







1 Department of Cardiology, Affiliated Hospital of Jiangnan University, 214122 Wuxi, Jiangsu, China
Abstract
Myocardial ischemia-reperfusion (I/R) injury refers to cell damage that occurs as a consequence of the restoration of blood circulation following reperfusion therapy for cardiovascular diseases, and it is a primary cause of myocardial infarction. The search for nove therapeutic targets in the context of I/R injury is currently a highly active area of research. p70 ribosomal S6 kinase (S6K1) plays an important role in I/R induced necrosis, although the specific mechanisms remain unclear.
This study aims to explore the effects of inhibiting S6K1 on myocardial I/R injury and its potential mechanisms.
A rat myocardial I/R model was created and treated with the S6K1-specific inhibitor PF-4708671. Hematoxylin-eosin (H&E) staining was applied to evaluate the pathological changes in cardiac tissues. 2,3,5-triphenyltetrazolium chloride (TTC) staining was used to measure the area of myocardial infarction (MI). Left ventricular systolic pressure (LVSP), left ventricular end-diastolic pressure (LVEDP), the maximum rate of increase in left ventricular pressure (+dp/dtmax), and the maximum rate of the decrease in left ventricular pressure (-dp/dtmax) were measured using ultrasonic echocardiography. The expression levels of cardiac troponin-1 (cTn-1), lactate dehydrogenase (LDH), creatine kinase MB (CK-MB), and aspartate aminotransferase (AST) were determined by enzyme-linked immunosorbent assay (ELISA). Terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) staining and propidium iodide (PI) staining were used to examine the apoptosis and necrosis of myocardial tissues. The expressions of apoptotic-related proteins, and key molecules of necrosis were detected by western blot. The relationship between S6K1 and receptor-interacting protein kinase 3 (RIP3) was analyzed by immunoprecipitation.
Inhibition of S6K1 reduces I/R-induced myocardial tissue damage, improves myocardial function, and inhibits myocardial tissue necrosis (p < 0.05). In addition, RIP3 is a direct target of S6K1, and activation of RIP3 blocked the protective effect of the S6K1 inhibitor PF-4708671 against myocardial I/R injury (p < 0.05).
Inhibition of S6K1 protects against myocardial I/R injury by down-regulating RIP3, suggesting that targeting S6K1 may offer a novel approach for intervention in myocardial I/R injury.
Keywords
- necrosis
- ischemia-reperfusion injury
- myocardial infarction
- PF-470867
- RIP3
- S6K1
Acute myocardial infarction (AMI) ranks among the primary contributors to illness and death, affecting approximately 17.6 million individuals worldwide [1]. According to the China Disease Report (2020), the cardiovascular disease-related mortality rate is approximately 45%, and nearly 23 million individuals are expected to have AMI by 2030 [2]. AMI is ischemia caused by decreased blood flow, leading to myocardial cell damage [3]. It is generally believed that the most effective treatment for ischemic heart disease is to re-establish blood flow and reoxygenation in the ischemic myocardial tissue [4]. However, the process of restoring blood flow and reoxygenating tissues can trigger inflammation, platelet activation, and excessive oxidative stress, leading to more severe microstructural destruction known as myocardial ischemia/reperfusion (I/R) injury [5, 6]. Therapeutic strategies for patients with myocardial I/R injury are currently being actively developed to minimize the time from portal to primary percutaneous coronary intervention. In addition, various alterations have been suggested, such as enhancements in early reperfusion efforts, advancements in percutaneous coronary intervention methods, adjustments to the formulation of reperfusion fluids, and administration of free radical scavengers and antithrombotic and antiplatelet agents. However, practical strategies for preventing myocardial I/R injury are still limited [7, 8]. Thus, it is important to study the pathogenesis of myocardial I/R injury and find new targets for intervention strategies for myocardial I/R injuries.
The mechanistic target of rapamycin complex 1 (mTORC1) and its downstream effector, p70 ribosomal S6 kinase (S6K1), are key regulators of energy homeostasis [9, 10]. mTORC1/S6K1 regulates various physiological processes, including cell proliferation, apoptosis, protein synthesis, and glucose homeostasis, which are tightly regulated according to the availability of substrates [10, 11]. mTORC1/S6K1 pathway plays a crucial role in many human diseases [12]. In particular, the mTORC1/S6K1 signaling pathway is closely related to cell necrosis and autophagy [13, 14]. Furthermore, it has been shown that S6K1 plays a crucial role in I/R injury [15]. PF-4708671 (an S6K1 inhibitor) relieves blood-brain barrier (BBB) damage and reduces the myocardial infarction (MI) area in early I/R [16]. However, the mechanism of the protective effect of S6K1 inhibition on myocardial I/R injury still needs to be further investigated.
The occurrence of myocardial I/R injury involves various factors and mechanisms,
such as intracellular calcium overload, energy metabolism disturbances,
programmed cell death, and inflammatory responses. These elements are
interconnected and influence one another [17]. Necroptosis is a form of
programmed death that distinguishes itself from traditional cell necrosis and
apoptosis. This process is inflammatory in nature and is marked by cell swelling
and the rupture of the cell membrane, leading to cell disintegration. As a
result, cellular contents are released, which triggers both innate and adaptive
immune responses [18]. Receptor-interacting protein kinase 1 (RIP1) is a
significant contributor to cell survival, apoptosis, and inflammatory responses.
