











1 Department of Cardiology, The Second Affiliated Hospital of Chongqing Medical University, 401336 Chongqing, China
2 Department of Cardiovascular Medicine, Southwest Hospital, Army Medical University, 400038 Chongqing, China
3 Emergency Department, The Second Affiliated Hospital of Chongqing Medical University, 401336 Chongqing, China
4 Department of High Altitude Physiology and Pathology, College of High Altitude Military Medicine, Army Military Medical University, 400037 Chongqing, China
Abstract
Myocardial ischemia-reperfusion (I/R) injury and coronary microcirculation dysfunction (CMD) are observed in patients with myocardial infarction after vascular recanalization. The antianginal drug trimetazidine has been demonstrated to exert a protective effect in myocardial ischemia-reperfusion injury.
This study aimed to investigate the role of trimetazidine in endothelial cell dysfunction caused by myocardial I/R injury and thus improve coronary microcirculation.
The myocardial I/R mouse model was established, and trimetazidine was administered for 7 days before myocardial I/R model establishment. Echocardiography, 2,3,5-triphenyltetrazolium chloride (TTC) staining, hematoxylin-eosin (H&E) staining, and thioflavin S staining were applied to assess myocardial injury and microvascular function. Additionally, the oxygen-glucose deprivation/reperfusion (OGD/R) model was developed in endothelial cells to simulate myocardial I/R injury in vitro. Griess reaction method, immunofluorescence, and western blotting (WB) were employed to detect the expressions of nitric oxide (NO), platelet endothelial cell adhesion molecule-1 (CD31) and vascular endothelial (VE)-cadherin, zonula occludens protein 1 (ZO-1), occludin, vascular endothelial growth factor (VEGF) and adenosine monophosphate (AMP)-activated protein kinase (AMPK) signaling-related proteins in endothelial cells and mouse cardiomyocytes. AMPK pathway inhibitor compound C was used for further mechanism validation.
Our research demonstrated that trimetazidine can alleviate myocardial pathological injury and cardiac function injury during myocardial I/R. Trimetazidine was observed to improve microvascular reflux phenomenon and microvascular function and barrier injury in myocardial I/R and OGD/R models. Additionally, the expressions of AMPK signal-related proteins were found to be inhibited in myocardial I/R and OGD/R models, which were then activated in mice administered trimetazidine. However, the effects of trimetazidine on endothelial cell function and barrier damage were attenuated after co-treatment with compound C and trimetazidine.
Trimetazidine ameliorated myocardial I/R-induced CMD by activating AMPK signaling.
Keywords
- trimetazidine
- coronary microcirculation dysfunction
- myocardial infarction
- AMPK signal
Myocardial infarction occurs mainly due to the sudden complete blockage of coronary arteries caused by factors such as atherosclerotic plaque rupture and thrombosis, resulting in the interruption of blood flow to the myocardium and subsequent myocardial cell necrosis [1]. Myocardial ischemia is often a precursor to myocardial infarction and reperfusion is a crucial treatment measure after myocardial infarction [2, 3]. However, reperfusion may also cause injury, which known as myocardial ischemia/reperfusion (I/R) injury [4]. Coronary microvascular dysfunction (CMD) plays an important role in the entire process. It is not only related to the occurrence and development of myocardial infarction but also significantly impacts myocardial reperfusion [5]. During myocardial infarction, the microcirculation is damaged due to thrombosis, vascular spasm, and endothelial cell injury [6]. After reperfusion, even if the large coronary arteries are reopened, the microcirculation may still have problems such as no reflow or slow flow, which affects the effectiveness of reperfusion therapy and increases the risk of adverse events such as myocardial infarction expansion, arrhythmia, and heart failure [7, 8]. CMD is characterized by structural and functional abnormalities of the coronary microvasculature [9]. Coronary microvascular endothelial damage leads to thrombosis and microvascular occlusion, which is one of the key causes of CMD [10, 11]. It is therefore crucial to develop effective strategies for improving coronary microvascular endothelial injury in order to improve the treatment of CMD and myocardial I/R injury.
Trimetazidine, an antianginal drug, has been employed in clinical practice for decades. Recently, trimetazidine has been evidenced to exhibit cardioprotective effects by regulating energy metabolism [12]. Furthermore, trimetazidine has been shown to attenuate pyroptosis in myocardial I/R injury by targeting Gasdermin D (GSDMD) [13]. Early trimetazidine treatment with percutaneous coronary intervention for ST-segment elevation myocardial infarction could reduce myocardial infarction size [14]. Chen and colleagues have elucidated that trimetazidine treatment can alleviate cardiac fibrosis and improve left ventricular dysfunction [15]. These findings suggest that trimetazidine plays a protective role in myocardial injury. However, the question of whether trimetazidine can reduce endothelial cell damage during myocardial I/R remains unanswered.
Collectively, this study aimed to figure out whether trimetazidine can maintain the function of coronary endothelial cells and reduce myocardial I/R injury as well as to discuss its regulatory mechanism. It also aimed to provide a theoretical reference for the selection of drugs for the treatment of CMD and the development of clinical application of trimetazidine.
Male C57BL/6 mice (10 weeks) obtained from the Experimental Animal Center at Yangzhou University were raised for 1 week in a SPF animal laboratory with controlled environmental conditions (at 21–23 °C, 60%–65% humidity). The mice were grouped into Control, Sham, I/R, I/R+Vehicle (10% DMSO) and I/R+trimetazidine groups, with 9 mice in each group. Mice in I/R group were intraperitoneally injecting with 1% pentobarbital sodium (40 mg/kg) for anesthetization and fixed in the supine position. After connected with ventilator, the left chest of the mouse were exposed. The left anterior descending coronary artery (LAD) was ligated with a 7-0 ophthalmic suture to completely block the blood flow. Myocardial ischemia was diagnosed when mice presented with st segment elevation and high-amplitude T waves on the ECG. After 30 min of occlusion, the ligature was released and reperfusion was implemented for 24 h. Mice in the Sham group were opened thorax and pericardium, but were not subjected to ligation. Mice in I/R+trimetazidine group or I/R+Vehicle group were orally treated with either trimetazidine (20 mg/kg/d, Cat HY-B0968A, MCE, Monmouth Junction, NJ, USA) or vehicle before I/R injury for 7 days. All mice were sacrificed at the end of the experiment by cervical dislocation. All animal experiments were approved by the ethical committee of Zhaofenghua Biotechnology [IACUC-20221009-03].
An animal ultrasound imaging system (Vevo 3100, VisualSonics, Canada) was applied to assess left ventricular end-diastolic diameter (LVEDD) and left ventricular end-systolic diameter (LVESD). Left ventricular ejection fraction (EF) as well as left ventricular fractional shortening (FS) was determined by computer algorithms.
2,3,5-triphenyltetrazolium chloride (TTC) staining was applied for the
estimation of myocardial infarct size. The frozen hearts were cut transversally
into 1-mm slices and then exposed to 1 % TTC (Cat G3005,
Servicebio, Wuhan, China) at 37 °C for 10 min.
Image Pro Plus software (Version 6.0, Media Cybernetics, Silver
Springs, MD, USA) was employed to express infarct volume. For statistical
analysis, we calculated the infarct area of the heart at each level and then
multiplied the sum of the infarct area of each heart by the thickness (1 mm) to
obtain the total infarct volume. The size of the infarct area was calculated as
Infarct volume % = (infarct volume / total volume)
Heart tissues were fixed with 4% paraformaldehyde (Cat MKCL5723, Sigma, St. Louis, MO, USA) and paraffin-embedded after 24 hours. Then, the tissues were sectioned into 5-µm sections and were deparaffinized and rehydrated. The sections were stained with hematoxylin solution (Cat 170103, shzychem, Shanghai, China) for 10 min, followed by differentiation in 1% ethanol hydrochloride. The sections were then stained in eosin staining solution (Cat 17372-87-1, shzychem) for 2 min. Finally, the sections were dehydrated and sealed with neutral gum, the results were observed under a microscope.
