


 , Jun Wei 1,2,*
, Jun Wei 1,2,*
1 The First College of Clinical Medical Science, China Three Gorges University, 443000 Yichang, Hubei, China
2 Department of Neurology, Yichang Central People’s Hospital, 443003 Yichang, Hubei, China
Abstract
Multiple sclerosis (MS) is a chronic autoimmune disorder marked by neuroinflammation, demyelination, and neuronal damage. Recent advancements highlight a novel interaction between iron-dependent cell death, known as ferroptosis, and gut microbiota, which may significantly influences the pathophysiology of MS. Ferroptosis, driven by lipid peroxidation and tightly linked to iron metabolism, is a pivotal contributor to the oxidative stress observed in MS. Concurrently, the gut microbiota, known to affect systemic immunity and neurological health, emerges as an important regulator of iron homeostasis and inflammatory responses, thereby influencing ferroptotic pathways. This review investigates how gut microbiota dysbiosis and ferroptosis impact MS, emphasizing their potential as therapeutic targets. Through an integrated examination of mechanistic pathways and clinical evidence, we discuss how targeting these interactions could lead to novel interventions that not only modulate disease progression but also offer personalized treatment strategies based on gut microbiota profiling. This synthesis aims at deepening insights into the microbial contributions to ferroptosis and their implications in MS, setting the stage for future research and therapeutic exploration.
Keywords
- ferroptosis
- gut microbiota
- multiple sclerosis
- neuroinflammation
- therapeutic targets
Multiple sclerosis (MS) is a chronic autoimmune condition affecting the central nervous system (CNS), where the immune system attacks the myelin, a crucial protective covering of nerve fibers [1]. This pathology leads to inflammation, demyelination, and neurodegeneration, manifesting as lesions or plaques primarily in the brain as well as spinal cord [2]. Globally, around 2.8 million people suffer from MS [3]. Clinically, MS presents with a wide array of symptoms that vary greatly among individuals depending on the location and extent of CNS involvement; these include sensory disturbances like numbness and tingling, motor issues such as muscle weakness and spasticity, visual impairments, cognitive deficits, and emotional disturbances such as depression and anxiety [4, 5, 6, 7]. MS progresses mainly in several forms, each with distinct progression patterns, with the most common being relapsing-remitting MS (RRMS), along with Primary Progressive MS (PPMS) and secondary progressive MS (SPMS) [8]. Treatment primarily involves disease-modifying therapies (DMTs) [9] designed to lower relapse rates and decelerate disease advancement, with emerging therapies like hematopoietic stem cell transplantation (HSCT) [10] showing promise. Managing MS is complex, requiring a multidisciplinary approach to address the diverse and debilitating symptoms associated with the disease.
Recent research has demonstrated a significant correlation between gut microbiota and various neurodegenerative diseases [11], particularly noting its importance in MS [12]. In patients with MS, there is a marked dysbiosis in the gut microbiome, manifesting as alterations in bacterial diversity and composition [13, 14]. This includes a decrease in beneficial bacterial groups like those within the Firmicutes phylum, coupled with an increase in potentially detrimental groups such as the bacteroidetes phylum [15, 16]. Such changes in the gut’s microbial environment have profound implications for the progression and activity of MS. For instance, diminished production of short-chain fatty acids (SCFAs), such as butyrate, may lead to compromised gut barrier function and heightened systemic inflammation [17, 18]. Additionally, shifts in specific microbial populations have been linked to inflammation markers, cytokine levels, and the frequency of relapses in MS patients [14].
Ferroptosis is a form of programmed cell death different from apoptosis and necrosis, primarily marked by excessive lipid peroxide accumulation [19]. This cell death mechanism is closely associated to iron metabolism and reactive oxygen species (ROS) [20]. Recent research suggests that the dysregulation of iron homeostasis and heightened oxidative stress in neural cells may induce ferroptosis, leading to the degeneration of neurons and oligodendrocytes—the cells responsible for producing myelin [21, 22]. Gut microbiota influences ferroptosis through several mechanisms [23, 24]. Microbial metabolites such as SCFAs and bile acids, along with siderophores produced by some bacteria, regulate iron metabolism crucial for ferroptosis. These metabolites can modulate systemic iron availability, thereby influencing ferroptosis sensitivity [25, 26]. Moreover, the gut microbiota impacts lipid metabolism and antioxidant defenses, both vital in the ferroptosis process [26, 27].
This review examines the interplay between gut microbiota and ferroptosis in MS, proposing that this relationship could unlock new therapeutic avenues and deeper understanding of disease mechanisms. As recent studies illuminate the impact of gut microbial dysbiosis on systemic immune responses and neurological health, the potential modulation of ferroptosis through microbial interactions offers a compelling perspective [28, 29].
Ferroptosis is a specialized form of regulated cell death, characterized by its unique dependency on iron and the accumulation of lipid peroxides [19]. This type of cell death has emerged as a crucial area of interest in medical research due to its potential implications in various diseases, including cancer [30], neurodegeneration [31], and ischemic injury [32]. Unlike apoptosis or necrosis, ferroptosis is primarily driven by the catastrophic failure of the cell’s lipid repair mechanisms, leading to lethal, oxidative damage.
The concept of ferroptosis was first introduced in 2012 by Dixon et al. [19], who identified it as a distinct, iron-dependent form of non-apoptotic cell death [19]. This discovery has since spurred extensive research into the molecular pathways that regulate ferroptosis and their potential therapeutic applications [33]. The process is marked by an accumulation of iron and the formation of ROS, particularly within the lipid structures of cell membranes [34].