Additionally, it has been recognized as a key factor in the process of
necroptosis [19]. Receptor-interacting protein kinase 3 (RIP3) along with its
substrate, mixed lineage kinase domain-like protein (MLKL), are essential
molecules in the necroptosis process. A crucial aspect of this pathway involves
the movement of phosphorylated MLKL to the inner portion of the cell membrane,
which subsequently leads to the disruption of membrane integrity [20]. Earlier
research suggests that N
Therefore, this research involved the creation of a model for ischemia-reperfusion injury, which was subsequently treated with PF-4708671, a specific inhibitor of S6K1. Relevant mechanism experiments confirmed that RIP3 was a direct target of S6K1. In addition, further studies showed that S6K1 inhibition reduced myocardial I/R injury by down-regulating the phosphorylation of RIP3 and thus played a protective role. These findings have revealed the potential function of inhibition of S6K1, suggesting that inhibition of S6K1 might become a new target for intervention strategies during myocardial I/R injuries.
Thirty 10-week-old Sprague Dawley male rats, weighing 200
The following five groups were randomly divided to all rats: (1) Sham group: The rats underwent a sham operation. (2) Ischemia-reperfusion (I/R) group (I/R model rats): The rats underwent an ischemia-reperfusion operation. (3) I/R+saline group: I/R model rats were intraperitoneally administrated with saline (1:10, HBPT033, Qingdao Haibo Biological, Qingdao, China). (4) I/R+PF-4708671 group: I/R model rats were intraperitoneally administrated with 75 mg/kg PF-4708671 (ZY6M2271, Shanghai Zeye Biotechnology Co., Ltd., Shanghai, China) (PF-4708671 was injected intraperitoneally for 1 day before ischemia). (5) I/R+PF-4708671+RIP3 group: Administer an intraperitoneal injection of 50 mg/kg RIP3 activator to the I/R+PF-4708671 treatment group of rats (HY-144828, MedChemExpress, Middlesex County, NJ, USA) (RIP3 activator was injected intraperitoneally at the beginning of the ischemia–reperfusion operation). PF-4708671 was dissolved in a saline solution as previously described [16].
The model for MI/R injury was developed following the previously outlined methodology [23]. Briefly, the rats were anaesthetized with 3% isoflurane (132220, Klamar, Shanghai, China) using a gas inhalation anesthesia machine (ABS-6A, YUYAN INSTRUMENTS, Shanghai, China). Following tracheotomy, a 7-0 nylon suture was used to ligate the left anterior descending artery (LAD), and a silicone tube (outer diameter = 86 mm) was inserted 1 mm below the ligation to induce myocardial ischemia. The success of the occlusion was confirmed by ligating the distal ventricle until it appeared white. After 30 minutes of occlusion, the silicone tube was removed to facilitate blood reperfusion for a duration of 2 hours, the restoration of myocardial color after reperfusion indicates that the I/R model has been successfully established [24, 25]. Then, 2 hours after surgery, anesthesia was provided by intraperitoneal injection of 1% pentobarbital sodium (40 mg/kg) (20090512, SINOPHARM, Beijing, China). Cervical dislocation: cervical dislocation is performed manually and euthanasia is achieved in approximately 10–15 seconds. The heart tissue and blood samples were collected for pathological evaluation and biochemical analysis, respectively.
Heart tissues were fixed in 4% formaldehyde (P6148, Sigma-Aldrich, St. Louis, MO, USA) for 48 h, dehydrated in an alcohol gradient series, and paraffin-embedded. Tissues were cut into approximately (~5-µm slices) and subsequently stained with hematoxylin (ST2023, Saint-Bio, Shanghai, China) for 3 minutes, followed by eosin for 30 seconds. Stained tissue sections were sealed with neutral resin and stored at room temperature. Heart tissue pathological assessments were performed under a light microscope (BX43, Olympus, Tokyo, Japan).
Myocardial infarction (MI) areas were measured by 2,3,5-triphenyltetrazolium
chloride (TTC, BB-44549, Bestbio, Shanghai, China) staining. The collected hearts
were quickly sectioned into 5 to 6 thin transverse slices and incubated in 1%
TTC for 30 min at 37 °C to delineate the infarct area from viable
myocardium. The MI region was imaged by a digital camera (v30, Epson, Suwa city, Japan) and quantified using
Image J v1.8.0 software (NIH, Madison, WI, USA). Infarct size (%) was calculated
using the following equation: Infarct myocardium area/(infarct myocardium area +
normal myocardium area)
After 30 minutes of occlusion, two hours of reperfusion were performed.
Echocardiography was performed in mice 3 days after I/R to detect cardiac
function. Isoflurane (0.5–1.5% in oxygen) anesthesia lasts 10–15 min and
positioned on an orbital system to ensure that their body
temperature was maintained at 37 °C
Blood were taken and spun at 5000 rpm for 10 minutes. Plasma samples were collected, and 20 µL of each sample was reacted with 200 µL of the working solution to identify cardiac troponin-1 (cTn-I), lactate dehydrogenase (LDH), creatine kinase-MB (CK-MB), and aspartate aminotransferase (AST) were measured using ELISA kits (cTn-I, yb-E12746, Shanghai Yubo Biotechnology Co., Ltd., China; LDH, SP12758; CK-MB, SP12933; AST, SP12839, Wuhan Saipei Biotechnology Co., Ltd., Wuhan, China).
