1. Introduction
The most important components of the signaling system involved in the regulation
of the functional activity of the male and female reproductive system are
gonadotropins, such as luteinizing hormone (LH), chorionic gonadotropin (CG) and
follicle-stimulating hormone (FSH), and their receptors. LH and CG, produced by
gonadotrophs of the adenohypophysis (LH, and sulphated CG), as well as the embryo
and placenta during the first trimester of pregnancy (hyperglycosylated and
classical CGs), are endogenous ligands of one G protein-coupled receptor (GPCR),
which is accordingly called the LH/CG receptor (LHCGR). They bind to a
high-affinity orthosteric site located in the ectodomain of this receptor. This
is not a common case for GPCRs, since most receptors in this superfamily are
activated by a single orthosteric agonist, but for a number of GPCRs, most
notably polypeptide hormone receptors, there are two or more orthosteric site
ligands, as has been demonstrated for the melanocortin receptors [1], orexin
receptors [2], apelin receptor [3], and N-formyl-peptide receptor [4].
In this case, the multiplicity of ligands can be due to various variants of
proteolysis or modification of the prohormonal molecule, a striking example of
which is the site-specific proteolysis of pro-opiomelanocortin, as well as
cross-interaction, which well illustrates the interaction of different orexin
isoforms with their receptors. The multiplicity of ligands is even more
characteristic of receptor tyrosine kinases, which is most clearly demonstrated
by the receptors of the insulin family peptides [5], the receptors of the
epidermal growth factor receptor family [6] and different isoforms of vascular
endothelial growth factor (VEGF) receptor [7].
LH and human CG (hCG) bind to LHCGR with high affinity, causing a wide range of
physiological responses, which is due to the functional interaction of activated
LHCGR with a large number of transducer proteins, through which the regulation of
many intracellular targets is carried out. Despite the fact that the regulatory
effects of LH and hCG are realized through the same receptor, and the
high-affinity orthosteric site responsible for their binding has a similar
structure and topology, the specificity of activation of intracellular cascades
by gonadotropins, and, accordingly, the cellular response caused by them can vary
significantly [8, 9, 10, 11, 12, 13, 14, 15, 16, 17]. This has a very definite biological significance, taking
into account the different physiological roles of LH and hCG in humans and some
other mammals. No less interesting is the fact that each of these hormones has a
large number of isoforms that differ in their specific activity, and this is
largely determined by the characteristics of the glycosylation of their
molecules, which significantly affects the binding of gonadotropin to LHCGR and
the bias of signal transduction [18, 19, 20, 21, 22, 23]. Finally, the response to gonadotropin
may be regulated at the level of LHCGR through post-translational modifications
of the receptor, as well as through receptor complex formation, including
heterodi(oligo)merization with other receptors, most notably the FSH receptor
(FSHR) [14, 24, 25, 26, 27, 28, 29, 30]. Glycosylation of gonadotropins and the processes of complex
formation and modification of LHCGR are the most important factors in the control
of LH/CG-mediated signal transduction in target cells, and they are based on
allosteric mechanisms that determine both the affinity of the gonadotropins for
LHCGR and the stability and pattern of activated conformations of LHCGR
responsible for selective signal transduction to intracellular effectors.
However, other, less studied factors in this aspect, such as the physiological
state of the organism, as well as the pathological processes, including
lipotoxicity, endoplasmic reticulum stress and overproduction of reactive oxygen
and nitrogen species, also have a significant impact on LH/CG-mediated signal
transduction, disrupting post-translational modification and “maturation” of
LHCGR and other signal proteins. In animal models of type 1 and type 2 diabetes
mellitus with hyperglycemia, insulin signaling dysfunctions, redox imbalance and
increased inflammatory processes, we and other authors have shown a significant
decrease in both LHCGR gene expression and the number of functionally
active LHCGRs on the surface of testicular and ovarian cells, which led to
weakened LH/CG signaling and impaired steroidogenesis [31, 32, 33, 34, 35].
All these factors, to one degree or another, influence the lipid composition of
membranes, the ionic and amino acid composition in the intra- and intercellular
environment, and the availability and activity of adapter and regulatory proteins
capable of forming complexes with LHCGR. It is known that cholesterol and
phospholipids, some simple ions (Na, Mg, Zn, Mn,
Cl, and others), amino acids and their derivatives (Tyr, Phy, Trp, Leu,
Ile, homocysteine, agmatine and others) can function as allosteric modulators of
GPCRs [36, 37, 38, 39, 40, 41, 42]. Their regulatory, modulating effect on the activity of LHCGR and
other components of LH/CG-stimulated cascades cannot be excluded, although strong
evidence for this has not yet been obtained. Autoantibodies to gonadotropins and
LHCGR, the formation of which has now been proven, can act as endogenous
allosteric regulators [43, 44, 45, 46, 47]. They are able to function as allosteric
regulators of LHCGR, which have their intrinsic activity, and to modulate the
effects of gonadotropins. It should be noted that with regard to other GPCRs,
there are numerous works on the allosteric effects of autoantibodies to GPCRs and
their key role in the development of autoimmune diseases [48, 49, 50, 51].
Thus, there are many mechanisms and targets of allosteric regulation of LHCGR
and the signaling pathways realized through it, which indicates the possibility
of fine-tuning the intensity and selectivity of LH/CG-induced signal
transduction. This tuning depends on the physiological status of the target cell,
the pattern of LH and CG glycoforms, the composition and ratio of
LHCGR-containing complexes, the activity of other signaling cascades modulating
LHCGR activity, and the presence of autoantibodies to gonadotropins and LHCGR.
Such a variety of allosteric influences is predetermined by the existence of a
large number of allosteric sites in the LHCGR, as has been shown for other class
A GPCRs. These sites can be localized in various loci of the receptor molecule,
including in the extracellular loops (ECLs) and the external entrance to the
transmembrane tunnel, in the internal cavity of this tunnel, on the outer surface
of the transmembrane domain (TMD), which is in contact with the lipid bilayer of
the membrane, and in the intracellular loops (ICLs) and in the cytoplasmic
entrance to the transmembrane tunnel [42] (Fig. 1). Accordingly, it becomes
possible to develop site-directed allosteric ligands that will have a different
profile of pharmacological activity, being both modulators of the effects of
gonadotropin and having their intrinsic agonistic or antagonistic activity. As is
known, allosteric regulators can reduce (negative allosteric modulator, NAM) or
increase (positive allosteric modulator, PAM) the affinity and/or effectiveness
of an orthosteric agonist, do not directly affect these parameters, but modulate
other allosteric effects (silent allosteric modulator, SAM), exhibit its
intrinsic activity as a full agonist, inverse agonist or neutral antagonist in
the absence of an orthosteric agonist, as well as combine the activity of a full
agonist or antagonist with the activity of PAM (ago-PAM, PAM-antagonist) or NAM
(ago-NAM) [42, 52, 53, 54, 55, 56] (Table 1). Since allosteric sites functionally
interact not only with the orthosteric site, but also with each other, forming a
multidirectional network of such interactions, the activity profile of the
allosteric ligand can be very complex and cannot be described within the
framework of the proposed classification, which is to a certain extent true for
some low-molecular-weight (LMW) allosteric regulators of LHCGR.