Thioflavin S (Th-S) staining was employed to measure no-reflow areas [16, 17]. Two hours after the end of the cardiac function test, mice were restrained and injected with 200 µL of 1% Th-S (Cat 1326-12-1, Sigma) was via the tail vein. After 2 min, the mice were anaesthetised and sacrificed (by cervical dislocation), and the heart tissue was removed and fixed in 4% paraformaldehyde overnight. The heart tissue was cut into sections with a thickness of 1 mm and sequentially placed on the slides. The results were captured by virtue of a stereo microscope at 359 nm (M205 FCA, Leica, Wetzlar, Germany).
Nitric oxide (NO) level in tissues or cells was estimated using Griess reagent Kit (Cat ml076941, mlbio, Shanghai, China). Briefly, the heart tissues were homogenized at 4 °C by adding 10-fold volume of RIPA lysate (Cat BL504A, Biosharp, Hefei, China) to the tissue. Tissue samples were then centrifuged at 10,000 g for 15 min at 4 °C, and the supernatant was collected for tissue NO detection. A total of 0.1 mL of tissue or cell supernatant was added to 1 mL of Griess solution. After 15 min, the OD value was determined with the application of a microplate reader (VARIOSKAN LUX, Thermo Scientific, Waltham, MA, USA) at 540 nm.
Primary mouse heart microvascular endothelial cells (HCMECs) were extracted from the mouse. Briefly, the hearts of mice were collected in a sterile environment after mice were euthanized and stored in cold PBS. The hearts were trimmed to eliminate excess tissue and major blood vessels, then cut into small fragments measuring 1–2 mm3. These tissue fragments underwent enzymatic digestion with a combination of collagenase and trypsin at 37 °C for 30 minutes while being gently shaken. The digestion process was halted by adding endothelial cell growth medium containing 10% FBS. The resulting cell suspension was passed through a cell strainer (40–70 µm) and the filtered cells were centrifuged at 1500 rpm for 10 minutes. The cell pellet was resuspended in endothelial cell medium (ECM, Cat 1001, Sciencell, Carlsbad, CA, USA) with 10% FBS (Cat 2068801CP, Thermo Fisher, Waltham, MA, USA) and 1% penicillin-streptomycin (Cat C0222, Beyotime, Shanghai, China) for 3 d at 37 °C with 5% CO2. The isolated endothelial cells were identified by detection of the endothelial cell marker platelet endothelial cell adhesion molecule-1 (CD31). The cells were passaged every two and a half days. All cell lines were validated by short tandem repeats (STR) profiling and tested negative for mycoplasma. Cells were all cultured in a humidified incubator at 37 °C and 5% CO2.
The HCMECs were incubated without glucose and 1% O2-94% N2-5% CO2conditions for 4 h. Then, normal medium was replaced and the cells were cultivated under normal conditions for 6, 12 or 24 h. Varying concentrations of trimetazidine (20, 40, and 80 µM, Cat HY-B0968A, MCE, Monmouth Junction, NJ, USA) were applied for the pre-treatment of cells for 24 h prior to oxygen-glucose deprivation/reperfusion (OGD/R) stimulation. The Adenosine Monophosphate (AMP)-activated protein kinase (AMPK) inhibitor compound C (1 µM, Cat HY-13418A, MCE, Monmouth Junction, NJ, USA) was employed for the pre-treatment of cells for 48 h prior to OGD/R stimulation.
The myocardial tissue sections were deparaffinized and permeabilized using 0.1%
Triton x-100 (Cat 1139ML500, BioFROXX, Germany) for 10 min. Then, the sections
were blocked with 5% BSA (Cat 4240GR500, BioFROXX, Germany) for 1 h at room
temperature. Meanwhile, after different treatment, mouse heart microvascular
endothelial cells (HCMECs) were fixed with 4% paraformaldehyde (Cat MKCL5723,
Sigma, St. Louis, MO, USA) for 20 min and then treated with 0.1% Triton x-100
(Cat 1139ML500, BioFROXX, Germany) for 20 min. After being washed, cells were
blocked in 5% BSA (Cat 4240GR500, BioFROXX, Germany) for 30 min at room
temperature. Subsequently, the sections and cells were exposed to primary
antibodies specific to CD31 (1:100, Ab24590, Abcam, Cambridge, MA, USA), vascular
endothelial (VE)-cadherin (1:100, AF6265, Affinity, Jiangsu, China) and zonula
occludens protein 1 (ZO-1) (1:100, Cat AF5145, Affinity, Jiangsu, China) at 4
°C, followed by cultivation with fluorescein isothiocyanate (FITC) (150
µL) conjugated goat anti-rabbit IgG (Cat GB21301, Servicebio, Wuhan, China)
at room temperature for 1 h. Hoechst33258 staining (Cat C1017, Beyotime,
Shanghai, China) or DAPI staining solution (Cat C1005, Beyotime, Shanghai, China)
was employed for the staining of sections. The photographs were imaged with
Olympus X73 fluorescence microscope (Tokyo, Japan). The number of positive cells
expressing the target protein was analyzed using ImageJ software 1.49 (NIH, Bethesda, MD, USA). The percentage
of cells expressing both CD31 and VE-cadherin was calculated as
CD31+VE-cadherin+-positive area (%) = (number of cells expressing both
CD31 and VE-cadherin/total number of cells)
The heart tissues and endothelial cells were homogenized RIPA
lysates (Cat BL504A, Biosharp, Hefei, China) to obtain total protein. BCA assay
kits (Cat P0012, Beyotime, Shanghai, China) were applied for the quantification
of protein concentration. 30 µg of protein for was added
into 12% SDS-polyacrylamide gels and transferred to PVDF
membranes (Cat IPVH00010, Millipore, Billerica, MA, USA). The
membranes were sealed with 5% BSA (Cat 4240GR500, BioFROXX, Germany) and
subsequently subjected to primary antibodies at 4 °C
overnight. Then the membranes were exposure to secondary antibodies (goat
anti-rabbit IgG, 1:5000, ab150077, Abcam, Cambridge, MA, USA) for
2 h at RT. The results were visualized using an ECL kit (Cat WBKLS0100, Millipore, Billerica, MA, USA) and
analyzed with ImageJ software. The primary
antibodies used in this study included endothelial nitric oxide
synthase (eNOS) (1:1000, 27120-1-AP, Wuhan, China),
phosphorylated eNOS (p-eNOS) (1:1500, AF3247, Affinity, Jiangsu, China),
endothelin-1 (ET-1) (1:1000, 12191-1-AP,
Proteintech, Wuhan, China), ZO-1 (1:5000, 21773-1-AP,
Proteintech, Wuhan, China), Occludin (1:1000,
#91131, CST, Danvers, MA, USA), VE-cadherin (1:1000,
27956-1-AP, Proteintech, Wuhan, China), AMPK (1:2000, 10929-2AP, Proteintech,
Wuhan, China), phosphorylated Adenosine Monophosphate (AMP)-activated protein
kinase (p-AMPK) (1:1500, AP1002, ABclonal, Wuhan, China),
kruppel-like factor 4 (KLF4) (1:1000, ab241666, Abcam, Cambridge, MA, USA),
peroxisome proliferator-activated receptor delta
(PPAR
The CCK-8 assay (Cat PR645, Dojindo, Kumamoto, Japan) was performed to detect the cell proliferation capacity. Cells that inoculated into 96-well plates were cultivated for 24 h. Following indicated treatment, added 10 mL of CCK-8 solution into each well and incubate for additional 2 h. A microplate reader (VARIOSKAN LUX, Thermo Scientific, Waltham, MA, USA) was applied to determine the OD at 450 nm.
Apoptosis was assessed using the double staining with Annexin V-FITC Apoptosis Detection Kit (Cat KCD-T2008, KCT Technology Corporation, Shenzhen, China). Following indicated treatment, 400 µL binding buffer was applied to suspend cells, after which were the exposure to Annexin V-FITC (5 µL) for 10 min and propidium iodide (PI, 10 µL) for 15 min in the dark. Cells were subsequently analyzed using flow cytometry (FACSAria™III, BD Biosciences, San Jose, CA, USA).