APO-BrdU™ TUNEL assay kit (A23210, Thermo Fisher, Mountain View, CA, USA) was utilized to conduct TUNEL staining for the identification of apoptotic cells. Tissue sections (~5 µm) were permeabilized in 10% Triton for 10 min and stained with TUNEL staining solution for 1 h. 4,6-dianmidino-2-phenylindole (DAPI) staining (PM11055, PERFEMIKER, Shanghai, China) was performed on each section for a duration of 15 minutes at room temperature. Subsequently, the slices were examined and imaged using a fluorescence microscope (Ti2-U, Nikon, Tokyo, Japan).
Extracted proteins were subjected to denaturation in a loading buffer and
boiled. The samples (~40 µL) were resolved on sodium
dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to
polyvinylidene fluoride (PVDF) membranes (1620177, Bio-rad, Hercules, CA, USA). The membranes
were probed with antibodies against RIP3 (ab226297, 1:500, Abcam, Cambridge, UK),
p-RIP3 (phospho T231 + S232) (ab222320, 1:500, Abcam, Cambridge, UK), S6K1
(ab32529, 1:500, Abcam, Cambridge, UK), p-S6K1 (phospho T229) (ab59208, 1:500,
Abcam, Cambridge, UK), Bcl-2 (ab32124, 1:500, Abcam, Cambridge, UK), Bax
(ab32503, 1:500, Abcam, Cambridge, UK), cleaved-caspase3 (ab32042, 1:500, Abcam,
Cambridge, UK), caspase3 (ab32351, 1:500, Abcam, Cambridge, UK), RIP1 (ab202985,
1:500, Abcam, Cambridge, UK), P-MLKL (ab196436, 1:500, Abcam, Cambridge, UK),
MLKL (ab184718, 1:500, Abcam, Cambridge, UK) and
Following the blood collection, the hearts were preserved in 4% paraformaldehyde (P6148, Sigma-Aldrich, USA), buried in paraffin, and then cut into measuring 3–4 µm, which were placed on slides. The resulting sections were subsequently stained with DAPI (PM11055, PERFEMIKER, Shanghai, China)/propidium iodide (PI) (HY-D0815, MedChemExpress, NJ, USA) in accordance with the provided protocol. Observations and photographic documentation were conducted using a fluorescent microscope (Ti2-U, Nikon, Tokyo, Japan).