Ferroptosis can be triggered by a variety of factors, including the inhibition of glutathione peroxidase 4 (GPX4), a critical enzyme that safeguards cells against oxidative damage [35]. The involvement of multiple pathways, including the mevalonate and transsulfuration pathways, introduces additional complexity into the regulation of ferroptosis [36, 37]. This form of cell death is distinguished by two main pathways: the classical, or GPX4-dependent pathway, and the non-classical (GPX4-independent) pathway that involves other regulatory mechanisms, highlighting its multifaceted nature in cell biology and its potential significance in therapeutic strategies (Table 1, Ref. [35, 38, 39, 40, 41, 42, 43]).
| Pathway | Main molecules involved | How to promote or inhibit ferroptosis | |
| GPX4-dependent | GPX4 | Inhibit: GPX4 employs glutathione to reduce lipid hydroperoxides to non-toxic lipid alcohols, preventing lipid peroxidation. | [35] |
| GPX4-independent | FSP1 | Inhibit: FSP1 reduces coenzyme Q10 to ubiquinol, enhancing the antioxidant defense against lipid peroxidation in cellular membranes. | [38] |
| p53 | Promote: p53 inhibits SLC7A11 to reduce glutathione synthesis and increases SAT1 and GLS2 expression, which boosts lipid peroxidation. | [39, 40] | |
| Inhibit: p53 blocks DPP4 activity and induces CDKN1A/p21, which mitigates oxidative stress and lipid peroxidation. | |||
| DHODH | Inhibit: DHODH reduces ubiquinone to ubiquinol in the mitochondrial inner membrane, preventing mitochondrial lipid peroxidation and cellular damage. | [41] | |
| GCH1/BH4 | Inhibit: GCH1 synthesizes BH4, which supports the integrity of phospholipids with polyunsaturated fatty acyl chains, thus preventing their peroxidation and subsequent cell death. | [42] | |
| NCOA4 | Promote: NCOA4 facilitates ferritinophagy, increases the cellular labile iron pool and exacerbates lipid peroxidation. Through phosphorylation by ATM kinase, NCOA4 enhances its interaction with ferritin, leading to ferritin degradation in lysosomes. | [43] |
GPX4, glutathione peroxidase 4; FSP1, ferroptosis suppressor protein 1; DHODH, dihydroorotate dehydrogenase; GCH1, GTP cyclohydrolase 1; BH4, tetrahydrobiopterin; NCOA4, nuclear receptor coactivator 4; SLC7A11, solute carrier family 7 member 11; SAT1, spermidine/spermine N1-acetyltransferase 1; GLS2, glutaminase 2; DPP4, dipeptidyl peptidase 4; CDKN1A, cyclin dependent kinase inhibitor 1A; GCH1, GTP cyclohydrolase 1; ATM, ATM serine/threonine kinase.
GPX4 is a key enzyme that regulates oxidative stress and lipid peroxidation, playing a crucial role in preventing ferroptosis [35]. GPX4 utilizes glutathione (GSH), a major cellular antioxidant, to convert lipid hydroperoxides into non-toxic lipid alcohols and transforms free GSH into oxidized glutathione (GSSG) [44, 45]. This reaction is vital for preserving cellular membrane integrity and shielding cells from oxidative damage.
The inhibition of GPX4 is a key factor in triggering ferroptosis. Specific compounds, including ras-selective lethal small molecule 3 (RSL3) and ML162, target and bind to the active site of GPX4, effectively blocking its enzymatic function [46, 47]. This inhibition results in the accumulation of lipid peroxides, triggering the ferroptotic process by amplifying lipid peroxidation cascades throughout cellular membranes.
Moreover, the genetic downregulation or ablation of GPX4 further elucidates its critical role in cell survival. Knockdown or genetic deletion of GPX4 in experimental models, such as conditional knockout mice, has shown to result in increased susceptibility to ferroptosis [48, 49]. These models have illustrated that the absence of GPX4 gives rise to severe pathological outcomes including acute renal failure [50] and neurodegeneration [51] due to rampant lipid peroxidation and ensuing ferroptosis. This susceptibility highlights the critical role of GPX4 in defending cells from oxidative stress and maintaining cellular viability.
Additionally, research has also pointed to the interaction between GPX4 inhibition and other ferroptosis regulators. For example, the availability of GSH, which is essential for GPX4’s peroxidase activity, is regulated by system Xc-, a cystine/glutamate antiporter [52]. When system Xc- is inhibited, it leads to a reduction in cystine absorption and a subsequent decrease in GSH, indirectly suppressing GPX4 activity, which promotes ferroptosis [53]. This interdependence illustrates the complex network of pathways regulating ferroptosis and emphasizes the complex role of GPX4 as a therapeutic target.
The non-classical pathway of ferroptosis does not depend on GPX4 but involves other regulatory mechanisms and enzymes [54]. One such pathway involves the protein ferroptosis suppressor protein 1 (FSP1), originally identified as AIF family member 2 (AIFM2) [55]. FSP1 acts independently of the glutathione-dependent antioxidant system, utilizing nicotinamide adenine dinucleotide phosphate (NADPH) to reduce ubiquinone to ubiquinol [38]. This reduction process serves as a potent lipophilic radical-trapping antioxidant, reducing lipid peroxidation and subsequently inhibiting ferroptosis [56]. This pathway highlights the role of alternative redox systems in controlling lipid peroxidation, independent of the classic glutathione system.
Another significant GPX4-independent mechanism is the p53-mediated pathway [57, 58]. p53, widely recognized for its role in tumor suppression, has been shown to regulate ferroptosis via several pathways, not just by modulating solute carrier family 7 member 11 (SLC7A11) [39]—a critical element of the cystine/glutamate antiporter which controls the cellular uptake of cystine and glutathione synthesis—but also by affecting lipid peroxidation through its impact on lipoxygenases such as arachidonate 12-lipoxygenase, 12S type (ALOX12) and arachidonate 15-lipoxygenase (ALOX15) [59, 60]. The latter is regulated via spermidine/spermine N1-acetyltransferase 1 (SAT1), enhancing the cell’s susceptibility to ferroptosis [61]. Notably, p53 can both promote and inhibit ferroptosis: it promotes this form of cell death by inhibiting anti-ferroptotic factors like SLC7A11 and boosting pro-ferroptotic factors including SAT1 and glutaminase 2 (GLS2) [39]. Conversely, it can act protectively by blocking dipeptidyl peptidase 4 (DPP4) activity and inducing cyclin dependent kinase inhibitor 1A (CDKN1A)/p21 expression, thereby aiding cell survival under stress [40]. This dual role highlights the intricate balance p53 maintains in cellular health and stress responses, positioning it as a potential target for therapies in conditions marked by abnormal ferroptosis, such as various cancers [30].