The In Vitro Vascular Permeability Assay (96-well) kit (Cat ECM642,
Merck Millipore, Billerica, MA, USA) was employed for cell permeability
detection. Briefly, 125 µL ECM medium was added into the upper chamber to
wash the chamber, and then the chamber was placed at room temperature for 15 min.
Then 100 µL mouse vascular endothelial cells (1
HCMECs angiogenesis was observed using µ-slide Angiogenesis (Cat
190807/4, ibidi, Martin rader, Germany). The µ-slide Angiogenesis
was placed into a sterile petri dish, and 10 µL of Matrigel (Cat
356231, Corning, Corning, NY, USA) on ice was added to the wells of the
µ-slide using a sterile syringe. The µ-slide was then
placed into an incubator for 30 min to allow the gel to solidify. Subsequently,
50 µL of cell suspension at the density of 2
Student’s t-test was applied while differences among three or more
groups were demonstrated with one-way ANOVA followed by Tukey’s post hoc test (GraphPad Prism 8, La Jolla, CA, USA)).
p less than 0.05 was supposed to indicate statistical significance. All
data were exhibited in the format of mean
The echocardiography results showed that compared with the Sham group, EF and FS were greatly decreased, and LVESD was significantly increased after I/R modeling, while there were no significant changes in the LVEDD, indicating that modeling was successful. Following trimetazidine treatment, EF and FS were evidently increased (Fig. 1A,B). TTC staining was utilized for the estimation of myocardial ischemic area. The results showed that by contrast with Sham group, the heart of mice had obvious ischemia after I/R modeling, which was then decreased by trimetazidine treatment, with significant difference (Fig. 1C). The observation of the pathological changes in hearts was implemented with hematoxylin-eosin (H&E) staining. It was discovered that the morphology of cardiomyocytes in the Control group and the Sham group was intact, with normal tissue structure. The myocardium of the I/R group was disorganized, infiltration by numerous inflammatory cells, and a considerable number of myocardial cell necrotic changes, accompanied by hyperplasia of fibrous tissue. The pathological changes observed in the I/R+Vehicle and I/R groups were found to be similar. Following the pre-treatment with trimetazidine, the infiltration of inflammatory cells and necrotic cardiomyocytes were markedly reduced (Fig. 1D).
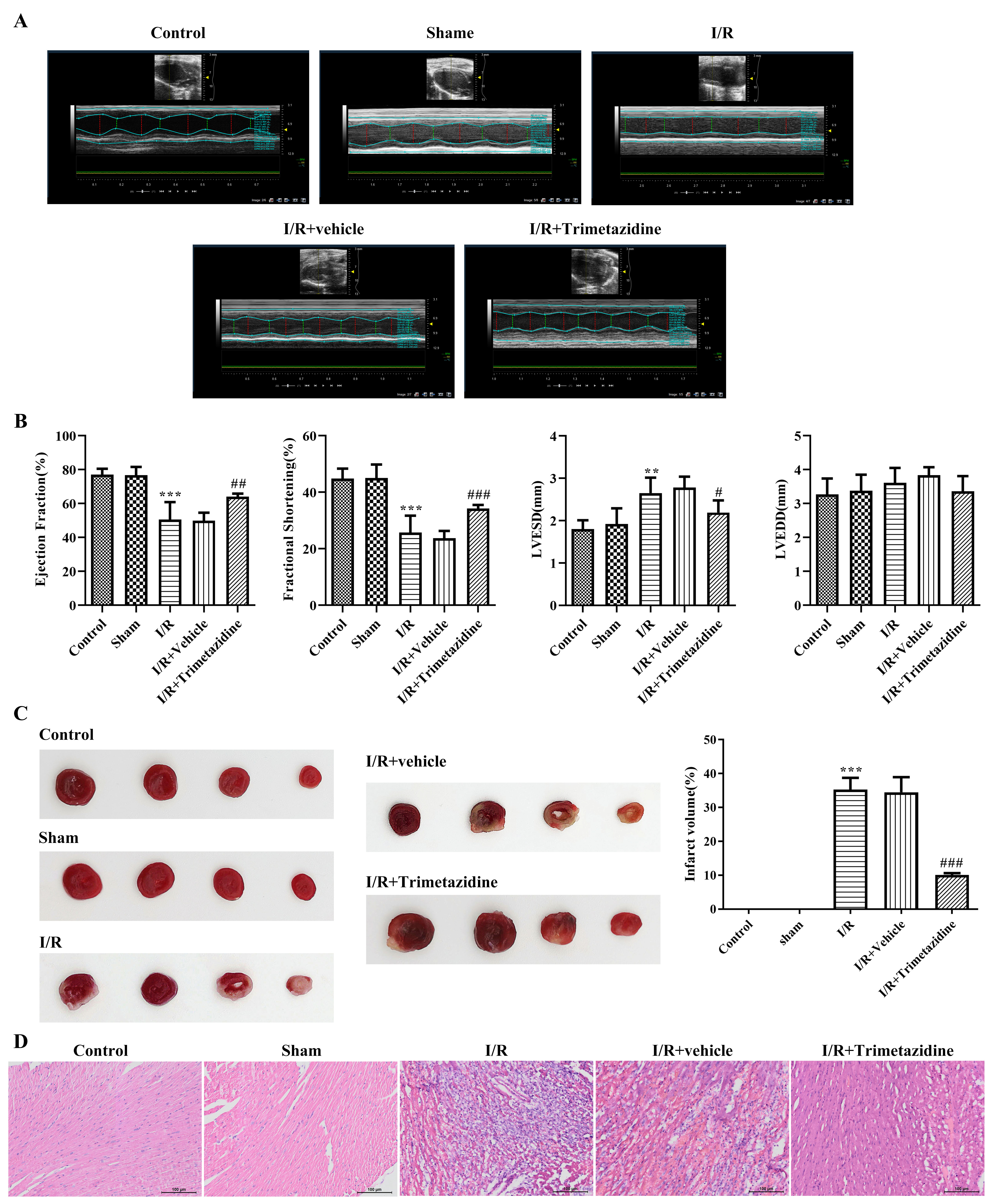 Fig. 1.
Fig. 1.
Trimetazidine alleviates myocardial pathological injury and
cardiac function injury during myocardial ischemia/reperfusion (I/R). (A)
Cardiac function by echocardiography, n = 6. (B) Quantitative analysis
of cardiac function by echocardiography, n = 6.
(C) Myocardial ischemic area was detected by 2,3,5-triphenyltetrazolium
chloride (TTC) staining, n = 3. (D) H&E staining was used to observe the
pathological changes of the heart (scale bar: 100
µm), n = 3. **p
Myocardium reflux was evaluated with Th-S staining. The image showed that the bright blue fluorescent area represented the myocardium that had restored blood supply after reperfusion, while some myocardium failed to restore blood flow smoothly and formed no-reflow phenomenon, which is depicted in the figure as a black area. The blood flow was normal in the Control group and Sham group, and there was no distinct diversity between I/R+Vehicle group and I/R group. Trimetazidine pre-treatment can effectively reduce the area without reflux (Fig. 2A). The detection results of NO expression within myocardial tissue exhibited that NO level in myocardial tissue was markedly descended in I/R group, which was subsequently ascended after the intervention with trimetazidine (Fig. 2B). No distinct difference can be found in CD31 and VE-cadherin expression between Control and Sham group. Relative to Sham group, the expressions of CD31 and VE-cadherin in I/R group were remarkably reduced, and there was no distinct difference between I/R+Vehicle group and I/R group. The expressions of CD31 and VE-cadherin were greatly elevated after the pre-treatment with trimetazidine (Fig. 2C).