Frozen heart tissues were lysed in non-denaturing lysis buffer (20 mM, pH = 8.0, Tris HCl, 100 mM NaCl, 1 mM EDTA, 0.5% Nonidet P-40) supplemented with Calbiochem® Protease and Phosphatase Inhibitor Cocktails (Calbiochem, Merck, Darmstadt, Germany). Then the samples were pre-cleared with protein G-immobilized agarose for 1 h at 4 °C, followed by incubation with S6K1 or RIP3 antibodies at 4 °C overnight. The samples were then subjected to SDS-PAGE/immunoblotting. The primary antibodies against S6K1 (1:1000, AB 32529, Abcam, Cambridge, UK), RIP3 (1:1000, AB 226297, Abcam, Cambridge, UK), and rabbit IgG, Isotype Control (1:1000, Ab172730, Abcam, Cambridge, UK) were added to the experiment. Goat anti-rabbit IgG H&L (HRP) (1:1000, ab6721, Abcam, Cambridge, UK) was used as the secondary antibody.
Each experiment was repeated three times in this study, and the results were expressed as means and standard deviations (SD). Statistical analysis of the data was conducted using GraphPad Prism 7 software (Dotmatics, San Diego, CA, USA). Multigroup comparisons were conducted using one-way analysis of variance (ANOVA) with Tukey’s post hoc test, while comparisons between two groups were performed using the t-test. Normal distribution data were subsequently analyzed using the Bonferroni test. A p-value of less than 0.05 was regarded as statistically significant. Unless indicated otherwise, all n values pertain to mice and are specified in the figure legends.
First, a western blot experiment showed that p-S6K1 (Thr389/Thr412) in the
I/R+PF-4708671 group was markedly down-regulated compared with the other three
groups. The results showed that PF-470867 inhibited S6K1 activity (Fig. 1A)
(p
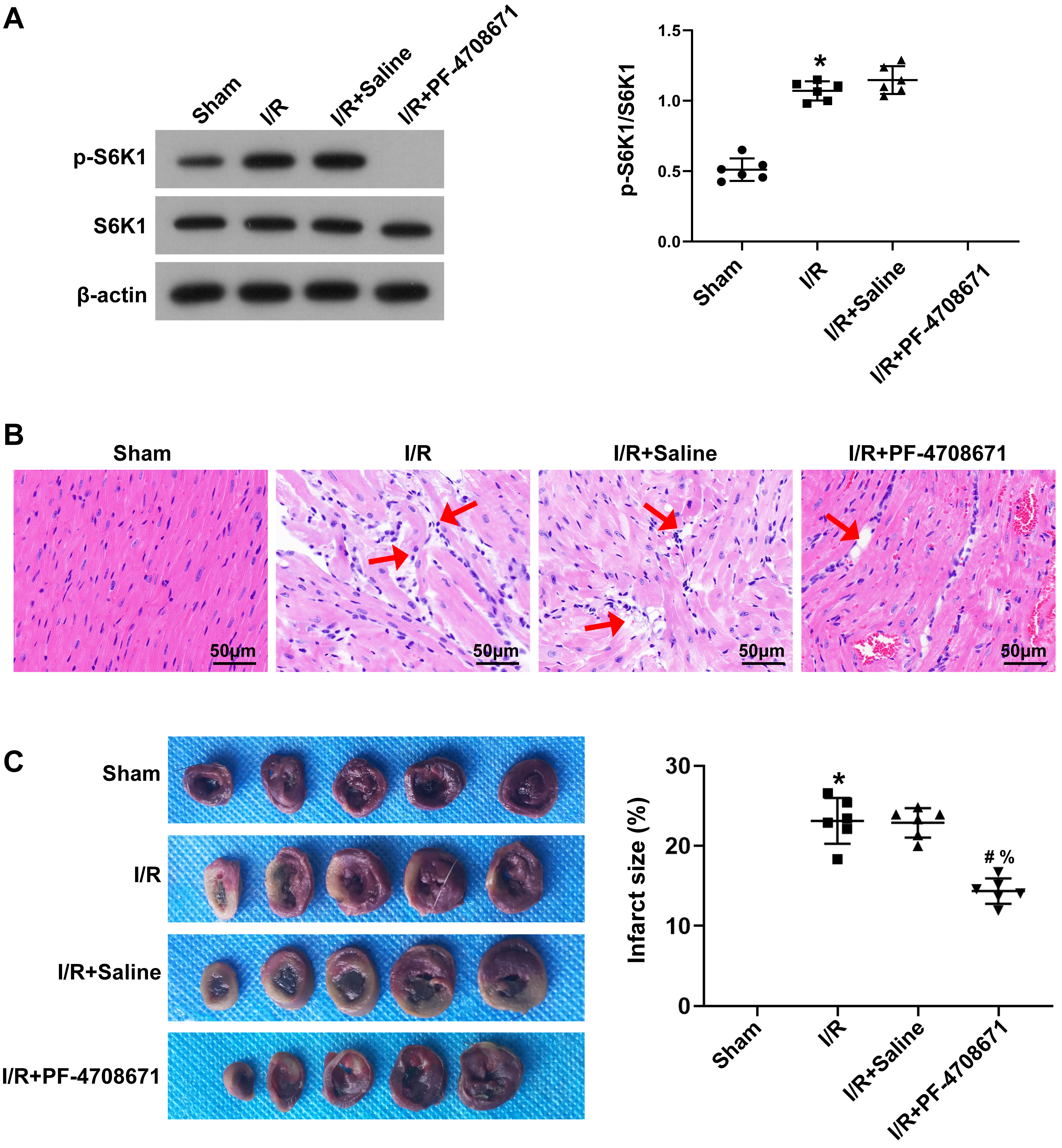 Fig. 1.
Fig. 1.
Inhibition of S6K1 mitigated I/R-induced myocardial injury. (A)
Expression of p-S6K1; S6K1 was detected by western blotting (n = 6 rats each).
(B) H&E staining of myocardial tissue (n = 6 rats each). Red arrows, interstitial edema and inflammatory infiltration. Scale bars, 50
µm. (C) Myocardial infarct size was assessed by TTC staining (n = 6 rats
each) (*p
Echocardiography was used to determine LVSP, -dp/dtmax, LVEDP, and +dp/dtmax
levels to evaluate the effect of S6K1 inhibition on myocardial function in I/R
rats. As shown in Fig. 2A, compared with the sham group, the expression of LVSP
and
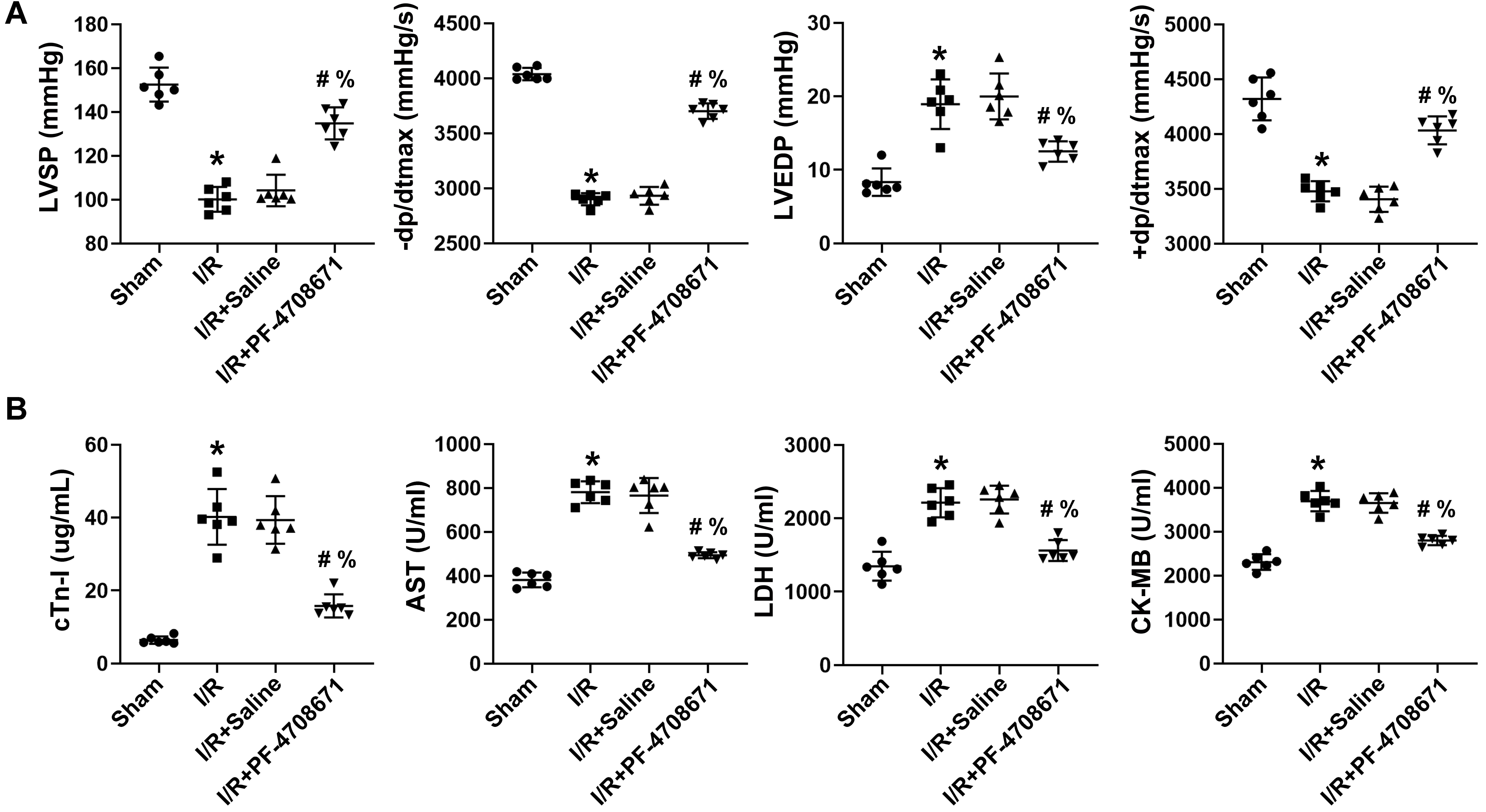 Fig. 2.