Beyond the FSP1 and p53-mediated pathways, several other GPX4-independent mechanisms significantly impact ferroptosis. These include the dihydroorotate dehydrogenase (DHODH) pathway, which involves mitochondrial redox activities affecting lipid peroxidation [41]; the GTP cyclohydrolase 1 (GCH1)/tetrahydrobiopterin (BH4) pathway, crucial for reducing oxidative stress and blocking lipid peroxidation [42]; and the regulation of iron storage and mobilization via ferritin and the autophagy-related protein nuclear receptor coactivator 4 (NCOA4), which controls the availability of reactive iron crucial for ferroptosis execution [43].
The inhibition of GPX4 is a fundamental characteristic of ferroptosis, essential for preventing lipid peroxidation by reducing lipid hydroperoxides into non-toxic lipid alcohols [35]. Beyond GPX4 inhibition, ferroptosis is also recognized by its iron dependence and extensive lipid peroxidation, marking it as a distinct type of cell death driven by iron-catalyzed oxidative stress and lipid damage [19].
Iron is fundamentally involved in ferroptosis, amplifying oxidative stress through various biochemical mechanisms. Its primary action is through the Fenton reaction, where ferrous iron (Fe2+) catalyzes the transformation of hydrogen peroxide into highly reactive hydroxyl radicals [62]. These radicals initiate the peroxidation process of polyunsaturated fatty acids (PUFAs)-rich phospholipids in cellular membranes, a cornerstone of ferroptosis [63].
In addition to the Fenton reaction, iron stimulates lipid peroxidation indirectly by the activation of lipoxygenases (LOXs) [45, 64]. These iron-dependent enzymes, such as arachidonate 15-lipoxygenase (ALOX15), catalyze the oxygenation of PUFAs in membrane phospholipids, forming lipid hydroperoxides [65]. These lipid hydroperoxides are the immediate precursors to toxic lipid peroxides, which further propagate the lipid peroxidation chain reaction, exacerbating cellular damage. The presence of available free iron, therefore, is a key determinant in the progression of ferroptosis due to its essential role in these enzymatic processes.
Research also highlights the importance of iron in regulating various signaling pathways and gene expression profiles associated with oxidative stress responses and mitochondrial function [66, 67, 68]. These studies further link iron metabolism to ferroptotic sensitivity by demonstrating how disruptions in iron handling can predispose cells to undergo ferroptosis, underlining the intricate relationship between iron metabolism and cellular health.
Lipid peroxidation is not only a defining feature of ferroptosis but also its main execution pathway [69]. It refers to a destructive process mediated by free radicals that oxidize PUFAs [70]. This oxidative process damages cellular membranes and further harms cellular structures and functions by releasing various toxic metabolic products, such as malondialdehyde and 4-hydroxynonenal [71]. The process of lipid peroxidation unfolds through two main pathways: enzymatic pathway and non-enzymatic pathway.
The enzymatic pathway relies predominantly on LOXs, a group of iron-dependent enzymes catalyzing the formation of lipid hydroperoxides from PUFAs in membrane phospholipids [72]. For example, ALOX15 is critical in ferroptosis, initiating the production of lipid hydroperoxides, which are precursors to more toxic lipid peroxyl radicals [73]. On the other hand, the non-enzymatic pathway is largely driven by the Fenton reaction, where hydrogen peroxide is converted into highly reactive hydroxyl radicals [63]. These radicals attack PUFAs in the cellular membranes, triggering a chain reaction that produces lipid alkoxyl and lipid peroxyl radicals. These species further promote lipid peroxidation by accumulating ROS [74]. Moreover, the conjugate acid of superoxide can also trigger lipid peroxidation, especially in the acidic microenvironments within cells, thereby accelerating the lipid peroxidation process [75].
Interestingly, research indicates that lipid peroxidation often initiates within the endoplasmic reticulum (ER) membrane before affecting the plasma membrane [70]. This progression suggests a systematic sequence of membrane degradation during ferroptosis, with the ER’s extensive membrane surface, rich in PUFAs, being particularly vulnerable to oxidative stress [76, 77, 78]. This intricate interplay of biochemical reactions underscores lipid peroxidation as a key factor in ferroptosis, profoundly impacting cell viability and disease pathology.
Ferroptosis is characterized by distinctive morphological alterations at both the cellular and subcellular levels, which are crucial for its identification and distinction from other types of cell death (Fig. 1). Ferroptosis, in contrast to apoptosis or necrosis, shows distinct changes indicative of its oxidative and iron-dependent characteristics.