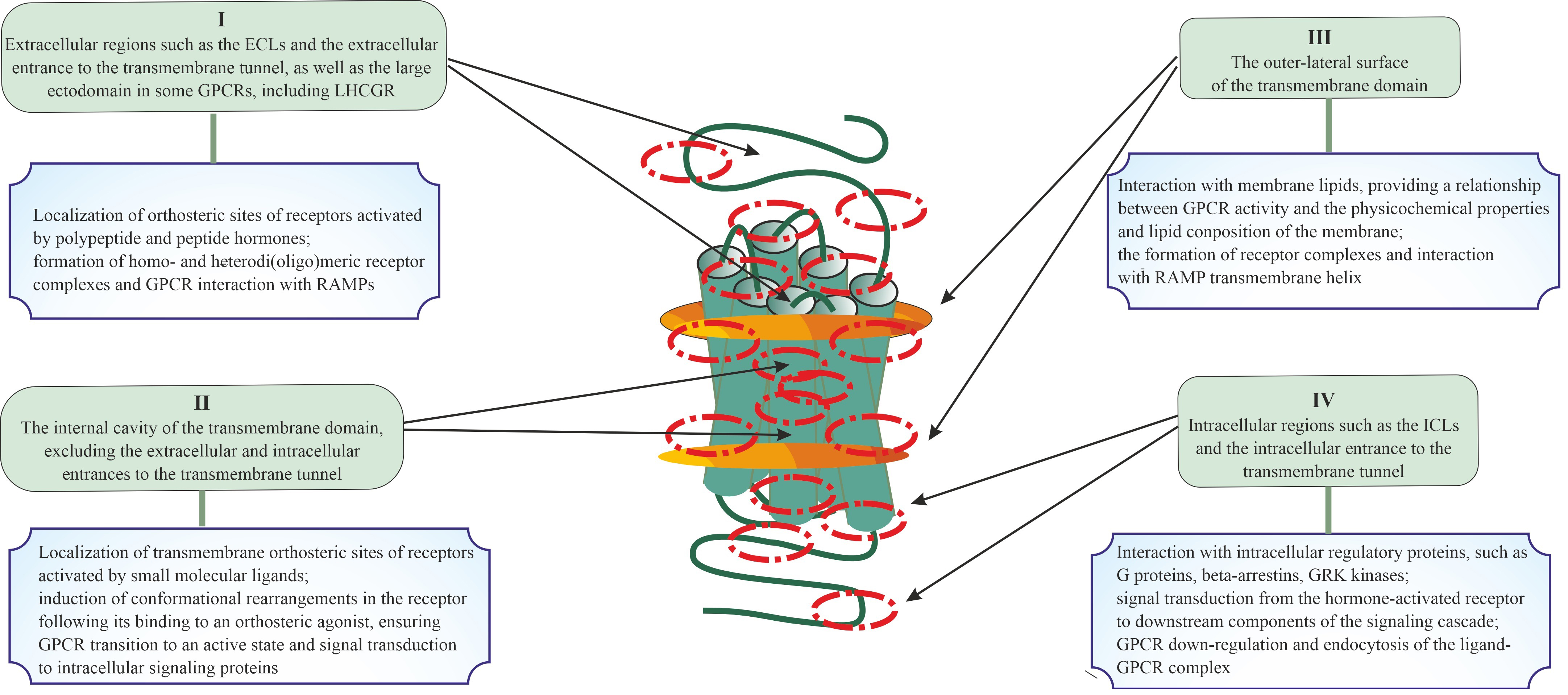 Fig. 1.
Fig. 1.
Topologically distinct regions and domains of G protein-coupled
receptors (GPCRs) in which allosteric sites can be located, and the role of these
regions and domains in the signal transduction and the formation of di- and
oligomeric complexes. Four possible localizations for allosteric sites of GPCRs
are presented, including extracellular regions and/or domains (I), the
transmembrane tunnel of the TMD (II) and its side surfaces that contact plasma
membrane lipids (III), and cytoplasmic regions and/or domains (IV). The main
functions of these topologically different regions or domains of GPCR in signal
transduction and in the formation of various complexes (between GPCR protomers or
between the GPCR protomers and other signal and adapter proteins) are presented,
which indicates the possible role of allosteric sites located in them in the
implementation of these processes. At the same time, taking into account the
cross-talk between allosteric and orthosteric sites, as well as between
allosteric sites, this role can vary greatly and be much broader and more
diverse. ECLs, extracellular loops; LHCGR, luteinizing hormone/chorionic
gonadotropin receptor; RAMPs, receptor activity-modifying proteins; ICLs,
intracellular loops; GRK, G protein-coupled receptor kinase; TMD, transmembrane domain.
Table 1.
Pharmacological profile of ligands of G protein-coupled
receptors (GPCRs) allosteric sites, including allosteric modulators of the
effects of orthosteric agonist, allosteric regulators with their intrinsic
activity and with a combination of their intrinsic and modulating activity.