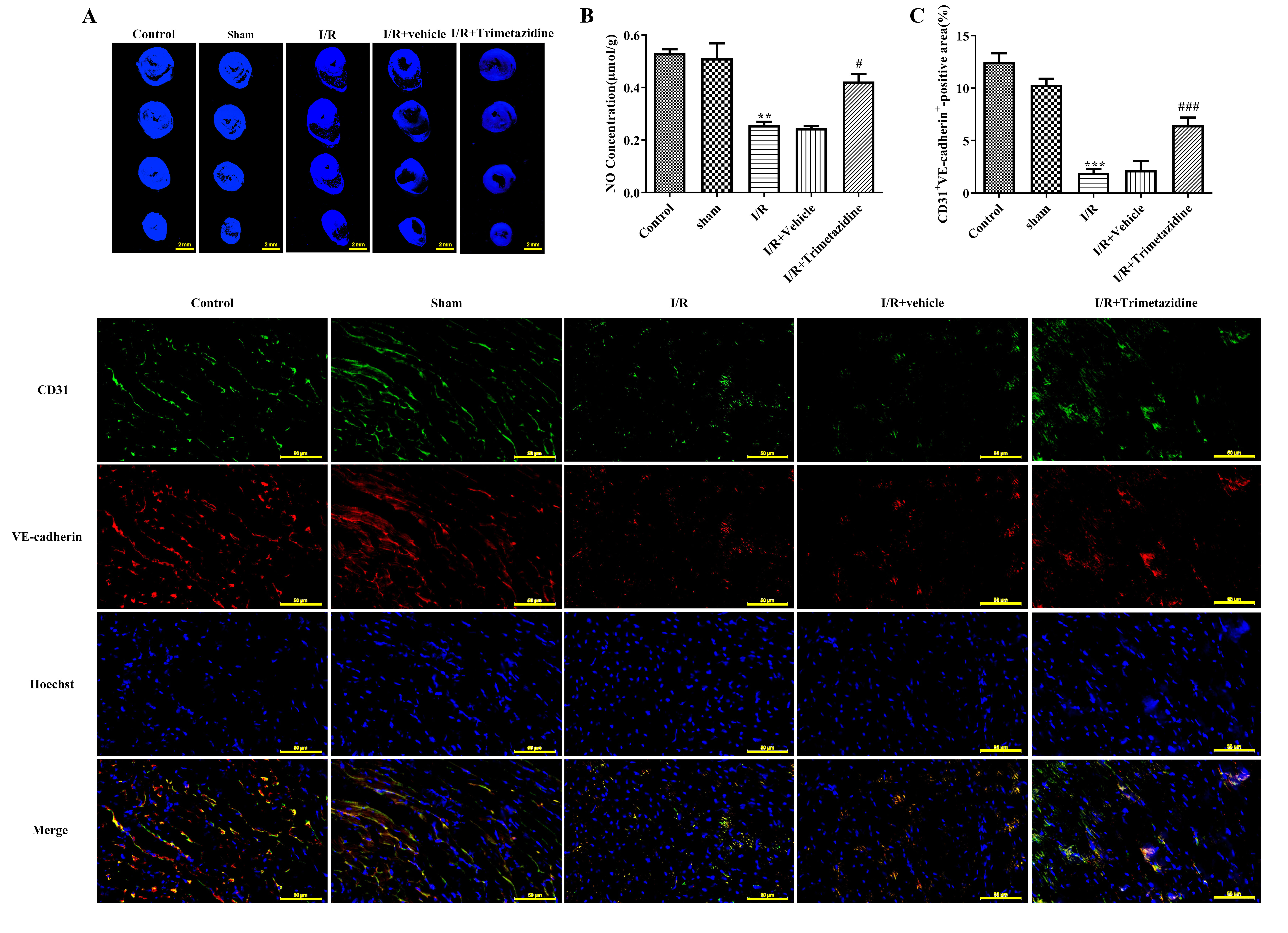 Fig. 2.
Fig. 2.
Trimetazidine improves microvascular reflux, microvascular
function and barrier injury during myocardial I/R. (A) Myocardium reflux was
measured by thioflavin S (Th-S) staining (scale bar: 2 mm). (B) The nitric oxide
(NO) expression in myocardial tissue was detected by Griess reagent Kit. (C)
Immunofluorescence (IF) was used to detect platelet endothelial cell adhesion
molecule-1 (CD31) and vascular endothelial (VE)-cadherin expression (scale bar:
50 µm). n = 3, **p
In addition, trimetazidine increased the expressions of ZO-1 in the heart tissue of mice compared with the I/R+Vehicle group and I/R group (Fig. 3A). Results obtained from WB revealed that by contrast with Control group and Sham group, the expressions of eNOS, p-eNOS, ZO-1, Occludin and VE-cedherin in I/R group were conspicuously decreased, while ET-1 expression was significantly elevated, which were then reversed following the intervention of trimetazidine (Fig. 3B). Moreover, we examined the activation of PI3K/AKT, a signaling pathway upstream of eNOS. The results showed that the ratio of p-PI3K/PI3K and p-AKT/AKT was decreased in the I/R group, but was restored after the pre-treatment with trimetazidine (Fig. 3C).
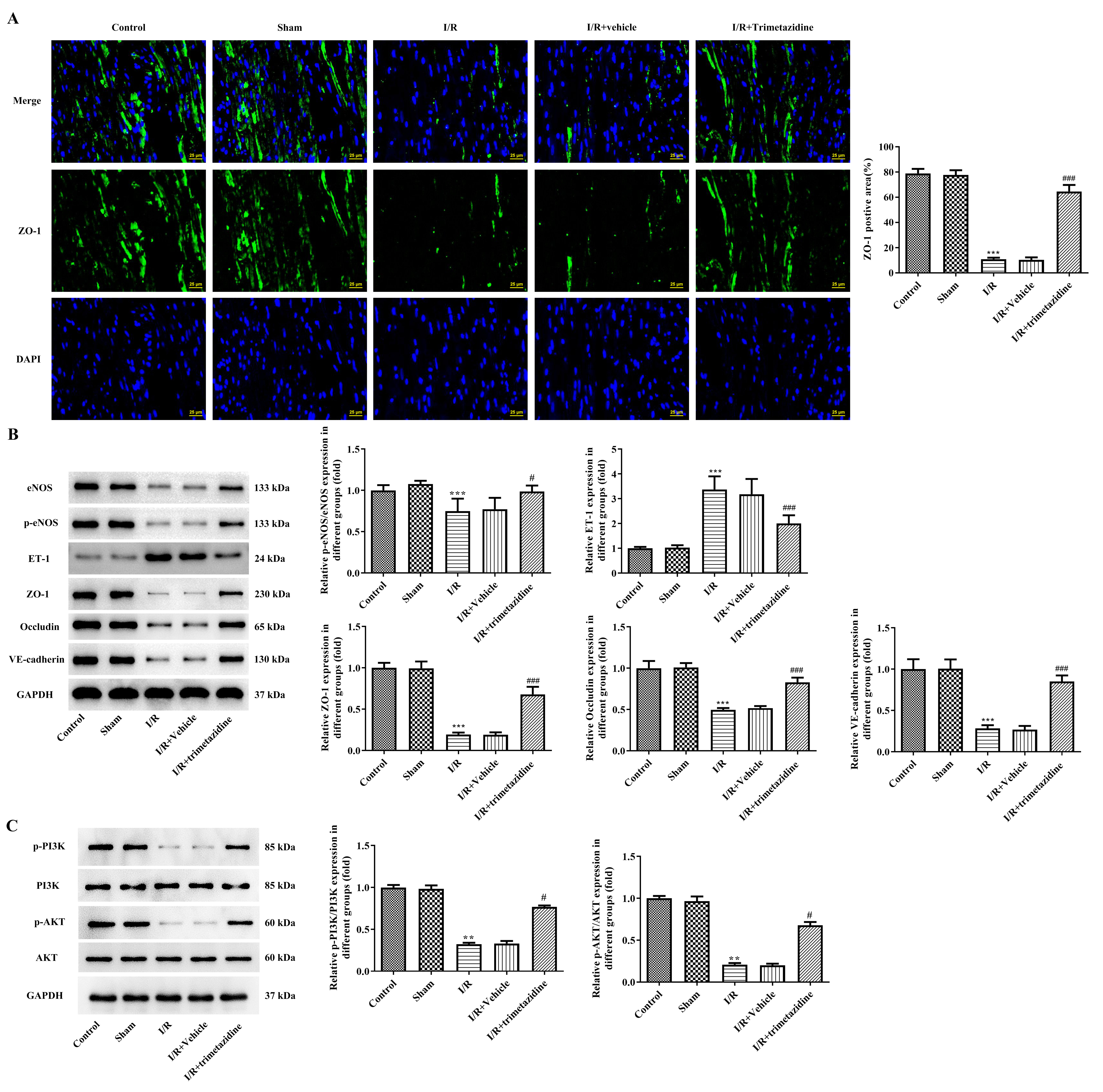 Fig. 3.
