occludin);
increased matrix metalloproteinase-9 (MMP-9) activity; and reduced expression of
Pdgfr
These alterations result in BBB disruption and NV uncoupling, processes that correlate strongly with cognitive decline trajectories in patients with diabetes. Animal models of diabetes demonstrate that PC loss precedes BBB leakage and cognitive impairment. Antioxidant treatments (e.g., topiramate, Mito-TEMPO) can mitigate these outcomes by preserving PC viability and mitochondrial function [118, 125].
Notably, while retinal PCs demonstrate rapid deterioration under hyperglycemic
stress, brain PCs exhibit delayed but equally consequential degeneration. This
temporal disparity may reflect intrinsic differences in metabolic flexibility or
ROS-scavenging capacity, as observed in comparative studies of PC apoptosis and
ROS dynamics [126]. The hippocampus appears particularly vulnerable, with
increased oxidative markers and early changes in PCs markers such as
PDGFR
These findings position brain PCs as pivotal metabolic sensors within the NVU, capable of translating systemic disturbances into localized cerebrovascular dysfunction. While short-term adaptations exist, sustained hyperglycemia and insulin resistance ultimately overwhelm PC defenses, shifting the balance toward maladaptive remodeling, NV instability, and cognitive decline.
As the brain transitions through the arc of aging, PCs — long considered silent sentinels of vascular stability — emerge as active participants in the NV response to time. Their roles in maintaining BBB integrity, modulating cerebral blood flow, and coordinating cellular crosstalk do not simply erode; they undergo qualitative transformation. Aging reveals a paradox in PC biology: the same plasticity that enables PCs to adapt in youth may, with time, become a vector for vulnerability.
At the structural level, age-associated PC loss is particularly pronounced in
hippocampal and cortical regions, where vessel coverage becomes fragmented and
capillary rarefaction emerges as a hallmark of cerebrovascular decline [128].
This anatomical disintegration is paralleled by impaired clearance of neurotoxic
metabolites, such as A
Molecularly, aged PCs shift toward a senescent, reactive phenotype. They exhibit
mitochondrial instability, heightened oxidative stress, and upregulation of
inflammatory signaling pathways including TGF-
Functionally, this transformation has profound consequences. Dysregulated PCs
contribute to cerebral hypoperfusion, NV uncoupling, and ultimately, the erosion
of homeostatic control over the brain’s internal milieu [131, 132]. Importantly,
PC damage can be measured via soluble PDGFR
In the context of brain aging, PCs assume a dual role, functioning both as key regulators of early NV organization and contributors to age-related vulnerability. This developmental-to-degenerative trajectory highlights a broader principle, namely, that molecular and structural programs established during development, when disrupted, may predispose the NVU to functional decline. Recognizing this temporal continuum provides not only mechanistic understanding but also a conceptual framework linking neurodevelopmental integrity to susceptibility to neurodegenerative processes.
As our understanding of PC biology deepens, so does the recognition that these cells are not only biomarkers of cerebrovascular health but are also potential targets for therapeutic modulation. Recent years have seen the emergence of innovative strategies aimed at restoring or enhancing PC function in neurological disease. These approaches reflect a conceptual shift — from viewing PC dysfunction as an irreversible consequence of pathology to positioning it as a modifiable node within the NV network.
PC transplantation represents one such frontier. Preclinical studies suggest that exogenous PCs may reconstitute damaged microvascular environments, integrating into host vasculature, promoting angiogenesis, and restoring BBB integrity in ischemic and neurodegenerative contexts [135]. While challenges remain in sourcing, scalability, and immunocompatibility, this strategy offers a compelling avenue for cellular repair of the NVU.
Parallel efforts have focused on targeted modulation of PC signaling,
particularly via PDGFR
Exosome-mediated interventions represent a rapidly evolving therapeutic platform. Engineered exosomes can selectively deliver microRNAs, peptides, or small molecules across the BBB, directly modulating PC function. PC-derived exosomes enriched in microRNA 210, for example, have been shown to enhance mitochondrial resilience and barrier stabilization via Janus kinase 1/signal transducer and activator of transcription 3 signaling in spinal injury models [137]. Such findings highlight the promise of PCs not only as targets but also as active vehicles for regenerative therapeutics.