| Ligand type |
Effect on the basal and stimulated GPCR activity |
Characteristics of GPCR binding and/or activation |
| Positive, negative, silent and biased allosteric modulators (PAM, NAM, SAM, and BAM) |
| PAM |
Increase in the affinity of an orthosteric agonist (OA) for GPCR and/or in the effectiveness of its action without affecting the constitutive activity of GPCR |
1/1/=1 |
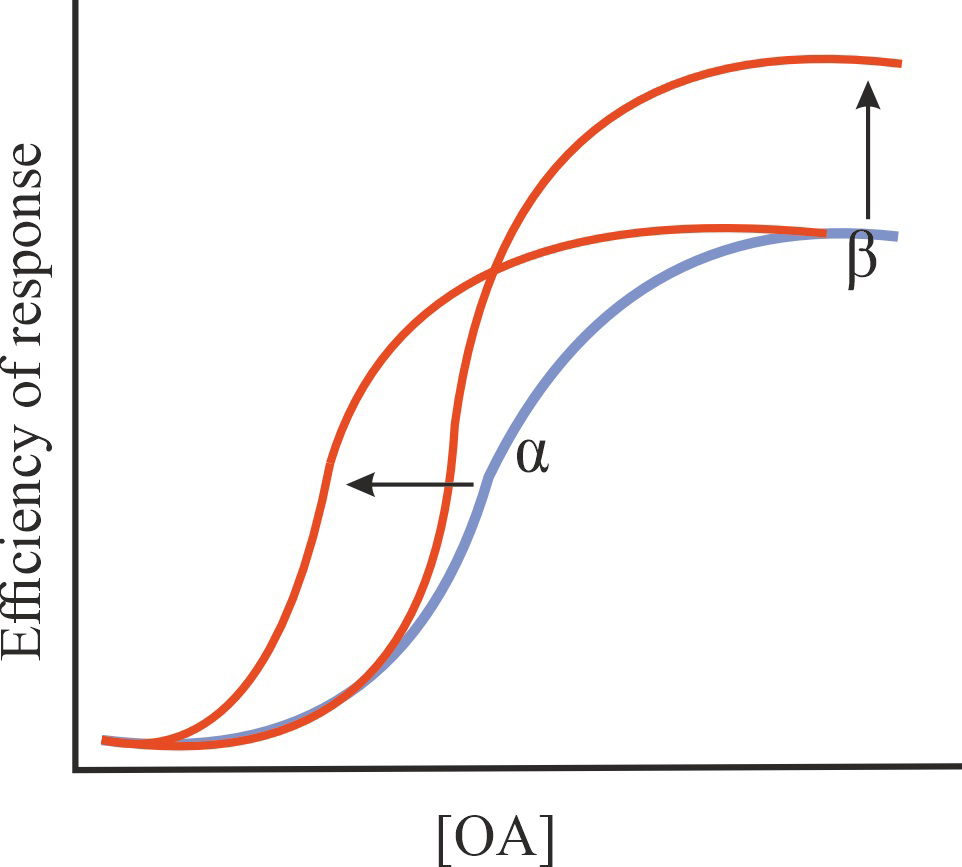 |
| NAM |
A decrease in the affinity of OA for GPCR and/or in the efficiency of its action without affecting the constitutive activity of GPCR |
1/1/=1 |
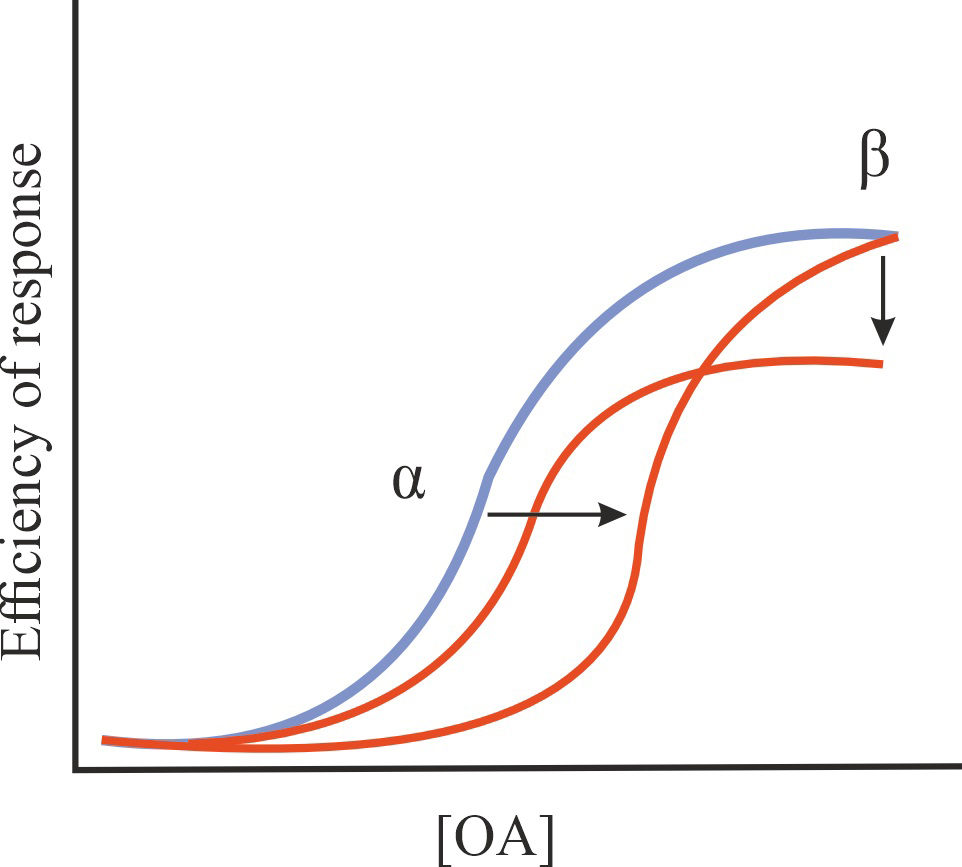 |
| SAM |
No effect on the affinity of OA for GPCR and (or) on the effectiveness of its action, possible change in some characteristics of the regulatory effect of allosteric agonists on GPCR activity (biased agonism, specificity of interaction with certain types of G proteins and -arrestins, formation of receptor complexes, availability of GPCR for allosteric regulators), no effect on the constitutive activity of GPCR |
=1/=1/=1 |
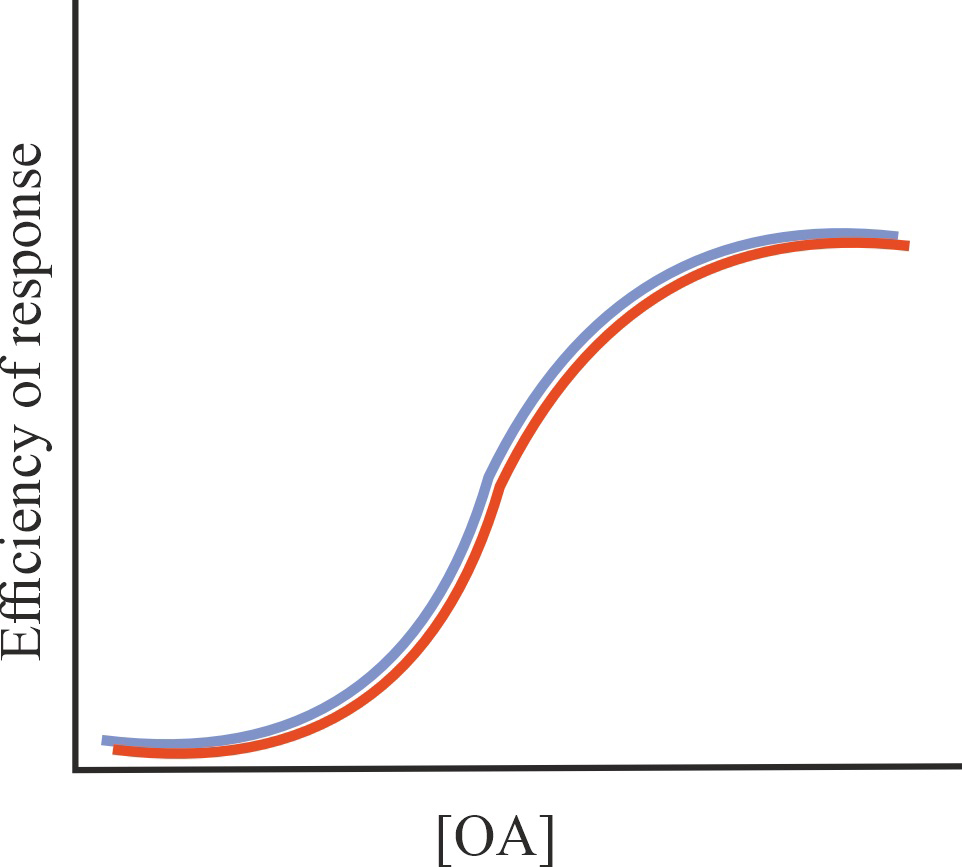 |
| BAM |
A change in the affinity of an OA for a GPCR and/or its efficiency of action, resulting in selective activation (positive allosteric modulator, PAM), inhibition (negative allosteric modulator, NAM) or modification (silent allosteric modulator, SAM) of a certain signaling cascade or a certain pattern of signaling cascades, which provides bias in OA action |
The values of , and vary depending on the specific cascade and the nature of the modulating allosteric action |
| Allosteric regulators with intrinsic activity, such as full (Full Ago) or inverse allosteric agonists (Inv Ago) and neutral allosteric antagonists (Ant) |
| Full Ago |
Stimulation of GPCRs in the absence of OA or other allosteric agonist, no effect on OA’s affinity for GPCRs or its effectiveness |
=1/=1/1 |
| Characterized by preferential activation of a certain signaling cascade (biased agonism), and more moderate stimulation of GPCR compared to OA |
| Inv Ago |
Reduction in constitutive GPCR activity in the absence of OA, as well as inhibition of OA- or allosteric agonist-stimulated GPCR activities |
=1/=1/1 |
| Characterized by preferential suppression of a specific OA-stimulated signaling cascade, as well as a selective influence on the pattern of active states of constitutively active GPCR; more moderate inhibition of constitutive and OA-stimulated GPCR activity as compared to orthosteric site inverse agonists |
| Ant |