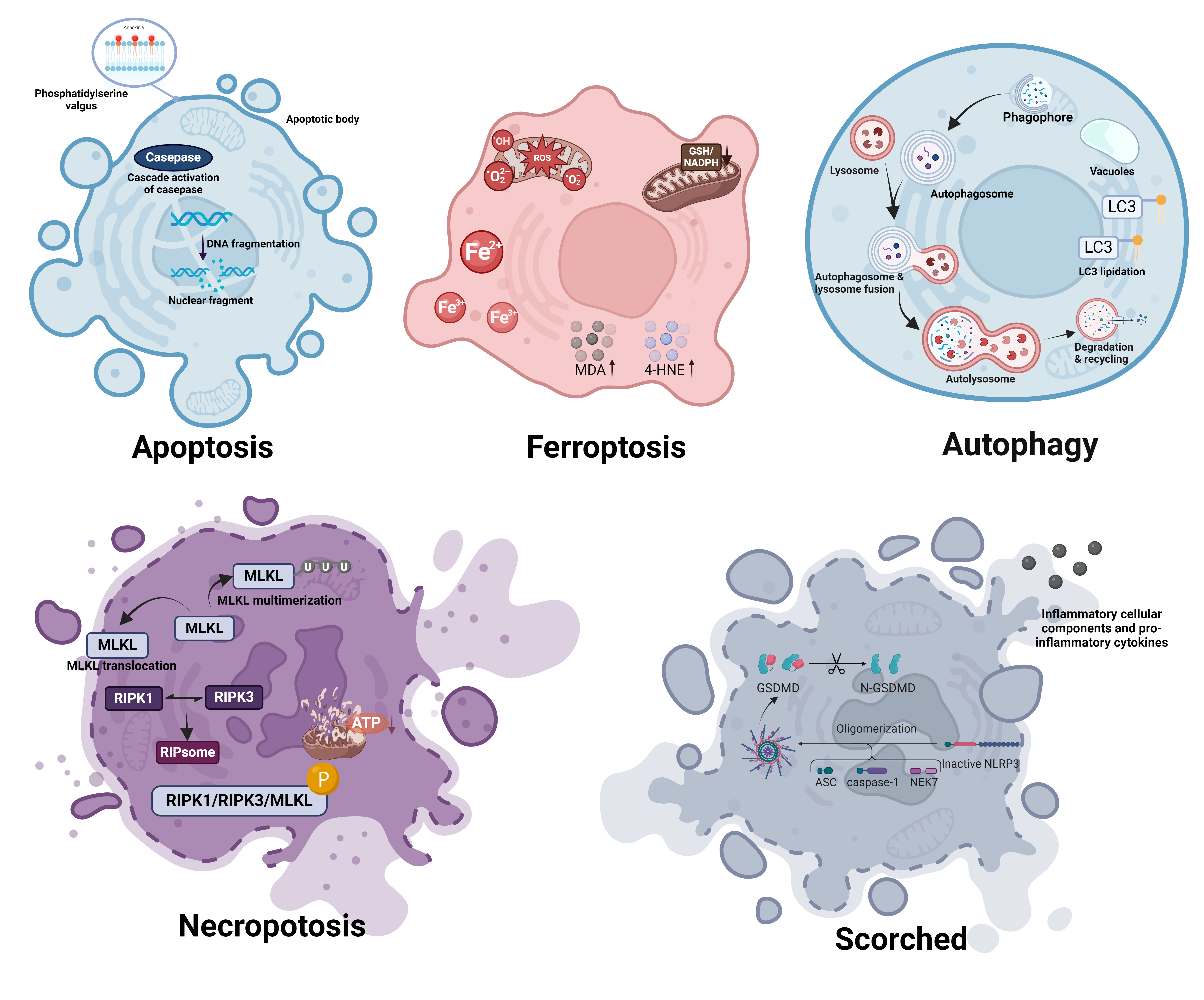 Fig. 1.
Fig. 1.
Morphological distinctions between ferroptosis and other forms of cell death. Apoptosis: Induced by internal or external signals leading to caspase cascade activation and nuclear fragmentation. Characteristically shows cell shrinkage, chromatin condensation, membrane blebbing, and the formation of apoptotic bodies without loss of membrane integrity until late stages. Ferroptosis: Triggered by iron-dependent lipid peroxidation, resulting in the accumulation of ROS and lipid peroxidation products such as MDA and 4-HNE, causing cell membrane damage. Characterized by reduced cell size, cell membrane rupture and blebbing, and mitochondrial shrinkage and abnormal morphology. Autophagy: Initiated in response to nutrient deprivation or cellular stress, involving the sequestration of cytoplasmic contents into double-membrane vesicles called autophagosomes. Features include extensive vacuolization of the cytoplasm, gradual degradation of cellular components within autolysosomes, and typically a lack of chromatin condensation. Necroptosis: Occurs through receptor-interacting protein kinase 1 and 3 (RIPK1, RIPK3) mediated phosphorylation of MLKL, leading to its oligomerization and membrane translocation. Morphologically marked by cell swelling, plasma membrane rupture, and release of cellular contents, leading to inflammation. Scorched: Characterized by the oligomerization and pore formation by gasdermin D proteins, leading to rapid cell swelling, plasma membrane rupture, and pronounced inflammatory responses. Often accompanied by cytoplasmic granulation and mitochondrial swelling. ROS, reactive oxygen species; Fe2+, ferrous ion; Fe3+, ferric ion; GSH, glutathione; NADPH, nicotinamide adenine dinucleotide phosphate; MDA, malondialdehyde; 4-HNE, 4-hydroxynonenal; LC3, microtubule-associated protein 1A/1B-light chain 3; MLKL, mixed lineage kinase domain-like protein; RIPK1, Receptor-Interacting Protein Kinase 1; RIPK3, Receptor-Interacting Protein Kinase 3; ATP, adenosine triphosphate; GSDMD, Gasdermin D; N-GSDMD, N-terminal domain of Gasdermin D; NLRP3, NOD-, LRR- and pyrin domain-containing protein 3; ASC, apoptosis-associated speck-like protein containing a CARD; NEK7, NIMA (never in mitosis gene a)-related kinase 7. (Created with BioRender.com).
At the cellular level, one of the most noticeable morphological changes associated with ferroptosis is the shrinkage of cell volume [79]. This is in contrast to the swelling that is typically observed in necrosis. Cells undergoing ferroptosis also display a formation of membrane protrusions, accompanied by a reduction in overall cell elasticity [80]. Plasma membrane shows signs of rupture and blebbing, indicating a loss of integrity [81, 82]. Unlike apoptosis, ferroptotic cells do not display chromatin condensation or nuclear fragmentation, and the nucleus remains normally sized [19]. Additionally, there is sometimes cytoplasmic swelling, and an increase in autophagic vacuoles is observed, pointing to a potential relationship between autophagy and ferroptosis [82, 83, 84].
At the subcellular level, the most striking features of ferroptosis are observed in the mitochondria, which undergo profound morphological changes. Mitochondria in ferroptotic cells are typically smaller, display enhanced membrane density, and show a reduction or complete loss of mitochondrial cristae [19, 50, 85]. These changes are indicative of disrupted mitochondrial function, which is essential for energy production and cellular metabolism. The altered mitochondrial morphology reflects the metabolic disturbances occurring during ferroptosis, such as impaired oxidative phosphorylation and energy depletion.