Fig. 2.
Inhibition of S6K1 improved myocardial function. (A) The LVSP,
LVEDP, +dp/dtmax, and -dp/dtmax levels of the sham, I/R, I/R + saline solution,
and I/R + PF-4708671 groups were measured (n = 6 rats each). (B) cTn-I, AST, LDH,
CK-MB levels of the above four groups were measured (n = 6 rats each)
(*p
Cardiomyocyte apoptosis was assessed by TUNEL staining, which showed a
pro-apoptosis effect following I/R induction compared to the sham group, which
was reversed by PF-4708671 treatment (Fig. 3A) (p
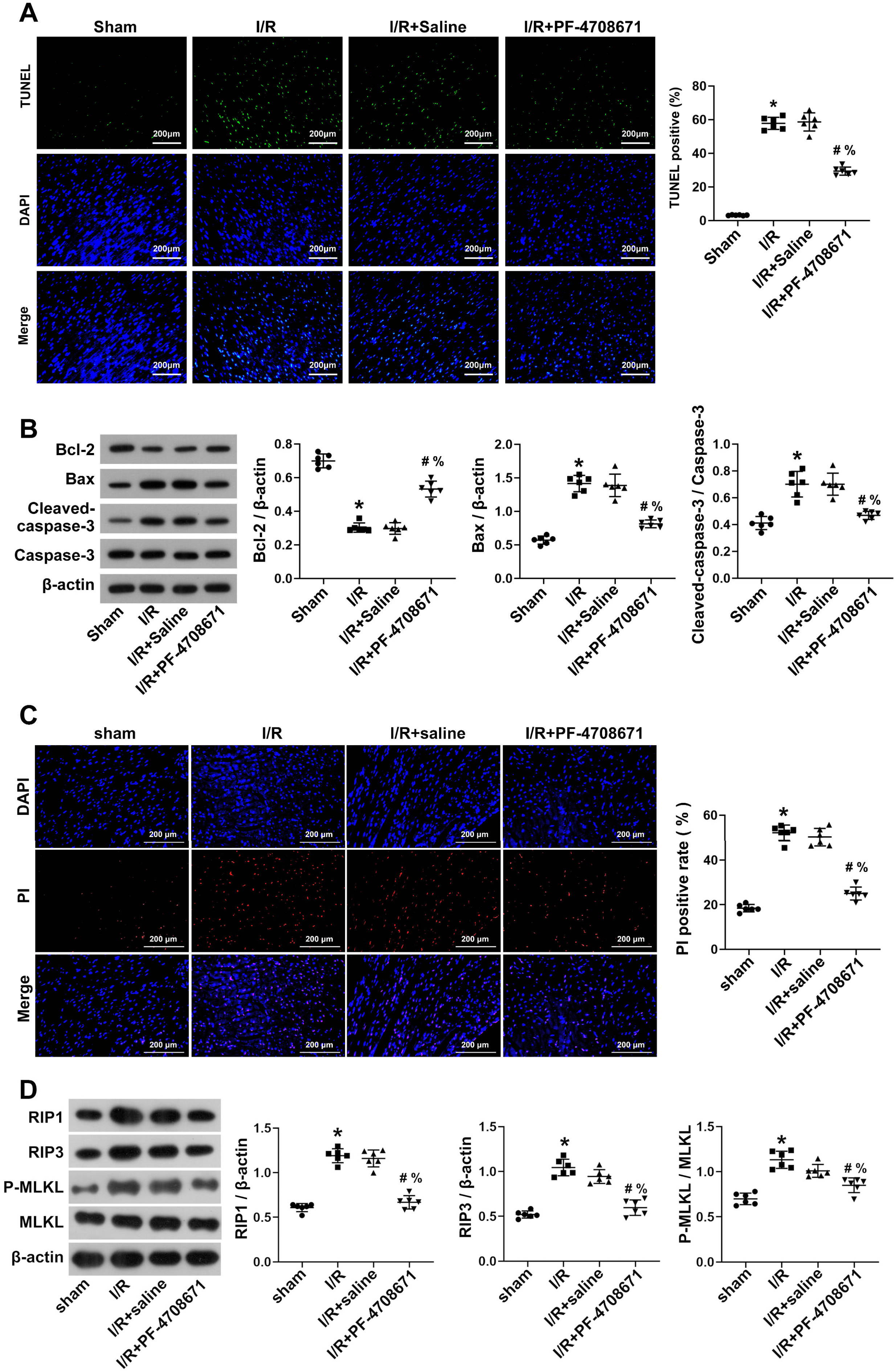 Fig. 3.