Beyond these primary strategies, nanotherapeutic platforms are emerging that co-modulate PCs and ECs signaling. For instance, simultaneous targeting of WNT pathways and PC depletion has been shown to transiently open the blood–tumor barrier, enhancing chemotherapeutic delivery to brain metastases [138]. These approaches offer unprecedented precision in modulating NV architecture for therapeutic gain.
PCs have emerged as indispensable cellular mediators that bridge neural and vascular biology throughout the lifespan. This review has traced their developmental trajectory from embryonic origins to specialized roles in the mature brain, revealing how their early establishment within the NVU predetermines their multifaceted functions in adulthood.
The developmental biology of PCs provides critical insights into their adult
capabilities. Their precise recruitment through PDGF-B/PDGFR
In the adult brain, PCs demonstrate a dual nature that is both protective and pathogenic. While essential for maintaining NV coupling and BBB integrity, they simultaneously contribute to disease processes when dysregulated. Their involvement spans neurodegenerative diseases, psychiatric conditions, and acute injuries like stroke, where their hypoxia sensitivity paradoxically limits their reparative potential. The discovery of L-PGDS expression patterns in PCs exemplifies how developmental markers may re-emerge in pathological contexts, suggesting conserved molecular programs across the lifespan.
Current research faces several critical challenges that require innovative solutions. The field needs more sophisticated tools to characterize PCs heterogeneity, monitor their dynamic functions in real time, and develop targeted interventions. Emerging technologies like high-resolution intravital imaging, single-cell omics approaches, and CRISPR-based manipulation offer promising avenues to overcome these limitations. Particularly crucial is the development of experimental models that better preserve the native interactions between PCs and other NVU components across developmental stages.
Looking forward, three interconnected priorities will shape PC research. First, establishing standardized approaches that account for developmental origins when studying adult PCs. Second, creating advanced organotypic models that maintain physiological PC–EC interactions. Third, translating fundamental insights about PC biology into clinically relevant applications. The convergence of developmental biology and clinical neuroscience perspectives will be essential for advancing our understanding of these versatile cells.
From their embryonic beginnings to their roles in aging and disease, PCs exemplify the profound interconnection between vascular and neural systems. Their proper characterization and manipulation hold exceptional promise for advancing both our fundamental knowledge of brain function and our ability to treat neurological disorders. As the field moves forward, integrating developmental principles with clinical insights will be crucial for unlocking the full therapeutic potential of these remarkable cells.
ANG1/2, angiopoietin 1 and 2; BBB, blood–brain barrier; BDNF, brain-derived neurotrophic factor; CNS, central nervous system; DAG, diacylglycerol; ECs, endothelial cells; FGF, fibroblast growth factor; HGF, hepatocyte growth factor; LIF, leukemia inhibitory factor; L-PGDS, lipocalin-type prostaglandin D synthase; LPS, lipopolysaccharide; NG2, neuroglial antigen 2; NGF, nerve growth factor; NVU, neurovascular unit; PC, pericyte; PDGF-BB, platelet-derived growth factor; PDGFR
UD originated and conceptualized the idea, conducted literature searches, wrote the manuscript (chapters “Functions of PCs in Adult Brain”, “Lipocalin-Type Prostaglandin D Synthase Expression in Brain PCs”, “Metabolic Stress Responses of Brain PCs under Hyperglycemia and Insulin Resistance”, “Age-Related Changes in PC Function”, “Emerging Therapeutic Strategies Targeting PCs” and “Conclusion), prepared Figs. 2,3. SV conducted literature searches, wrote the manuscript (chapters “Morphological Features and Tissue Distribution of PCs”, “Identification and Molecular Markers of PCs” and “The Development and Functional Role of PCs in Blood–Brain Barrier Formation”). DL originated and conceptualized the idea, conducted literature searches, wrote the manuscript (Abstract and chapters “Introduction” and “Leptomeninges in Brain Development”), prepared Figs. 1,4. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
We gratefully acknowledge the Institute of cytology and genetics for the opportunity to realize the project.
This research was funded by the Russian Science Foundation grant No. 24-25-00154.
The authors declare no conflict of interest.
During the preparation of this work the authors used the free version of DeepL Write and DeepSeek-V3 in order to check spell and grammar. After using this tool, the authors reviewed and edited the content as needed and takes full responsibility for the content of the publication.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.