Research has identified alterations in key genes associated with ferroptosis in patients with multiple sclerosis or in animal models, suggesting their potential role in the pathology of the disease [21, 86, 87, 88]. In the experimental autoimmune encephalomyelitis (EAE) model—a widely recognized mouse analog of MS—there is an upregulation of acyl-CoA synthetase long-chain family member 4 (ACSL4) [89]. ACSL4 is essential for integrating PUFAs into phospholipids that are susceptible to peroxidation [90]. Additionally, EAE mice show a reduction in the glutathione-dependent antioxidant defense, specifically the levels of system xC (xCT) and GPX4 [21]. This reduction is indicative of a decreased capacity to counteract oxidative stress, leading to enhanced lipid peroxidation and subsequent ferroptotic cell death [35].
Evidence from susceptibility Magnetic resonance imaging (MRI) has revealed elevated iron levels in deep gray matter structures of MS patients, correlating with increased disability and gray matter atrophy [91, 92, 93]. An autopsy study further substantiate these findings, demonstrating significant degeneration of deep gray matter associated with iron accumulation and oxidative damage [94]. In the same individuals, the accumulation of iron detected through susceptibility MRI also aligns with postmortem analyses, such as X-ray fluorescence and iron staining [95, 96]. Susceptibility MRI also indicates iron deposition near lesions where myelin loss occurs [97]. Both active and chronic lesions in MS show increased unstable iron (Fe2+), and the cerebrospinal fluid (CSF) of MS patients shows an elevated Fe2+/Fe3+ ratio [98]. Interestingly, while iron levels are elevated in deep gray matter structures and adjacent to lesions, there is a notable reduction of iron in normal-appearing white matter (NAWM). This reduction appears to correlate with disease duration, indicating a potential link between iron homeostasis and the progression of MS.
Increased lipid peroxidation is a notable feature in MS, evidenced by high immunoreactivity for E06 and 4-hydroxy-2-nonenal (4-HNE) in both active and chronic MS lesions, with the most pronounced signals detected in active lesions [98]. Additionally, CSF from MS patients show elevated levels of lipid peroxidation markers, including malondialdehyde (MDA) [99, 100] and oxidized phosphatidylcholine [101], compared to control groups. Serum analyses also reveal increased levels of MDA [102], lipid peroxides [103], and fluorescent lipid peroxidation products (PFLPP) [104] in MS patients. Furthermore, a study has indicated heightened markers of lipid peroxidation in urine samples from MS patients relative to controls [105]. Notably, increased lipid peroxidation was observed even before the onset of neurological symptoms in a study involving seven-week-old male Lewis rats with acute EAE [106], indicating that lipid peroxidation may play a crucial role early in the pathogenesis of MS.
Cellular alterations in the brains of MS patients reveal significant pathophysiological changes that may intersect with ferroptotic mechanisms. In the lateral geniculate nucleus, parvocellular neurons are smaller in MS brains (mean size: 226 µm2) compared to controls (230 µm2), with greater variation in size, indicating more atrophic neurons [107]. This atrophy may reflect similar cellular shrinkage seen in ferroptosis, characterized by reduced cell volume [79, 83]. Oligodendrocytes become dysfunctional in MS, leading to myelin damage and axonal degeneration, exacerbated by metabolic disruptions such as mitochondrial dysfunction [108]. Notably, studies show that mitochondria in axons of EAE spinal cord are shorter compared to those in naïve spinal cord, resembling the profound morphological changes observed in ferroptotic cells, where mitochondria are smaller and exhibit enhanced membrane density [19, 50, 85]. Dysfunction in the mitochondrial fusion/fission machinery, with a notable increase in Y-shaped mitochondria in EAE spinal cord axons [109] also parallels the disrupted mitochondrial morphology associated with ferroptosis. These changes suggest a potential convergence between the pathophysiological processes of MS and ferroptotic cell death.
Ferroptosis may contribute to MS through oxidative stress mechanisms driven by iron accumulation and lipid peroxidation [110, 111]. In MS, abnormal iron deposition within the central nervous system catalyzes the Fenton reaction, producing reactive hydroxyl radicals and leading to significant oxidative damage and cellular stress [21]. The enhanced lipid peroxidation of PUFAs within cellular membranes, compounded by an imbalance in iron homeostasis, further drives the ferroptotic process, undermining neuronal and oligodendrocyte integrity [72].
The inflammatory response in MS is intricately linked to ferroptosis through the iron dysregulation and the accumulation of lipid peroxides [112, 113]. Elevated iron levels and lipid peroxidation products within MS lesions suggest that iron-mediated oxidative stress perpetuates ongoing inflammation and tissue damage [98]. Moreover, ferroptosis can exacerbate neuroinflammation by releasing lipid peroxidation products and damage-associated molecular patterns (DAMPs), which activate microglia and other immune cells, thereby promoting a vicious cycle of inflammation and cell death [114]. The process also influences T-cell activation through the T-cell receptor signaling pathway, enhancing the autoimmune response and leading to further demyelination and neuronal damage [89].
Ferroptosis significantly contributes to the death of neurons and oligodendrocytes in MS [115]. Oligodendrocytes, responsible for myelinating axons, succumb to iron-induced oxidative stress and lipid peroxidation, resulting in their ferroptotic death [116]. This cell death contributes directly to the demyelination observed in MS [21]. Neurons also fall victim to iron toxicity and the resultant oxidative stress. The high metabolic demand of neurons makes them particularly vulnerable to disrupted mitochondrial function and oxidative damage, which are hallmarks of ferroptosis [117].