Fig. 3.
Inhibition of S6K1 activity ameliorated myocardial apoptosis and
necrosis in I/R rats. (A) Myocardial apoptosis was detected by the TUNEL
staining (n = 6 rats each). Scale bars, 200 µm. (B) Bcl-2, Bax,
cleaved-Caspase-3, and Caspase-3 in the myocardial tissue were detected by
western blotting (n = 6 rats each). (C) The necrosis of I/R rat cardiomyocytes
assessed by DAPI–PI staining (n = 6 rats each). Scale bars, 200 µm. (D)
Western blot analysis was used to assess the expression of key molecules for
necrosis in myocardial tissue (n = 6 rats each) (*p
To determine whether PF-4708671 improves necrosis in cardiomyocytes of I/R rats,
we conducted DAPI-PI staining and assessed necrosis markers. The DAPI–PI
staining results showed that compared with the sham group, the number of necrosis
cells in the I/R group was markedly increased, while the number of necrosis cells
decreased after treatment with PF-4708671 (Fig. 3C) (p
Concerning the relationship between RIP3 and S6K1, protein blot analysis showed
that after I/R induction, the expression of phosphorylation-S6K1/S6K1 and
phosphorylation-RIP3/RIP3 increased significantly compared to the sham group;
however, PF-4708671 treatment reversed the effects (Fig. 4A) (p
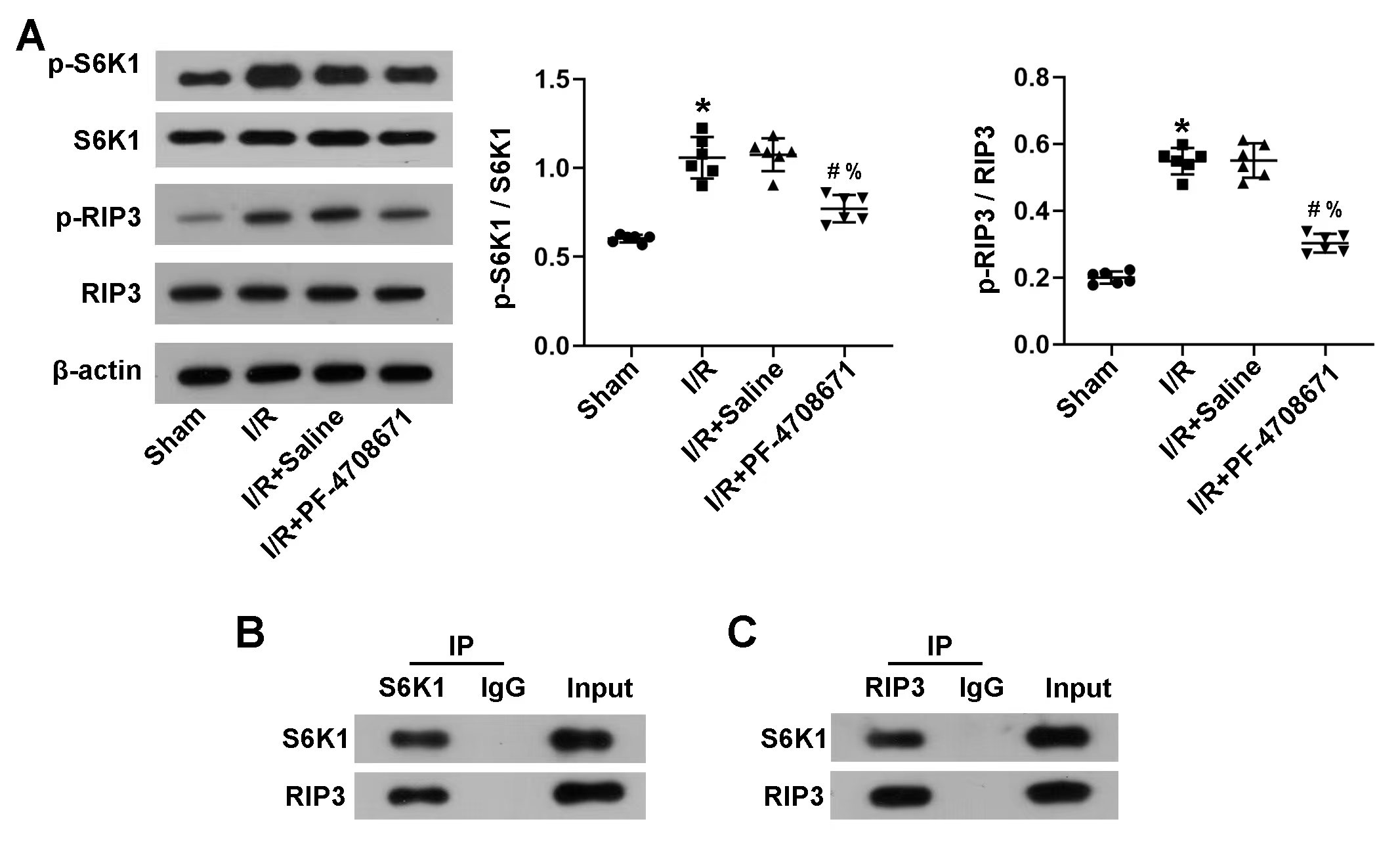 Fig. 4.