Inhibition of OA- or allosteric agonist-stimulated GPCR activities without affecting constitutive GPCR activity |
=1/=1/1 |
| Characterized by a moderately pronounced inhibitory effect on OA-stimulated signaling cascades, which does not lead to their blockade, typical of orthosteric site antagonists |
| Allosteric regulators with combined modulating and agonistic activity (Ago-PAM – full allosteric agonist-PAM; Ago-NAM – full allosteric agonist-NAM; Ant/PAM – neutral allosteric antagonist/PAM) |
| Ago-PAM |
Increase in the affinity of OA for GPCR and/or in the efficiency of its action, stimulation of basal GPCR activity in the absence of OA, possibly potentiation of the effect of OA on GPCR activity |
1/1/1 |
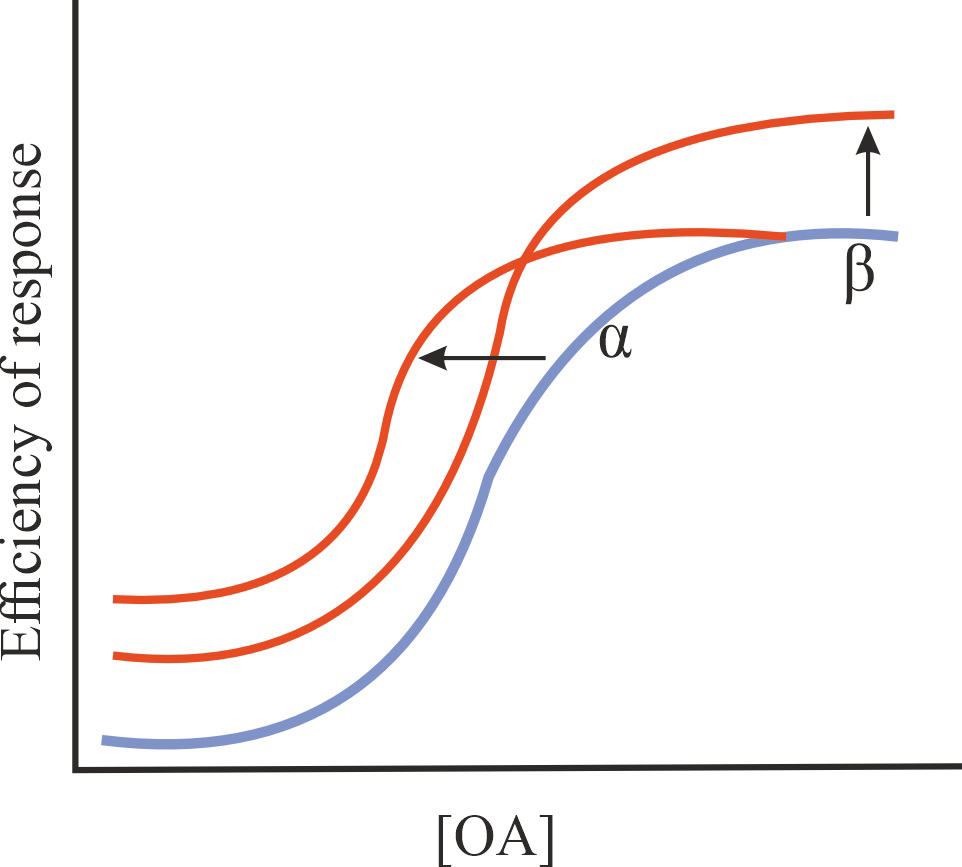 |
| Ago-NAM |
Reduction in the affinity of OA for GPCRs and/or in the efficiency of its action, stimulation of basal GPCR activity in the absence of OA |
1/1/1 |
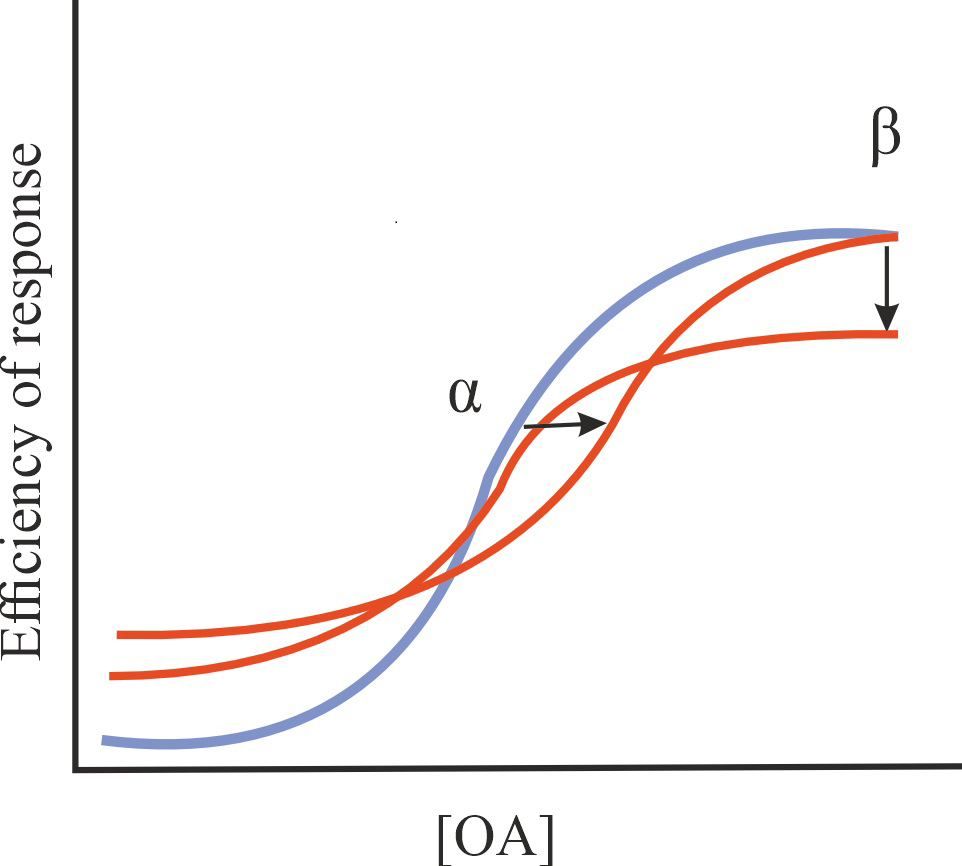 |
| Ant/PAM |
A decrease in the effectiveness of OA on GPCRs (antagonistic effect), but an increase in the affinity of OA for GPCRs (PAM effect) |
1/1/=1 |
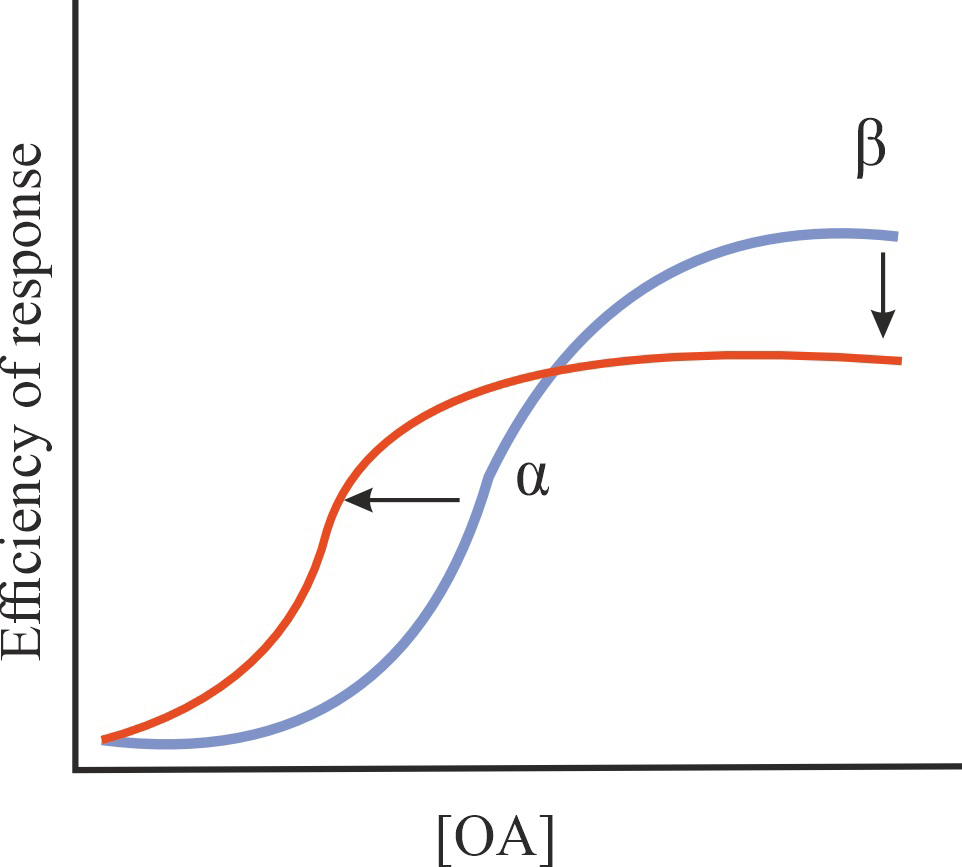 |
Note: – the factor of binding cooperativity between the OA
and allosteric modulator; – the operational factor of cooperativity
for quantitative evaluation of the effects of allosteric modulator on operational
efficacy of OA (receptor activation); – operational efficacy for the
complex of GPCR with allosteric ligand. The values of binding cooperativity
() and operational cooperativity () greater than 1 denote
positive cooperativity, and the corresponding values below 1 denote negative
cooperativity. The value 1 for operational efficacy () is normalized
since the absolute value of can vary widely depending on the basic
design parameters used. For BAM and other cases of biased allosteric regulation,
different variants of the values of , and are
possible, since in this case it is necessary to differentiate the factors being
assessed for each specific signaling cascade regulated by OA or allosteric
regulator. PAM, positive allosteric modulator; NAM, negative allosteric modulator; SAM, silent allosteric modulator.
In addition, given the multiplicity of signaling cascades activated by
orthosteric agonists through GPCRs, allosteric modulators can selectively enhance
or, conversely, attenuate only a certain signaling cascade, thereby demonstrating
biased allosteric modulator (BAM) activity [54, 56, 57]. Such modulators are
important for ensuring selectivity of action and achieving target physiological