Apoptosis and Ferroptosis: Apoptosis, a form of programmed cell death, involves
cell shrinkage, chromatin condensation, and DNA fragmentation [118]. Unlike
ferroptosis, which is driven by lipid peroxidation, apoptosis is primarily
controlled by caspase activation through either the intrinsic or extrinsic
pathways [119]. However, recent studies have suggested a connection between these
two pathways [120, 121]. Ferroptotic agents can trigger endoplasmic reticulum (ER)
stress leading to apoptosis through activation of pathways such as eukaryotic translation initiation factor 2 alpha kinase 3 (EIF2AK3, or PERK)-eukaryotic initiation factor 2 alpha (eIF2α)-activating transcription factor 4 (ATF4)-DNA damage inducible transcript 3 (DDIT3, or CHOP (
PERK-eIF2
Necroptosis and Ferroptosis: Necroptosis is another form of regulated cell death that is dependent on the receptor-interacting protein kinase (RIPK) pathway, particularly RIPK1 and RIPK3, and the mixed lineage kinase domain-like pseudokinase (MLKL) [123, 124]. Like ferroptosis, necroptosis is associated with oxidative stress and inflammation but is distinguished by its mechanism of cell membrane disruption [125, 126, 127]. Iron overload in ferroptosis can trigger mitochondrial permeability transition pore (MPTP) opening, exacerbating RIPK1 phosphorylation and enhancing necroptosis [128]. Additionally, heat shock protein 90 (HSP90) acts as a common regulator for both pathways by modulating receptor interacting serine/threonine kinase 1 (RIPK1, or RIP1) phosphorylation and suppressing GPX4 activation [129, 130], linking oxidative stress to both ferroptosis and necroptosis [131].
Pyroptosis and Ferroptosis: Pyroptosis is an inflammatory form of cell death driven by gasdermin proteins, which form pores in the cell membrane, causing cell lysis and the release of inflammatory cytokines [132]. The inflammasome pathway, which is central to pyroptosis, has been shown to interact with ferroptosis mechanisms [133, 134]. The activation of inflammasomes can lead to ROS production, which can promote lipid peroxidation and ferroptosis [134]. On the contrary, lipid peroxidation, a key feature of ferroptosis, can also promote pyroptosis by destabilizing cellular membranes and enhancing gasdermin-mediated cell lysis [135].
The gut microbiota, comprising the microbial communities residing in the human gastrointestinal tract, plays a critical role in host health [136]. These microbes are not only involved in digestion but also significantly influence the immune system’s functionality and the host’s susceptibility to diseases [137]. Recent research has illuminated complex interactions between the gut microbiota and various diseases, including MS [12], an autoimmune disorder affecting the central nervous system. The gut microbiota may influence the progression of multiple sclerosis through various mechanisms (Fig. 2).
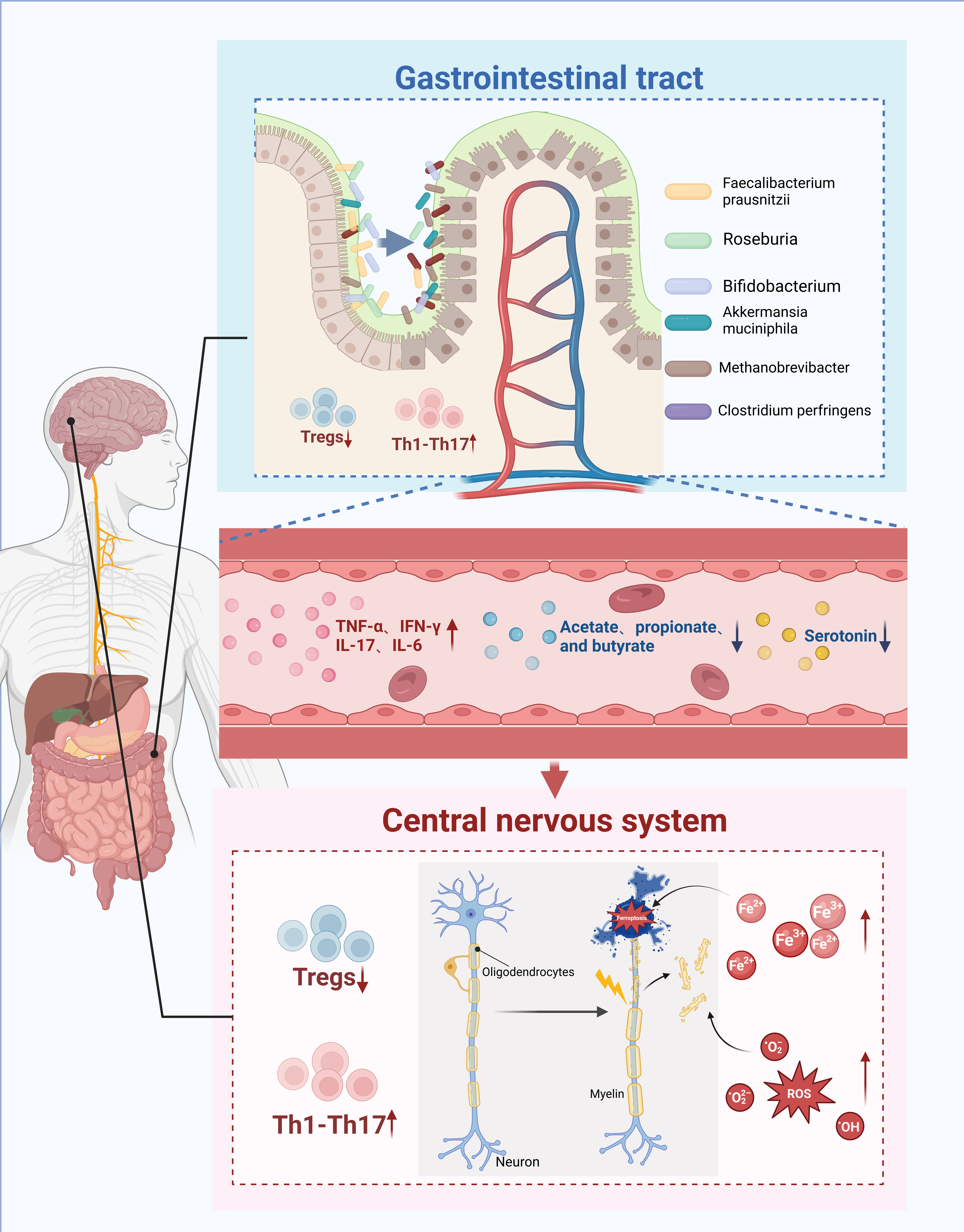 Fig. 2.