Fig. 4.
RIP3 was a direct target of S6K1. (A) p-S6K1, S6K1, p-RIP3, and
RIP3 were detected in the myocardial tissue by western blotting (n = 6 rats
each). (B,C) Co-immunoprecipitation was performed in the myocardial tissue. The
protein complex was downregulated by S6K1 (B) or RIP3 (C) or immunoglobulin G
(IgG), followed by immunoblotting with S6K1 and RIP3 antibodies, respectively.
IP, Immunoprecipitation. (*p
To clarify whether the activation of RIP3 can block the protective effect of
S6K1 inhibition on myocardium I/R injury, we treated I/R-induced myocardial
tissue with a RIP3 activator for subsequent experiments. H&E staining showed
that the rats in the I/R + PF-4708671 + RIP3 activator 1 group exhibited
significantly aggravated interstitial edema and inflammatory infiltration
compared with the I/R+PF-4708671 group (Fig. 5A). Subsequently, we carried out
TTC staining on I/R-induced myocardial tissue after treatment with RIP3
activator. The results showed that compared with the I/R+PF-4708671 group, the
infarct size increased after treatment with RIP3 activator (Fig. 5B) (p
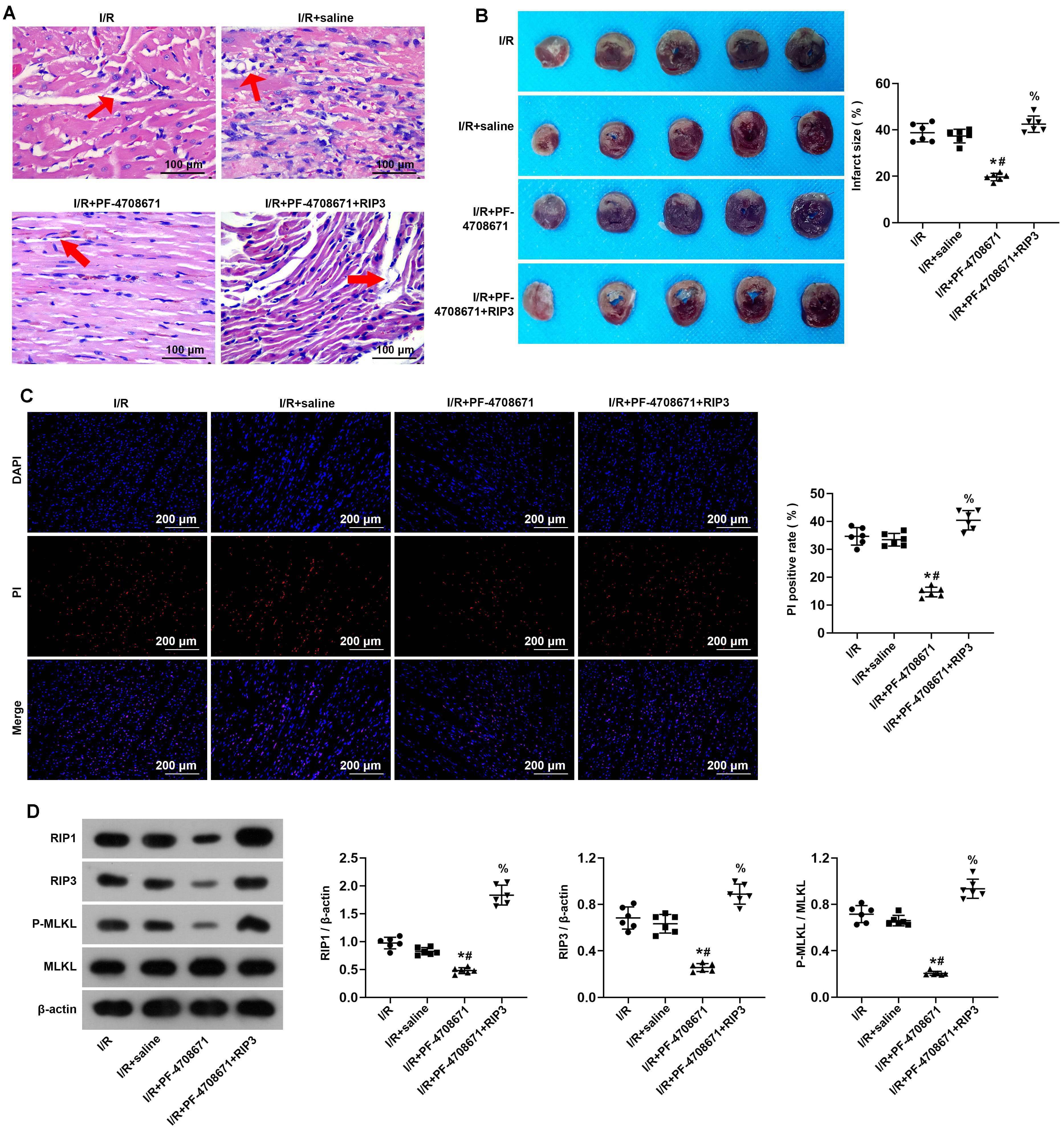 Fig. 5.
Fig. 5.