effects when using an orthosteric agonist that has low selectivity for
intracellular signaling. Allosteric regulators with their intrinsic activity,
full or inverse agonists, can also specifically regulate only a certain signaling
cascade, functioning as biased allosteric full or inverse agonists (Table 1).
This review is devoted to the analysis and discussion of the similarities and
differences in the regulatory effects of LH and various forms of hCG on the
activity of LHCGR and its signaling pathways, the role of glycosylation of LH and
CG and LHCGR complex formation in signal transduction, including its
heterodimerization with FSHR, as well as the possible contribution of
autoantibodies against gonadotropins and LHCGR in the control of LHCGR activity
and in the etiology and pathogenesis of reproductive dysfunctions. Along with
this, the review examines modern advances in the development of allosteric
regulators of LHCGR, including full and inverse LMW agonists and allosteric
modulators capable of interacting with the transmembrane allosteric site of LHCG.
The regulators with agonistic activity are of significant interest for the
correction of hypogonadotropic conditions and in the assisted reproductive
technologies (ARTs), while regulators with antagonistic activity may be in demand
in the treatment of hormone-dependent tumors and in contraception. The discussion
of the issues presented above is preceded by a brief description of the
structural and functional organization of gonadotropins and their receptors, the
molecular basis of their interaction, as well as the LHCGR signaling realized
through G proteins and -arrestins.
2. Structure of Luteinizing Hormone and Chorionic Gonadotropins
The LH is a -heterodimer with a molecular weight of about 30
kDa. LH secretion is carried out by gonadotrophs, specialized cells of the
adenohypophysis, and is controlled by the hypothalamic gonadotropin-releasing
hormone (GnRH) [58, 59, 60]. Along with GnRH, the synthesis and secretion of LH are
regulated by polypeptide factors such as kisspeptin, melanocortin peptides,
gonadotropin-inhibiting hormone, leptin, adiponectin, activins and inhibins, as
well as steroid hormones, corticosteroids and, by a negative feedback mechanism,
sex steroid hormones, primarily estrogens, and some growth factors and cytokines
[61, 62, 63, 64, 65, 66, 67, 68, 69]. These factors can act either at the level of control of GnRH release,
directly or indirectly affecting the activity of GnRH-expressing neurons, or
directly influence the synthesis and secretion of LH by pituitary gonadotrophs.