Fig. 2.
Gut microbiota regulation of MS through ferroptosis. Patients
with multiple sclerosis show dysbiosis of the gut microbiota and increased
intestinal permeability. This condition facilitates the polarization of T cells
towards a pro-inflammatory phenotype, specifically T helper 1 (Th1) cells and T helper 17 (Th17) cells.
There is a concomitant increase in pro-inflammatory cytokines, like tumor
necrosis factor-alpha (TNF-
Patients with MS show microbial dysbiosis characterized by disrupted gut microbiota. This includes a diminished presence of beneficial bacteria and a rise in potentially harmful bacterial species. A case-control study has identified differences in 61 bacterial species between MS patients and healthy individuals, with 31 species enriched in MS cases [14]. Beneficial bacteria such as Faecalibacterium prausnitzii, Roseburia, and Bifidobacterium, known for their anti-inflammatory properties and roles in maintaining gut health, are typically reduced. Conversely, potentially harmful bacteria such as akkermansia muciniphila, methanobrevibacter, and clostridium perfringens increase, promoting an inflammatory environment [13, 14, 138, 139].
Furthermore, the gut microbiota composition in MS patients varies with the
activity of the disease [14]. Certain bacteria that correlate with
pro-inflammatory cytokines such as Interleukin-17A (IL-17A) and tumor necrosis factor-alpha (TNF-
The gut barrier is a protective boundary in the gut that prevents harmful substances and pathogens from entering the body while allowing the absorption of water, nutrients, and electrolytes [141]. This barrier consists of a physical barrier formed by tight junctions between epithelial cells, a secretory barrier that includes antimicrobial peptides, mucus, and other fluids, and an immune barrier that comprises elements of both the innate and adaptive immune systems [142].
Patients with MS often have a genetic predisposition that makes them susceptible to gut barrier disruptions [143]. Such disruptions can be exacerbated by dysbiosis in the gut microbiota, which can provoke immune responses leading to further damage. For example, an imbalance between pro-inflammatory T helper (Th)1-Th17 cells and regulatory T cells can disrupt the tight junctions within the gut epithelium [144].
In patients with MS, alterations in the expression of proteins that form tight junctions in gut epithelial cells have been observed. This disruption increases gut permeability, permitting the translocation of bacteria and their products into the bloodstream. This, in turn, can trigger systemic inflammation and may have implications for the CNS [142, 145].
The gut microbiome is fundamental in training and developing the host’s immune system, with about 70–80% of immune cells residing in the gut [146, 147, 148]. This interaction is particularly crucial during early life as it aids in establishing proper immune maturation. Disruptions during this critical period can have long-term impacts on immune function [148, 149]. Intestinal immune function is underdeveloped in germ-free mice [150]. The influence of the gut microbiota extends beyond local mucosal immunity to affect systemic immune responses [148]. A diverse and healthy microbiota is associated with a well-functioning immune system. Disruption or imbalance in the gut microbiota, known as dysbiosis, is linked to various immune-mediated diseases and increased susceptibility to infections [151]. The microbiota produces metabolites and molecular patterns that regulate the immune system, including SCFAs and other compounds that can modulate immune cell functions [152, 153]. This relationship is reciprocal; the microbiota shapes the immune system, which in turn helps preserve the equilibrium of the gut microbiota [154].
Pro-inflammatory cytokines are integral to the pathogenesis of MS. These
cytokines, including interferon-gamma (IFN-
The regulation of iron homeostasis in the body is significantly influenced by the gut microbiota, which operates through various interconnected mechanisms [159]. Firstly, it affects iron absorption in the intestines; certain bacteria produce metabolites that enhance iron uptake, while others compete with the host for this essential mineral [159, 160, 161]. Moreover, the microbiota can modulate the production of hepcidin, the master regulator of iron metabolism [162]. Hepcidin manages iron absorption and recycling by targeting ferroportin, the primary cellular iron exporter, thereby influencing systemic iron levels [163]. Additionally, gut microbiota assists iron uptake by certain immune cells, notably regulatory T cells in the colon, which is crucial for maintaining intestinal immune tolerance [164].
These microbiota functions extend to influencing iron storage, with observations that germ-free mice show lower ferritin levels in their colonic regulatory T cells (Tregs) cells compared to normal counterparts [164]. Furthermore, some gut bacteria produce siderophores, iron-binding molecules that sequester iron, reducing its availability for absorption [165]. This complex interaction between microbiota and iron also involves the modulation of inflammation and gut barrier integrity, both of which impact iron metabolism [166]. Chronic intestinal inflammation, often influenced by microbiota, can result in anemia of inflammation, marked by decreased iron availability [167]. Importantly, this relationship is bidirectional, where iron levels change the microbiota’s composition and function, potentially causing dysbiosis and associated iron-related disorders [160, 168]. This intricate relationship might also extend to ferroptosis, an iron-dependent form of cell death, suggesting that disruptions in gut microbiota could influence cell survival and disease pathogenesis through iron dysregulation [169].
Patients with MS show significant alterations in metabolites that are closely associated with the gut microbiota [170]. Research has revealed that these changes in metabolites, particularly those involved in metabolic and immune pathways, may influence the pathophysiology of the disease (Table 2, Ref. [18, 171, 172, 173, 174, 175, 176, 177, 178]).
| Participants | Metabolite changes | ||
| MS | Control | ||
| 304 | 68 | Propionate↓ | [172] |
| 30 | 10 | Propionate↓ | [171] |
| 129 | 58 | Butyrate↓ | [18] |
| 20 | 15 | Acetate↓, Propionate↓, Butyrate↓ | [173] |
| 98 | 55 | Acetate↓, Propionate↓, Butyrate↓ | [174] |
| 34 | 34 | Acetate↓, Propionate↓, Butyrate↓ | [175] |
| 227 | 36 | Serotonin↓ | [176] |
| 60 | 12 | Serotonin↓ | [177] |
| 22 | 21 | L-tyrosine↓, L-isoleucine↓, L-tryptophan↓ | [178] |
| L-glutamic acid↑, L-valine↑ | |||
MS, multiple sclerosis; The symbols “
SCFAs such as acetate, propionate, and butyrate, which are produced by the gut microbiota through the fermentation of dietary fibers, are essential for maintaining gut health and modulating immune functions [179]. SCFAs possess anti-inflammatory properties and participate in immune regulation, including promoting Tregs and suppressing pro-inflammatory cells [180, 181]. It has been observed that levels of SCFAs, especially butyrate and propionate, are generally lower in MS patients [17, 182]. This reduction correlates with a decreased abundance of SCFA-producing bacteria in their gut microbiota [171]. Notably, lower serum levels of propionate have been documented in MS patients [14], and treatments with propionate have shown promise in inhibiting the development of EAE, by promoting the expansion of Tregs [172]. SCFAs have been found to regulate ferroptosis; therefore, changes in these metabolites may indirectly regulate the development of MS by affecting the ferroptosis pathway.