Fig. 3.
Trimetazidine improves microvascular reflux, microvascular
function and barrier injury during myocardial I/R. (A) IF was
used to detect zonula occludens protein 1 (ZO-1) expression (scale bar: 25
µm). (B) Western blot analysis was performed to detect the
expressions of endothelial nitric oxide synthase (eNOS), phosphorylated eNOS
(p-eNOS), endothelin-1 (ET-1), ZO-1, occludin and vascular
endothelial(VE)-cadherin. (C) Western blot analysis was performed to detect the
expressions of phosphatidylinositol 3-Kinase (PI3K), phosphorylated PI3K
(p-PI3K), Protein Kinase B (AKT), phosphorylated AKT (p-AKT). n = 3, **p
The result of WB demonstrated that the expressions of p-AMPK, KLF4 and
PPAR
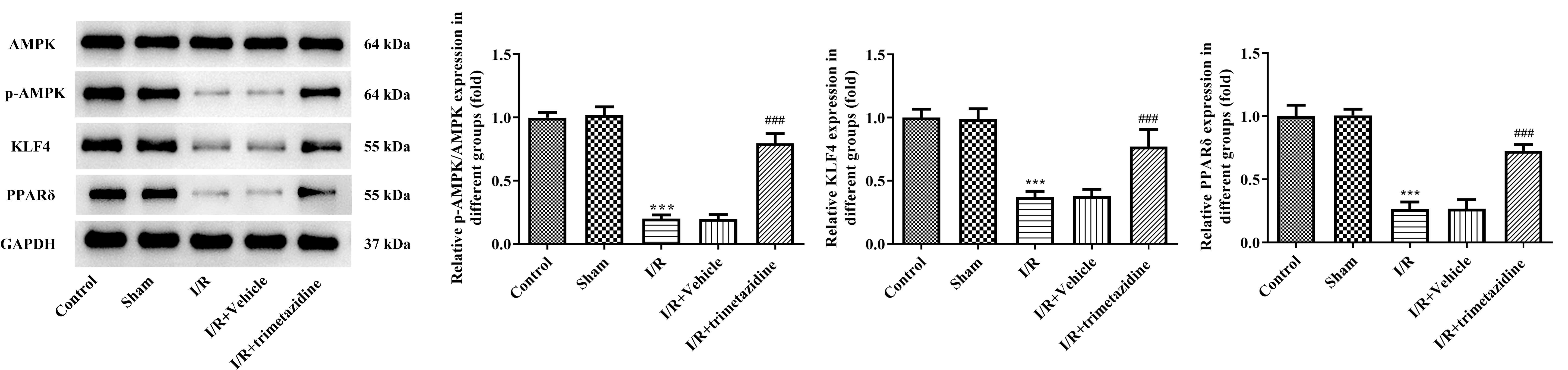 Fig. 4.
Fig. 4.
Trimetazidine regulates the expression of AMPK
signaling pathway during myocardial I/R. The expressions of adenosine
monophosphate (AMP)-activated protein kinase (AMPK), phosphorylated AMPK
(p-AMPK), kruppel-like factor 4 (KLF4) and peroxisome proliferator-activated
receptor delta (PPAR
The extracted mouse heart microvascular endothelial cells were shown in Fig. 5A. To characterize the isolated cell types, we assessed the expression of the endothelial cell marker CD31. The results showed that the extracted cells expressed CD31 (Fig. 5B), indicating that the extracted cells were endothelial cells. Different doses of trimetazidine (20, 40, and 80 µM) were adopted to pre-treat primary mouse heart microvascular endothelial cells, and it was discovered that trimetazidine had no significant effect on cell viability (Fig. 5C). Then CCK-8 was employed to evaluate the impacts of reoxygenation time in OGD/R model on cell viability, and it was found that the cell viability was injured with the extension of reoxygenation time. The reoxygenation time with cell viability in the range of about 50%–60% was selected, that is, OGD4/R24 was used as the hypoxia reoxygenation condition for further study (Fig. 5D). The cells were grouped into Control, OGD/R, trimetazidine (20 µM), trimetazidine (40 µM) and trimetazidine (80 µM). Results obtained from CCK-8 results presented that the viability of cell in OGD/R group was dramatically reduced by contrast with that in Control group, which was the partially revived by trimetazidine treatment with dose-dependent (Fig. 5E).
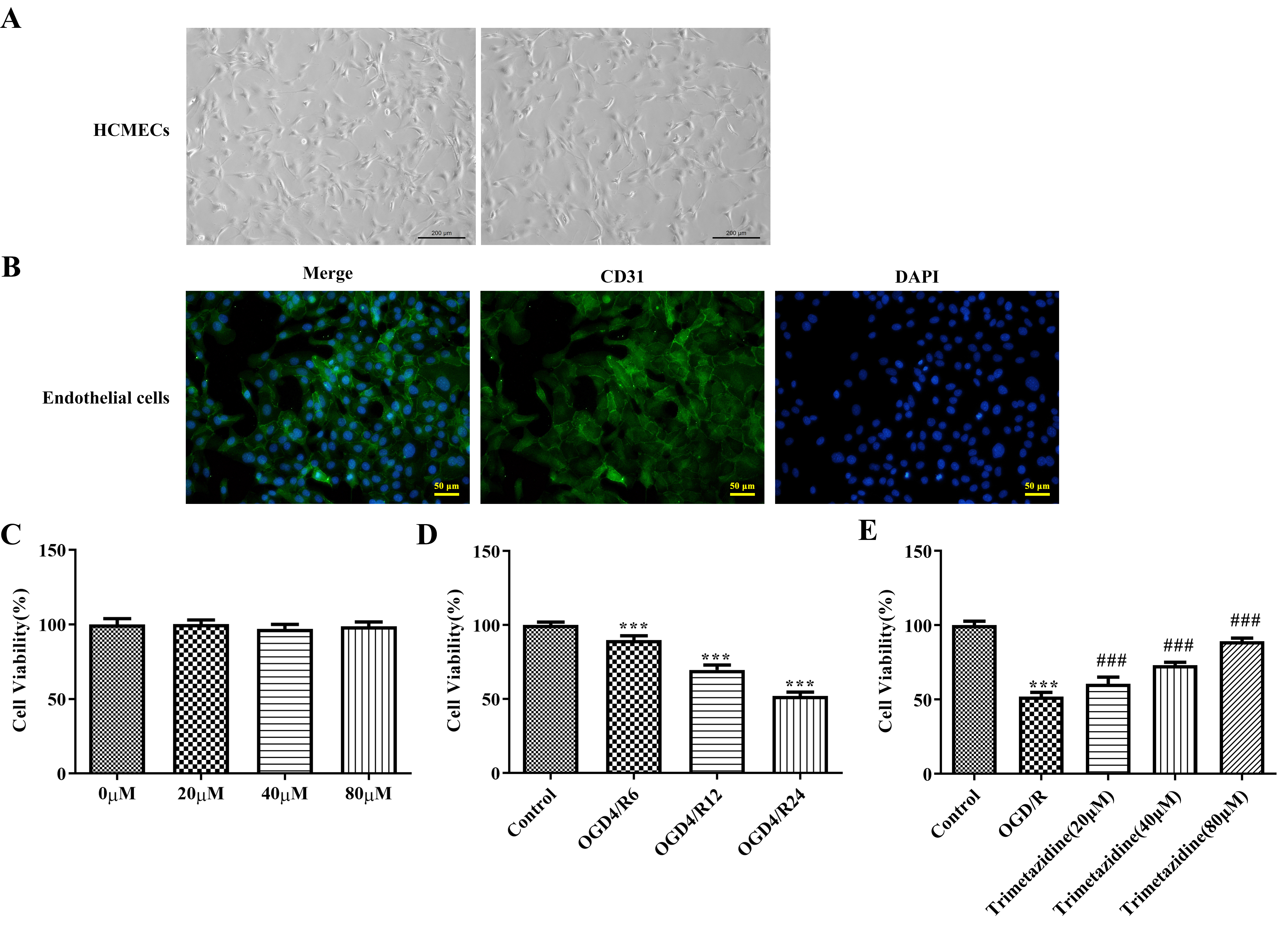 Fig. 5.