Along with this, some of these regulators can influence both the secretion of
GnRH and the production of LH, as shown, for example, for leptin [70, 71] and
gonadotropin-inhibiting hormone [72].
Among the isoforms of human CG (hCG), the isoform of hCG expressed in the
pituitary gland, the so-called sulphated hCG [73], and the classical hCG, which
has an extrapituitary origin, are known. Classical hCG is synthesized and
secreted by the embryo and placenta during the first trimester of pregnancy.
Sulphated hCG is produced by the gonadotrophs of both men and non-pregnant women,
and despite its small quantities, it is significantly more active than LH, and
thus makes a significant contribution to the total LH-like activity of the common
pool of gonadotropins [73]. A hyperglycosylated form of the hormone has also been
discovered, which is expressed at the early stages of embryo development, mainly
at the cytotrophoblast stage and significantly differs in functional properties
from LH and other forms of hCG [74, 75].
The -subunit is encoded by a single gene and is common to all
gonadotropins. It is a polypeptide with a length of 116–120 amino acid residues
(AARs) and is characterized by a high degree of homology of the primary
structure. Thus, when comparing the human -subunit with those of
monkeys, up to 98% identity of the amino acid sequence was shown, when comparing
with the -subunits of rat, mouse, bovine, pig, dog, cat and rabbit,
75–76% identity was found, and when comparing with orthologues of birds,
amphibians, reptiles, and fish only 70–73% identity was shown.
-Subunits vary greatly in primary structure and determine the type of
gonadotropin. When comparing the -subunits of human LH, hCG and FSH,
only 33% of identical AARs were identified. At the same time, cysteine residues,
which determine the 3D structure of -subunits and are responsible for
the formation of functionally active -heterodimer complexes,
are highly conserved. The degree of identity of the sequences 45–153
-LH and 29–139 human -hCG is significantly higher and amounts
to 83%, which indicates their structural and functional similarity and
determines the ability of these hormones to specifically bind to the same
receptor. The homology of the -subunits of LH or hCG varies among
different animal species, but on average is quite high [18, 76]. Thus, when
comparing human and monkey -LH, the identity of the primary structure
varies only from 95 to 99%, and when comparing human -LH with rat,
mouse, rabbit and cat -LH it decreases to 72–75%. Moreover, among
various species of primates, the homology of -LH and -CG is
noticeably higher than when compared with -LH of other animals, which
indicates a relatively late divergence of -LH and -CG in the
evolution of higher vertebrates.
In the 1990s, based on X-ray diffraction data, the 3D structure of the
heterodimeric hCG was first identified [77, 78], and subsequently the 3D
structure of the LH was established [79, 80]. The main structural characteristic
of the - and -subunits that make up gonadotropins is the
presence of intramolecular disulfide bonds in them. They connect segments of
- and -subunits that are distant from each other, causing them
to cross each other and form a rigid knot structure called a cystine knot.
Cystine knots are localized in the central part of the - and
-subunits and stabilize three loops (L1, L2, and L3) extending from the
center of the dimer. Two of them (L1 and L3) are rigid in structure and have the
shape of a hairpin (the so-called hairpin structures), while the L2 loop is more
flexible and is located on the opposite side from the center of the molecule [76, 79]. In a heterodimer, the - and -subunits are located
symmetrically with respect to each other, have an elongated shape and are
characterized by a large ratio of surface area to volume of the molecule. The
polypeptide, which corresponds to the C-terminal region of the
-subunit and extends beyond its central part, held together by cystine
knots, functions as a safety belt by wrapping around the antiparallel
-helices that form the L2 loop of the -subunit. The 3D
structure of the seat belt is stabilized by an intramolecular disulfide bond, the
formation of which involves cysteine residues, one of which is localized in the
L1-loop of the -subunit, and the other is closer to its
C-terminus (Cys and Cys in -hCG). It should be
noted that the segment that forms the central part of the seat belt of the
-subunit (the region 93–100 in -hCG) determines the
specificity of the interaction of gonadotropin with LHCGR. It is important that
in -LH and -hCG this segment has a net positive charge, which
determines its interaction with the negatively charged orthosteric site LHCGR
[79, 81]. The C-terminal segment 88–92 of the -subunit of hCG
is also involved in binding to LHCGR [82, 83].
Both - and -subunits of gonadotropins undergo
N-glycosylation because they contain asparagine-containing sites,
targets for N-glycosyltransferases, with the consensus motifs
Lys-Asn-(Val/Ile) or (Glu/Tyr)-Asn-His [18]. In the -subunit, common to
all gonadotropins, two such sites are localized (Asn, Asn), while
in the -subunit there are one (-LH, Asn) or two
(-hCG, Asn and Asn) sites for N-glycosylation.
The -subunit of FSH, like -hCG, also has two sites for
N-glycosylation (Fig. 2). The C-terminal extension of
-hCG also has four sites for O-glycosylation, including the
residues Ser, Ser, Ser, and Ser as targets. Sites
that are modified by N-glycans are located in all three loops (L1–L3)
of the - and -subunits [18, 19, 84, 85] (Fig. 2). The degree
of glycosylation, localization and structure of N-glycans (branching,
charge, etc.) in - and -subunits make a significant
contribution to the formation of gonadotropin heterodimeric complexes and their
stability, and also determine the binding characteristics and effectiveness of
gonadotropins, the bias of their signaling, and affect their pharmacokinetics
[18, 19, 85, 86] (see also Section 6).
 Fig. 2.
Fig. 2.
The N- and O-glycosylation of the
luteinizing hormone (LH) and human chorionic gonadotropin (hCG) subunits, as well
as the follicle-stimulating hormone (FSH) subunits shown for comparison. For
-LH, -FSH and -CG, glycoforms of
-glycoprotein hormone (-GPH) are presented, characteristic of
the molecules LH, FSH and classical (placental) hCG. All gonadotropin subunits
show sites for N-glycosylation, including asparagine residues. In the
C-terminal part of the -subunits of human and equine CG, sites
for O-glycosylation, including serine and threonine residues, are also
localized. The most typical structures of N-glycans characteristic of
the presented gonadotropins are presented. In the - and
-subunits of human and horse LH and CG, weakly branched (hybrid and
bi-antennary) N-glycans predominate, and in LH secreted by the pituitary
gland there is more terminal sulphated N-acetylgalactosamine (GalNAc),
and in CG secreted by the fetus and placenta, sialic acid residues predominate.
The - and -subunits of FSH contain a significant number of
more branched (three- and four-antennary) N-glycans enriched in sialic
acid residues. It should be noted, however, that under specific conditions (a
certain phase of the estrous cycle, age, pathological conditions, etc.),
N-glycans can differ significantly structurally, both between the same
types of subunits and between the - and -subunits that form
the dimeric gonadotropin complex. The terminal sialic acid residues are indicated
by yellow squares, and the terminal sulphated N-acetylgalactosamine
(GalNAc) residues are indicated by red circles. CG, chorionic gonadotropin; eCG,
equine chorionic gonadotropin.