Activation of RIP3 blocked inhibition of S6K1 effects on
myocardium ischemia/reperfusion injury. (A) Myocardial tissue was detected by
Hematoxylin-Eosin staining (n = 6 rats each). Red arrows, interstitial edema and inflammatory infiltration. Scale bars, 100 µm. (B) The
size of the myocardial infarction was assessed by TTC staining (n = 6 rats each).
(C) I/R rat cardiomyocyte necrosis assessed by DAPI-PI staining (n = 6 rats
each). Scale bars, 200 µm. (D) The expression of key molecules for necrosis
in myocardial tissue assessed by Western blot analysis (n = 6 rats each)
(*p
Myocardial I/R injury is an inevitable and malignant consequence of several cardiac operations, leading to myocardial necroptosis [26]. I/R injury contributes to cardiac damage through multiple mechanisms. Oxidative stress, apoptosis, and fibrosis are all intricately linked to I/R injury, potentially resulting in heart failure [27, 28]. Although progress has been made in the management of myocardial I/R injury, the potential mechanism of I/R-induced myocardial injury has not been fully elucidated. However, the mechanism of occurrence and development of myocardial I/R injury is of great clinical significance to pave the way for its early diagnosis and treatment.
mTORC1 is an atypical serine/threonine protein kinase that promotes cell growth by phosphorylating various proteins, including S6K1. The mTORC1-S6K1 pathway is overactivated in various cancers, including hematological malignancies, making it a key target for developing pharmacological inhibitors. However, inhibiting mTORC1 with rapamycin has positive and negative effects after cerebral ischemia-reperfusion [29, 30, 31]. These discrepancies suggest that rapamycin has more than one target of mTORC1; it can also inhibit mTORC2 and Akt activation [29]. In addition, it means that the mTORC1 downstream targets may play different roles in regulating cell apoptosis and autophagic cell death [32]. S6K1 is a downstream effector of mTORC1, and PF-4708671 is the first reported S6K1-specific inhibitor. Most studies have confirmed that PF-4708671 inhibits the effect of S6K1 during the ischemia-reperfusion process. For example, by using PF-4708671, the specific inhibitor of S6K1 to suppress the activity of S6K1, it can decrease the cortical infarct size of the cerebral cortex and reduce BBB disruption during I/R of rats undergoing middle cerebral artery occlusion (MCAO) surgery [16]. Lysophosphatidic acid increased infarct size and BBB permeability in the early stages of cerebral ischemia-reperfusion, which was reversed by F-4708671 pretreatment [25]. Inhibiting S6K1 may result in a reduction of infarct size and enhance the balance of O2 supply and consumption in specific microregions following cerebral ischemia-reperfusion [32]. In this study showed that inhibiting S6K1 improved the I/R-induced myocardial injury and myocardial function in rats; it also inhibited the increase in infarct size necrosis, exerting a protective effect on myocardial I/R injury.
Necroptosis is recognized as a new form of programmed cell death that significantly influences the regulation of various acute and chronic illnesses [33]. The onset and implementation of necroptosis rely on the activation of various kinases, such as RIP1, RIP3, and MLKL. When cells experience stimulation from either internal or external sources, these kinases can assemble into a complex known as necrotrophic apoptotic vesicles [34]. In the complex, RIP1 and RIP3 interact by binding via their receptor-interacting protein isoform interaction motifs, and they become activated through a sequence of autophosphorylation and trans-phosphorylation events. Once activated, RIP3 phosphorylates the pseudokinase structural domain of MLKL at threonine 357 and serine 358, resulting in the formation of phosphorylated MLKL, which is essential for activating MLKL [20]. In agreement with earlier research, we also verified that necroptosis plays a role in the pathological progression that occurs after myocardial I/R [35, 36]. Of note, our current study also indicates that inhibiting S6K1 activity improved myocardial necrosis in I/R rats. In addition, the apoptotic pathway is also influenced by PF-4708671, as inhibiting S6K1 activity reduces apoptosis in cardiomyocytes.
It has been reported that RIP3 is an important mediator of ischemia- and
oxidative stress-induced myocardial apoptosis and necroptosis [23].
Phosphorylated RIP3 mediates apoptosis and necroptosis during various abnormal
physiopathological processes [37]. In fact, RIP3 has become an important target
for multifaceted disease therapy [37]. In addition, RIP3 has been demonstrated to
be involved in various I/R injuries; for example, in renal I/R injuries,
MicroRNA-124 is reported to downregulate poly (ADP-ribose) polymerase 1 (PARP1),
which helps mitigate renal ischemia-reperfusion damage by blocking the
TNF
In summary, we created a rat myocardial I/R model and demonstrated that inhibiting S6K1 ameliorated I/R-induced myocardial injury and myocardial function in rats, reduced infarct size, and inhibited cardiomyocyte necrosis. Furthermore, RIP3 was the direct target of S6K1, and activation of RIP3 blocked the protective effect of the S6K1 inhibitor PF-4708671 against myocardial I/R injury. In conclusion, the present study suggests that inhibiting S6K1 protect against I/R-induced myocardial necrosis by downregulating RIP3, inhibiting S6K1 may be a new target for intervention strategies during myocardial I/R injury.
The data used to support the findings of this study are available from the corresponding author upon request.
HS and XYW designed the research study and wrote the first draft. JJS and JZ performed the research. JZ and YFG analyzed the data. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
The Ethics Committee of Affiliated Hospital of Jiangnan University granted approval for the study (JN. No20240630), in alignment with the “Guide for the Care and Use of Laboratory Animals” issued by the US National Institutes of Health.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.