Fig. 5.
Effects of trimetazidine with varying concentrations on the
viability of primary mouse heart microvascular endothelial cells induced by
oxygen-glucose deprivation/reperfusion (OGD/R). (A) Mouse heart microvascular
endothelial cells (scale bar: 200 µm). (B)
Endothelial cell marker CD31 immunofluorescence identification
(scale bar: 50 µm). (C–E) Cell Counting Kit-8 (CCK-8) was used to
detect the cell viability. n = 3,
***p
WB was selected as the method of estimation for AMPK signaling
pathway-associated proteins. The results indicated that the p-AMPK, KLF4 and
PPAR
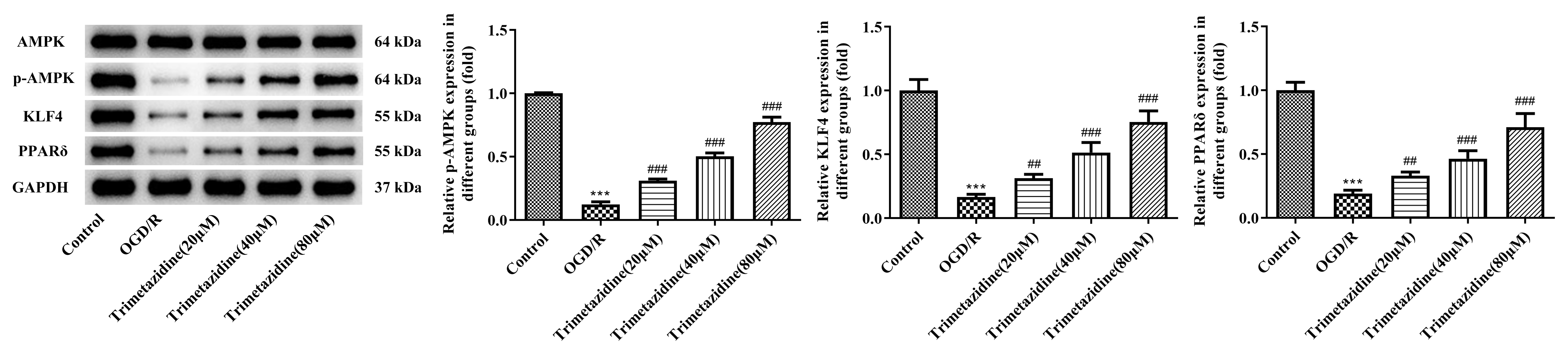 Fig. 6.
Fig. 6.
Effect of trimetazidine with varying concentrations on AMPK
signaling pathway in primary mouse heart microvascular endothelial cells induced
by OGD/R. Western blot assay was used to detect the expressions of AMPK
signaling pathway-related proteins. n = 3, ***p
Subsequently, we applied trimetazidine together with an AMPK inhibitor compound
C to the cells. Results obtained from WB elucidated that the decreased contents
of p-AMPK, KLF4 and PPAR
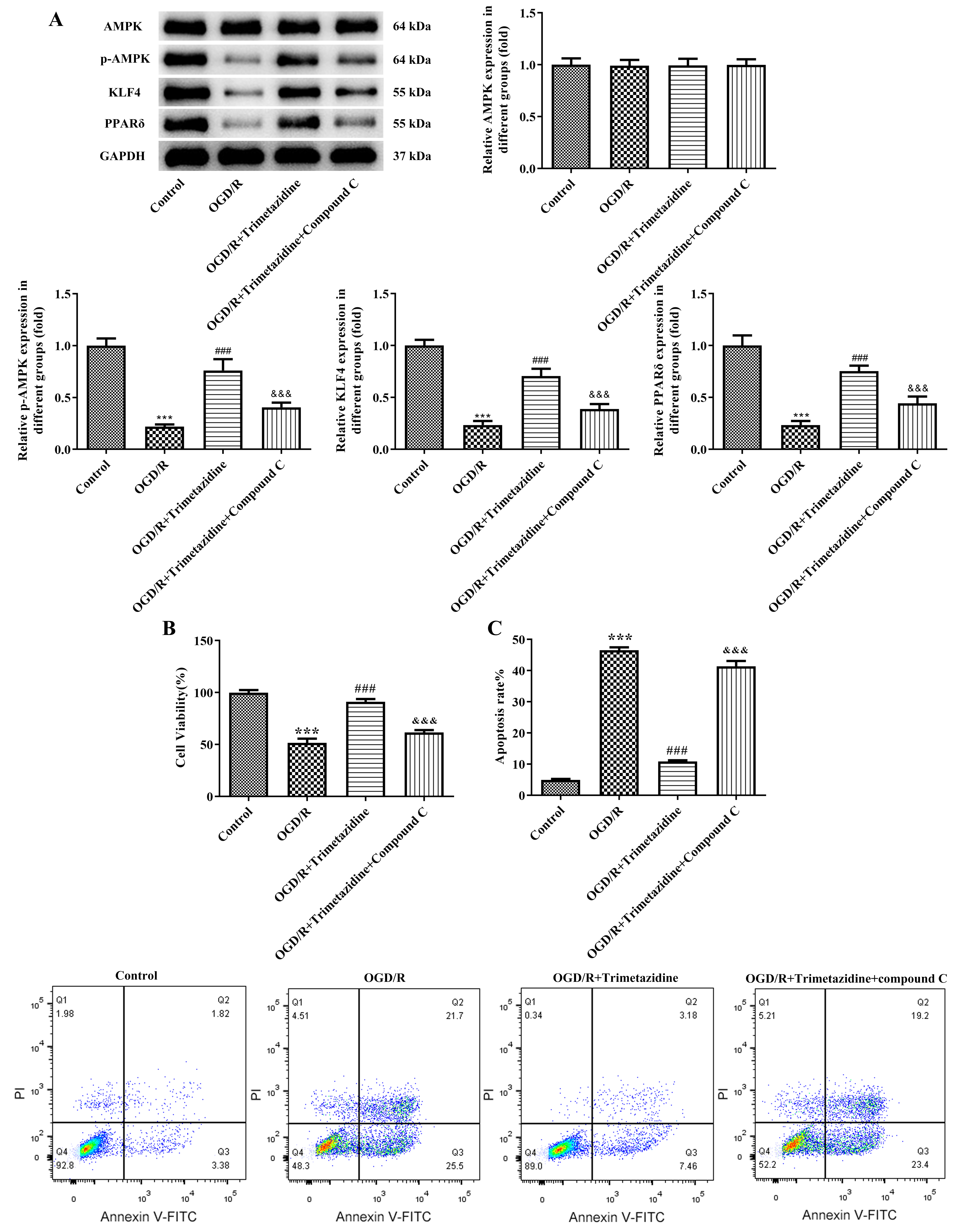 Fig. 7.
Fig. 7.
Trimetazidine alleviates the viability and the apoptosis of
OGD/R-induced primary mouse heart microvascular endothelial cells through AMPK
signaling pathway. (A) Western blot was used to detect the expressions of AMPK
signaling pathway-related proteins. (B) CCK-8 was used to detect cell viability.