A representative of a rather unusual group of gonadotropins with LH activity is
equine chorionic gonadotropin (eCG), which is encoded by a single gene and
combines the properties of LH and FSH when acting on the reproductive system of
various (non-equid) mammals [87, 88, 89, 90]. Unlike hCG, the -subunit of eCG
has only one site for N-glycosylation (Asn), but in the
C-terminal region it contains up to 11–12 sites for
O-glycosylation, and the targets of O-glycosyltransferases are
serine (Ser, Ser, Ser, Ser, Ser,
Ser, Ser, and Ser) and threonine residues (Thr,
Thr, Thr, and Thr) [91] (Fig. 2). The degree of
glycosylation of these sites ranges from 20 to 100%, and this is the reason that
-eCG is one of the most highly glycosylated proteins, in which the
glycosyl component accounts for up to 40% of the molecular weight. Along with
-eCG, the horse has -LH, which is also encoded by a single gene
and has dual specificity, activating both gonadotropin receptors, LHCGR and FSHR.
Like -eCG, -eLH has up to 11 sites for
O-glycosylation, but is glycosylated to a lesser extent (the degree of
glycosylation of these sites is from 10 to 77%) [87, 91, 92] (Fig. 2).
3. Structure of the Luteinizing Hormone/Chorionic Gonadotropin Receptor
and the Mechanisms of Its Binding to Gonadotropins
The LHCGR belongs to the superfamily of serpentine receptors functionally
coupled to heterotrimeric G proteins (GPCRs), and is included in the group
of the rhodopsin family, together with FSHR, thyroid-stimulating
hormone receptor (TSHR), and relaxin and insulin-like factor-3 (INSL3) receptors.
Gonadotropins bind specifically to the high-affinity orthosteric site of LHCGR,
which is formed by a large extracellular domain, similar to that observed in
other receptors of the group.
The LHCGR ecdomain includes up to 360 AARs and contains two structural
subdomains, the first of which includes 9 leucine-rich repeats (LRRs), while the
second is a large hinge region. connecting the LRR subdomain to the TMD. The main
structural elements of the TMD are seven hydrophobic transmembrane helices (TMs)
that form the transmembrane tunnel. At the N- and C-termini of
the hinge region there are two more LRR segments, LRR10 and LRR11, between which
the -helix Pro–Asn (hinge helix) and an extended hinge
loop [28]. At the C-terminus of the hinge region, where the
extracellular domain connects to the TMD, a small region of P10
(Phe–Tyr) is located. The hinge region finely regulates the
binding of gonadotropins to the ectodomain and ensures the signal transduction
generated by them to the TMD, largely determining the differences in LH- and
hCG-induced intracellular signaling [28, 93].
The surface of hCG, which is an -heterodimer complex,
contains clusters enriched with positively charged AARs that electrostatically
interact with clusters of negatively charged AARs that form the ligand-binding
surface of the LHCGR ectodomain. Using cryoelectron microscopy, it was shown that
both subunits, the hCG-specific -subunit and the -subunit
common to all glycoprotein hormones (-GPH), are involved in the
interaction with LHCGR. The C-terminal segment 92–106 of -hCG
specifically interacts with the residues Arg, Ser, Ala and
Tyr, located in LRR1, and with the Glu, located in LRR7, and the
residues Val and Gln of -hCG form contacts, respectively,
with the residues Gln and Arg located in LRR10. In turn, residues
Tyr, Tyr, and Ser of -GPH interact with residues
Tyr, Ile, Lys, and Tyr, located in LHCGR repeats
LRR4–LRR6 [28]. When hCG binds to the receptor, significant conformational
changes are observed in four segments: in a region localized in -hCG,
called the “seat belt”, which is responsible for stabilizing the
-heterodimer of hCG, as well as in -sheet structures
localized in -hCG (L2, L3) and -hCG (L3) [28, 94]. Despite the
similarity of the “seat belt” in the -hCG and -LH, there are
significant differences between the -sheet structures of these subunits,
which entails significant differences in the efficiency of their interaction with
LHCGR and in the ability to selectively activate intracellular signaling
cascades, first all due to the different pattern of their interaction with the
hinge region LHCGR [28, 93].
As in the case of a number of other representatives of class A GPCRs, during
gonadotropin-induced activation of LHCGR, a change in the superposition of the
TM6 helix and the interacting TM5 and TM7 helices in the TMD occurs, despite the
fact that gonadotropin binding occurs in the extracellular part of LHCGR. Changes
in the relative position of TMs and the configuration of the internal cavity of
the TMD are a trigger for conformational changes in the heterotrimeric G protein
associated with the cytoplasmic regions of the receptor. These conformational
changes promote the guanosine diphosphate (GDP)/guanosine triphosphate (GTP)
exchange in the G subunit of the G protein and weaken its association
with the G dimer, which leads to the activation of G
subunit and G dimer-dependent intracellular signaling
cascades.
When hCG binds to LHCGR, the C-terminal segment of the TM6 moves
outward (by 12.8 angstroms), and this is accompanied by a slight outward shift
(by 2 angstroms) of the TM5 helix and an inward shift (by 3.6 angstroms) of the
TM7 helix [28]. An assessment of the distance between different segments of the
TM6 and TM7 by Xinheng He and his colleagues [95] using molecular dynamics showed
that when LHCGR binds to hCG, the distance between them increases both in the
outer vestibule of the transmembrane tunnel and in its central part, and this is
accompanied by a significant increase in the volume of the internal cavity of the
transmembrane tunnel. As a result, in the hCG-bound LHCGR, the average distance
between the -carbon atoms of the Ala and
Lys residues, localized in the extracellular ends of the TM6 and
TM7, is 12.9 1.3 angstroms, while in the hormone-unbound receptor it is
significant in short, only 7.7 3.5 angstroms. The distance between the
-carbon atoms of residues Cys and Asn,
located in the central part of the TM6 and TM7, in hCG-bound LHCGR is 8.5
0.2 angstroms, which also, although to a small extent, exceeds this value in
hormone-free receptor (7.2 0.9 angstroms).
It should be noted that in the ternary complex of hCG–LHCGR–G protein,
the distance between the TM6 and TM7 is similar to that in the double complex of
hCG–LHCGR. It is important that the calculated volume of the internal cavity of
the TMD for the double (253.7 121.1 Å) and ternary (188.4
111.3 Å) complexes significantly exceeds that for the
hormone-free receptor (313.03 162.09 Å) [95]. In this case, the
entrance to the transmembrane tunnel expands to the greatest extent, which also
occurs when small ligands bind to the TMD of a large number of GPCRs [96]. The
result of an increase in the volume of the internal cavity of the TMD and
associated changes in the superposition of its TMs is a change in the
conformation of the cytoplasmic regions proximal to the membrane, belonging to
its second and third ICLs (ICL2, ICL3) and the cytoplasmic C-terminal domain.