Serotonin (5-HT), a neurotransmitter that regulates mood, cognition, and immune function [183], is typically reduced in MS patients, correlating with comorbidities such as depression and anxiety [184, 185]. Serotonin can modulate immune responses by acting on various immune cells, such as T cells and dendritic cells, influencing cytokine production and immune cell activity, crucial for the inflammatory processes in MS. Selective serotonin reuptake inhibitors (SSRIs) have demonstrated efficacy in reducing the severity of EAE by regulating immune responses [186, 187]. The gut microbiota has an influence on serotonin production in the intestines, thereby affecting systemic serotonin levels and immune functions [12, 158]. Changes in gut microbiota may impact serotonin generation, influencing systemic immune functions and potentially affecting MS progression [12, 188]. 5-HT has been identified as a regulator of ferroptosis [189]. Consequently, fluctuations in its endogenous levels may also impact the progression of MS through modulation of the ferroptosis.
Targeting Ferroptosis: Targeting ferroptosis offers a promising avenue for MS treatment. In the EAE mouse model, using ferroptosis inhibitors to target ferroptosis or reduce the expression of ACSL4 has shown to improve behavioral phenotypes, reduce neuroinflammation, reverse iron overload, and inhibit demyelination, ultimately preventing neuronal death [89, 190]. Treatment with the ferroptosis inhibitor Fer-1 markedly reverses oligodendrocyte death and demyelination [21]. In a preclinical model of RRMS, the ferroptosis inhibitor UAMC-3203 has been shown to delay relapses and enhance disease management, underscoring the potential of ferroptosis modulation in MS therapy [98].
Targeting the Gut Microbiota: Animal studies suggest that probiotic treatments can stimulate anti-inflammatory cytokine IL-10 production by T regulatory type 1 (Tr1) cells, reduce central nervous system inflammation, and decrease autoreactive T cell responses, thereby ameliorating EAE [191, 192]. In patients with MS, probiotics have been shown to increase the levels of beneficial taxa such as Lactobacillus while reducing MS-associated taxa like Akkermansia and Blautia [193]. MS patients treated with Fecal Microbiota Transplantation (FMT) for constipation not only experience alleviation of gastrointestinal symptoms but also improvement in neurological symptoms [194]. This evidence opens the door to personalized medicine approaches that could tailor microbiota-targeted therapies based on individual microbial profiles. By analyzing the unique microbiota compositions of MS patients, treatments could be customized to enhance beneficial bacteria and suppress harmful species, potentially leading to more effective management of symptoms in MS.
Supplementing Microbiota-Derived Products: Supplementing with microbiota-derived metabolites like SCFAs and tryptophan metabolites is another potential therapeutic approach. SCFAs have improved symptoms in EAE mice, whereas long-chain fatty acids (LCFA) have exacerbated them [195, 196]. Supplementing untreated MS patients with propionic acid (PA) as an adjunct to MS immunotherapy has shown promising results. After two weeks of PA intake, there is a significant and sustained increase in functional Treg cells, and a significant decrease in Th1 and Th17 cells. After three years of PA treatment, the annual relapse rate decreases, disability stabilizes, and brain atrophy is reduced [172]. Oral butyrate significantly inhibits lysolecithin-induced demyelination and enhances myelin regeneration, promoting oligodendrocyte differentiation [197]. Similarly, dietary supplementation with tryptophan metabolites has improved symptoms in EAE mice, while the use of antibiotics to suppress tryptophanase-positive bacteria, reducing tryptophan metabolite levels, has worsened EAE scores [198].
Optimal Timing for Therapeutic Interventions: Determining the optimal timing for therapeutic interventions targeting ferroptosis in MS involves several complex considerations. Ferroptosis has been shown to contribute to disease progression from early stages [98], suggesting that early targeting could be beneficial. Additionally, treatments with ferroptosis inhibitors have delayed relapse and ameliorated disease progression in preclinical models of relapsing-remitting MS [98], indicating value during active disease phases and in preventing progression. When considering combination with current therapies, inhibiting ferroptosis could supplement existing immunosuppressive strategies, though the optimal timing likely depends on coordination with these treatments. However, the long-term effects of inhibiting ferroptosis are not yet fully understood, requiring careful evaluation of safety profiles to determine the appropriate timing and duration of treatment [199]. Clinical trials will be crucial in determining the most effective treatment protocols targeting ferroptosis in MS.
This review has illuminated the intricate interplay between ferroptosis, an iron-dependent form of regulated cell death, and gut microbiota, underscoring their significant roles in the pathogenesis and progression of Multiple Sclerosis (MS). We have explored how gut microbiota-induced dysbiosis exacerbates ferroptotic processes by altering iron metabolism and inflammatory responses, thus impacting neurological health. Addressing these mechanisms, novel therapeutic strategies including probiotics, microbiota-derived metabolite supplementation, and ferroptosis inhibitors show promise in altering disease progression and improving clinical outcomes. Future research should aim to identify precise biological markers within these pathways and assess the long-term effects of such therapies on MS, paving the way for tailored and effective patient care.
JJ and JW contributed to writing and editing of this review. JW and JJ collected the information. Both authors read and approved the final manuscript. Both authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.