(C) Flow cytometry was used to detect cell apoptosis.
n = 3, ***p
Results obtained from WB exhibited a reduction in eNOS and p-eNOS contents and an ascending ET-1 expression in OGD/R group, which were then reversed following the pre-treatment of trimetazidine. However, compound C attenuated the effect of trimetazidine (Fig. 8A). Relative to the OGD/R group, trimetazidine pre-treatment led to a notable elevation in NO level, which was subsequently declined again following the administration of compound C (Fig. 8B). The results of WB assay showed that the ratio of p-PI3K/PI3K and p-AKT/AKT was increased after the pre-treatment with trimetazidine compared with the OGD/R group. Also, compound C attenuated the effect of trimetazidine (Fig. 8C). Results obtained from tubule formation presented that the tube number in OGD/R cells was remarkably decreased, which was then increased following trimetazidine pre-treatment. By contrast with the OGD/R+trimetazidine group, compound C administration diminished the tube number again (Fig. 8D). Relative to the OGD/R group, trimetazidine treatment greatly elevated the contents of CD31 and VEGF, which were subsequently reduced after the addition of compound C (Fig. 8E).
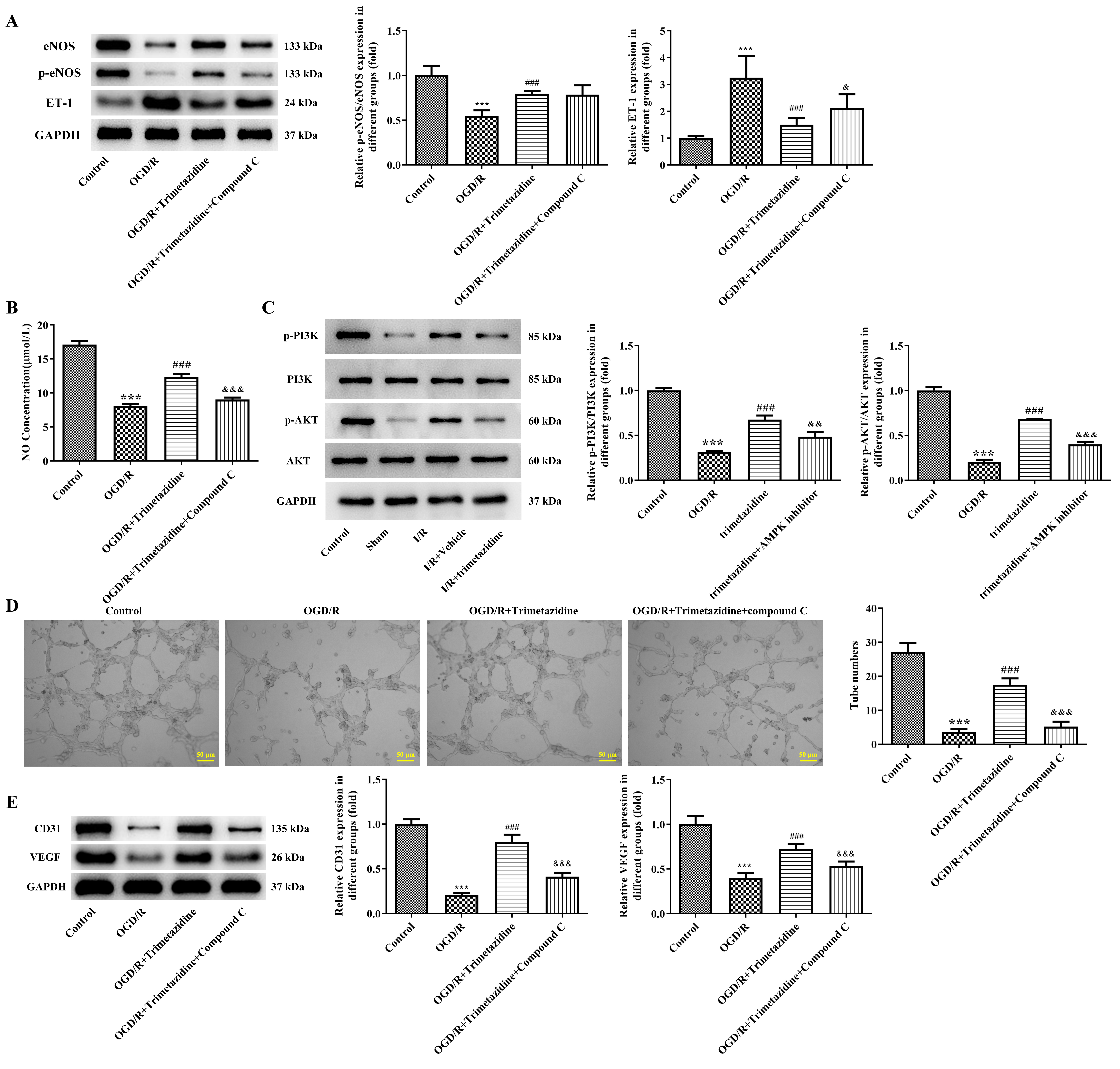 Fig. 8.
Fig. 8.
Trimetazidine alleviates functional impairment of OGD/R-induced
primary mouse heart microvascular endothelial cells through AMPK signaling
pathway. (A) The expressions of eNOS, p-eNOS and ET-1 were detected by western
blot. (B) The NO expression in cell supernatant was detected by the Kit. (C) The
expressions of p-PI3K, PI3K, p-AKT, AKT were detected by western blot. (D) The
number of tubules was detected by tubule formation assay (scale bar: 50
µm). (E) The expressions of Cd31 and vascular endothelial growth
factor (VEGF) were detected by western blot. n = 3, ***p
The endothelial cell permeability experiment demonstrated that the fluorescence value in OGD/R group was increased, which was then reduced following the pre-treatment of trimetazidine. However, the addition of compound C led to an increase in fluorescence values compared with the OGD/R+trimetazidine group (Fig. 9A). The expression of VE-cadherin was detected by IF to investigate the integrity of cell membrane and the results illustrated that VE-cadherin fluorescence brightness was dimmed in OGD/R group, indicating that cell membrane integrity was injured. Nevertheless, the damaged cell membrane integrity in OGD/R group was subsequently improved by trimetazidine pretreatment (Fig. 9B). Additionally, it was observed that the declined contents of ZO-1, Occludin and VE-cadherin in the OGD/R group were elevated following trimetazidine pre-treatment (Fig. 9C). The fluorescence intensity of ZO-1 was increased in trimetazidine group compared with the OGD/R group (Fig. 9D). However, compound C reversed the effect of trimetazidine on VE-cadherin, ZO-1 and Occludin expression.
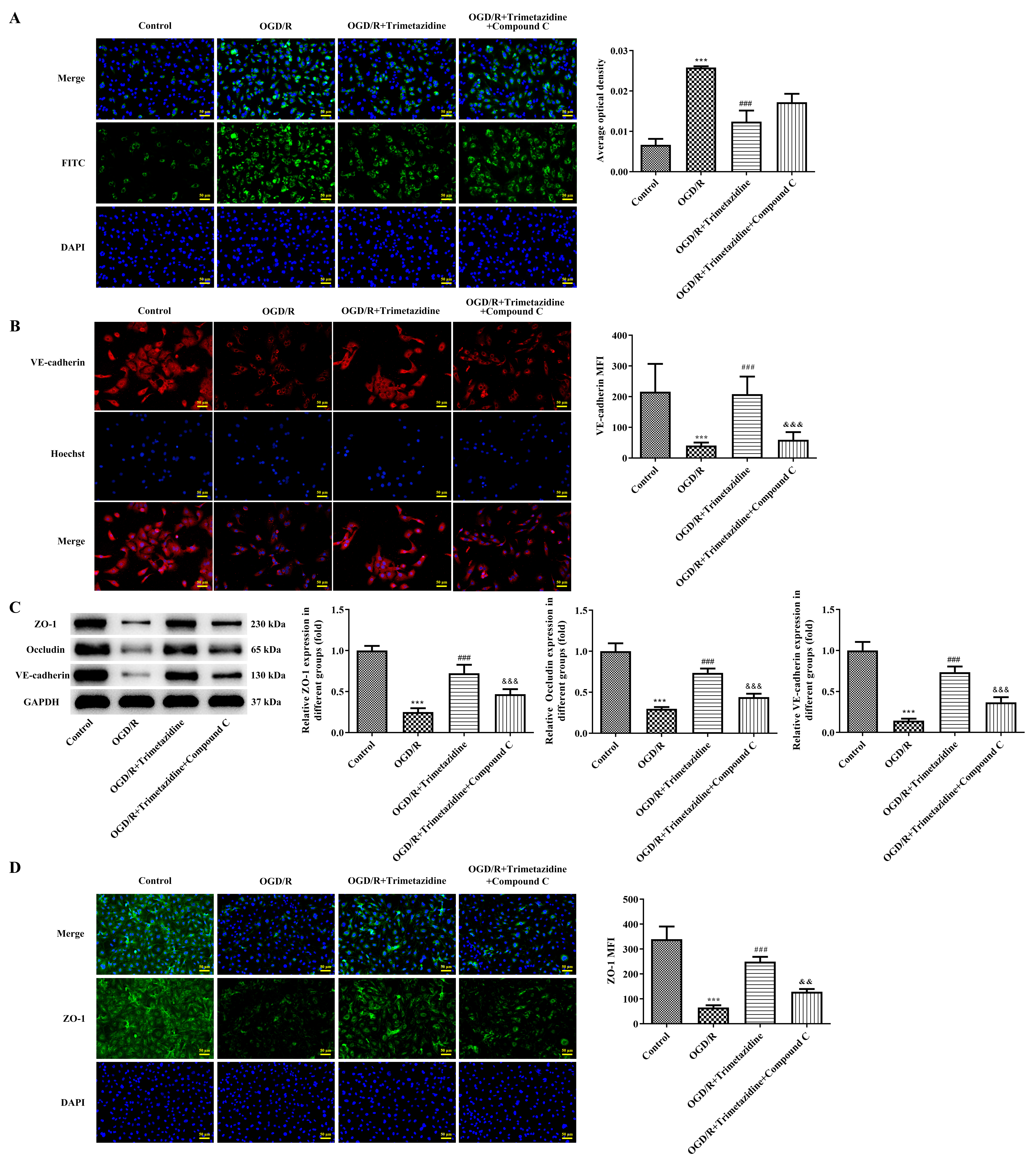 Fig. 9.
Fig. 9.
Trimetazidine alleviates barrier damage of OGD/R-induced primary
mouse heart microvascular endothelial cells through AMPK
signaling pathway. (A) The endothelial cell permeability experiment was used to
detect the cell permeability (scale bar: 50 µm). (B) The expression
of VE-cadherin was detected by IF to investigate the integrity of cell membrane
(scale bar: 50 µm). (C) Western blot detected the expressions of
ZO-1, Occludin and VE-cadherin. (D) IF was used to detect ZO-1 expression (scale
bar: 50 µm). n = 3, ***p
CMD is the underlying cause of ischemic heart disease. A reduction in coronary flow reserve is related to an increased risk of myocardial infarction size, a decline in left ventricular EF and FS, and impaired left ventricular remodeling [18, 19, 20]. The outcomes of percutaneous coronary intervention (PCI) treatment for patients suffering from microvascular dysfunction are not satisfied. However, reperfusion paradoxically contributes to additional tissue damage, which results in patients developing muscle damage and heart failure [21]. Therefore, maintaining normal microvascular function after myocardial infarction has emerged as a cruical element in the management of the condition.
CMD is characterized by abnormal structure and function of coronary microvessels with a vessel diameter within 500 µm, contributing to a disparity in blood and oxygen supply to the myocardium [9]. The disruption of coronary microvascular endothelium, platelet adhesion and inflammatory response promote the in-situ formation of thrombosis and microvascular occlusion, which are the primary causes of CMD [10, 11]. Therefore, effective improvement of coronary microvascular endothelial injury is very important to restore coronary microcirculation and enhance myocardial I/R.
The previous study has shown that trimetazidine belongs to a new class of drugs promoting myocardial metabolism, which can help to relax myocardium, suppress myocardial ischemia, and improve the cardiac function of patients [22]. trimetazidine exhibited protective effects on myocardial I/R by inhibiting excessive autophagy [23]. In addition, trimetazidine could improve rat myocardium after I/R injury by effectively alleviating myocardial apoptosis injury, which is of great value for myocardial ischemia and reperfusion treatment [24]. However, it remains unclear whether it can reduce the damage to endothelial cells during myocardial I/R. In our experiment, it was discovered that trimetazidine can alleviate myocardial pathological injury and cardiac dysfunction, improve microvascular reflux phenomenon and microvascular function and alleviate barrier injury during myocardial I/R. In cell experiments, trimetazidine could promote the viability and inhibit the apoptosis of OGD/R-induced primary mouse heart microvascular endothelial cells. Moreover, trimetazidine could alleviate the damage to the function and the barrier of OGD/R-induced primary mouse heart microvascular endothelial cells. It has been shown that trimetazidine could reduce endothelial dysfunction in ischemic heart disease patients [25]. Additionally, trimetazidine was evidenced to alleviate and prevent endothelial dysfunction caused by PCI in unstable angina pectoris patients [26]. The above-mentioned findings are consistent with our experimental investigation. Our outcomes revealed that trimetazidine could improve myocardial I/R injury-induced endothelial cell disfunction, thereby improving CMD.
Studies have shown that trimetazidine can stimulate the
expression of AMPK signal in cardiomyocytes [22, 27], and the activation of AMPK
can improve myocardial I/R injury [28] and promote eNOS expression in myocardial
I/R tissues [29]. Meanwhile, it has been reported that AMPK can mitigate
endothelial cell dysfunction resulting from elevated glucose levels by activating
downstream PPAR
Myocardial ischemia is caused by epicardial coronary artery stenosis or atherosclerotic disease affecting microcirculation. Trimetazidine promotes glucose oxidation which optimizes cellular energy processes in ischemic conditions. A study have demonstrated that trimetazidine treatment prior to elective percutaneous coronary intervention (PCI) reduced microvascular dysfunction by lowering postprocedural index of microcirculatory resistance (IMR) values [32]. There has been a lack of research into how trimetazidine enhances vascular ring function. This study is the first to examine the mechanisms by which trimetazidine alleviates microvascular dysfunction resulting from myocardial infarction and ischemia-reperfusion. The findings reveal that trimetazidine enhances the function and mitigates barrier damage in mouse heart microvascular endothelial cells by modulating the PI3K/AKT/eNOS/NO signaling pathway. Additionally, prior research has indicated that trimetazidine can stimulate AMPK signaling in cardiomyocytes [22, 27], which helps to alleviate myocardial ischemia-reperfusion injury [28]. In this study, we noted the activation of AMPK signaling in heart microvascular endothelial cells, and the protective effects of trimetazidine on these cells were significantly diminished when AMPK signaling was inhibited. This suggests that trimetazidine activates the AMPK signaling pathway not only in cardiomyocytes during ischemia-reperfusion but also in heart microvascular endothelial cells. Furthermore, we discovered that the AMPK signaling pathway inhibitor compound C notably reduced the protective effects of trimetazidine on the function and barrier integrity of heart microvascular endothelial cells and inhibited the activation of the PI3K/AKT/eNOS/NO signaling pathway. This indicated that trimetazidine’s regulation of the PI3K/AKT signaling pathway in mouse heart microvascular endothelial cells was mediated through the AMPK signaling pathway.
Trimetazidine could improve CMD induced by myocardial infarction by activating AMPK signal. Our study offers a theoretical basis for trimetazidine in the clinical treatment of CMD caused by myocardial infarction.
Original data of this study are available from the corresponding author on request.
XQ and YY designed the experiment research. XQ, PY and LJ performed the experiments. XQ performed formal analyses and wrote the original manuscript. PY and YY contributed to the conception and design of the work. All authors have revised the manuscript and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
All animal experiments were approved by the ethical committee of Zhaofenghua Biotechnology [IACUC-20221009-03].
Not applicable.
This research received no external funding.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.