These regions contain the main molecular determinants responsible for the
interaction of LHCGR with various types of G proteins and -arrestins.
The basis for changes in the superposition of TMs upon activation of LHCGR by
gonadotropin is a change in the interaction between the LRR subdomain and the
hinge region of the ectodomain, on the one hand, and the TMD, primarily the ECLs,
forming the outer vestibule of the transmembrane tunnel, on the other [95]. In
the absence of hormonal activation, the close interaction between the LRR
subdomain and the TMD ensures that the latter remains in an inactive state. After
binding to gonadotropin, the interaction of the LRR subdomain with the TMD is
weakened, as indicated by a significant increase in the distance between them. In
this case, the LRR subdomain moves into a vertical position relative to the TMD.
In turn, the hinge region, on the contrary, as a result of its rotation,
approaches the extracellular vestibule of the TMD, while simultaneously moving
away from the LRR subdomain [97]. This is indicated by the results of assessing
the mobility and structural changes in the LRR subdomain, hinge region and TMD of
LHCGR during its transition from the active to the inactive state using molecular
dynamics methods. Calculations show that upon activation of LHCGR, the distance
between the LRR subdomain and the TMD can increase on average from 60 to 88
angstroms, while the distance between the hinge region and the TMD decreases from
79 to 48 angstroms [97]. Of key importance in the association and functional
coupling of the ectodomain and the TMD of LHCGR are the interactions between the
helix of the hinge region of the ectodomain and the helix formed by the middle
part of ECL1, as well as the interactions between the C-terminal segment
P10 of the hinge region, which borders the upper part of the TMD bundle, with one
side, and the outer vestibule of the transmembrane tunnel formed by the
extracellular ends of helices TM1, TM2 and TM7 and all three ECLs, on the other
[28, 95, 98, 99]. A change in the nature of these interactions upon binding of
LHCGR to gonadotropin affects the relative position of the TM5, TM6 and TM7
helices and the conformation of the TMD as a whole, including its cytoplasmic
regions, thereby activating intracellular signaling.
Among the molecular determinants located in the LHCGR ectodomain that mediate
its activation by gonadotropin, the Tyr residue plays an extremely
important role, which undergoes sulfation at the hydroxyl group in the phenolic
ring and therefore acquires a negative charge. This residue is located in the
middle of the hinge region and is surrounded by negatively charged AARs, thereby
forming a cluster with a high negative charge density. The Tyr and its
neighboring residues Asp and Glu electrostatically interact with
positively charged hCG clusters, thereby controlling the relative position of the
hinge region and the TMD of LHCGR. Substitutions of these residues with other
AARs that disrupt the integrity of the anionic cluster prevent effective
interaction with gonadotropin and lead to a significant decrease in its ability
to activate LHCGR [100]. Another important determinant is Ser, which is
involved in the formation of the Pro–Asn helix, localized in the
N-terminal part of the hinge region. It mediates the interaction of
helix 272–280 with the ECL1 helix and thereby modulates the stability of the
complex between the ectodomain and the TMD. It is also suggested that the
hydroxyl group of Ser is capable of forming a hydrogen bond with
Asn, located in the highly conserved region P10 [28], which is often
considered as a tethered agonist for LHCGR [98]. It should be noted that a
structurally similar region is also localized in the corresponding TSHR locus,
which also functions as a tethered agonist.
Along with the full-length forms of LHCGR, shortened soluble forms that lack the
transmembrane domain are generated in men and women [101, 102]. The blood level
of the soluble LHCGR form negatively correlates with the success of embryo
implantation and is one of the risk factors for premature birth and miscarriage
[103]. Soluble LHCGR forms, localized in the cytosol, make a significant
contribution to the malignancy of adrenal cells [104]. These forms of the LHCGR
are capable of negatively influencing LH signaling by suppressing the effects of
LH and hCG, including the formation of inactive heterodimeric complexes with
protomers of the full-length LHCGR, and thus can be considered as negative
allosteric LHCGR modulators [105]. In addition, the protomers of the soluble
LHCGR form a complex with FSHR and prevent its translocation into the plasma
membrane, thereby reducing the number of surface FSHRs and weakening
FSH-dependent responses [106].
4. Common Principles of Organization and Functioning of
Gonadotropin-Regulated Signaling Cascades
4.1 Intracellular Signaling Cascades, Targets of LH and hCG
Specific binding of -heterodimers of LH and hCG to the
extracellular domain of LHCGR induces conformational rearrangements in it, which,
as noted above, affect the hinge region and the ECL2 and ECL3 contacting this
region. The relative position and conformational mobility of ECL2 and ECL3
directly affect the structure of the TMD and the interaction of ICL2 and
especially ICL3 with the G proteins and -arrestins. Using a multi-step
mechanism of conformational rearrangements, gonadotropins regulate intracellular
signaling cascades, both through the activation of various types of G proteins
associated with LHCGR, and by recruiting adapter and regulatory proteins,
including various isoforms of -arrestins, into complex with the
receptor. It is necessary to take into account the fact that LHCGRs embedded in
the membrane are capable of forming both homodimeric (homooligomeric) complexes
and heterodimeric (heterooligomeric) complexes with other GPCRs, including FSHR.
This will have a significant impact on the molecular mechanisms and selectivity
of signal transduction from the extracellular domain of LHCGR to intracellular
effector proteins. Transactivation of LHCGR is also important. It consists in the
fact that the hormone binds to one of the protomers of the di(oligo)meric
receptor complex, while signal transduction to intracellular effectors occurs
through another protomer, and the latter can be the protomer of another GPCR, for
example, FSHR.
Two pathways play a decisive role in the implementation of gonadotropin
signaling via LHCGR [12, 14, 15]. There are the cyclic adenosine monophosphate
(cAMP)-dependent pathway, including LHCGR–G protein–adenylyl cyclase
(AC)–cAMP–protein kinase A (PKA)/Exchange Protein directly activated by Cyclic
AMP (EPAC) family factors, and the phospholipase pathway, including
LHCGR–G protein–phosphoinositide-specific phospholipase C
(PLC)–diacylglycerol (DAG)/inositol-3,4,5-triphosphate (IP3)–protein
kinase C (PKC)/calcium signaling (Fig. 3). In cells of the reproductive system,
they participate in the gonadotropin-induced regulation of steroidogenesis, in
the control of proliferation, angiogenesis, apoptosis, autophagy and other
fundamental cellular processes [12, 14, 15]. In this case, -arrestin
pathways, which have been intensively studied recently, also play a significant
role. Through them, the cascade of mitogen-activated protein kinases (MAPKs) is
activated, the regulation of which can also be carried out through a G
protein-dependent pathway, including cAMP-activated PKA (Fig. 3). These signaling
pathways themselves and their regulation by gonadotropins with LH activity are
discussed below, with an emphasis on the differences in the signaling properties
of LH and hCG.
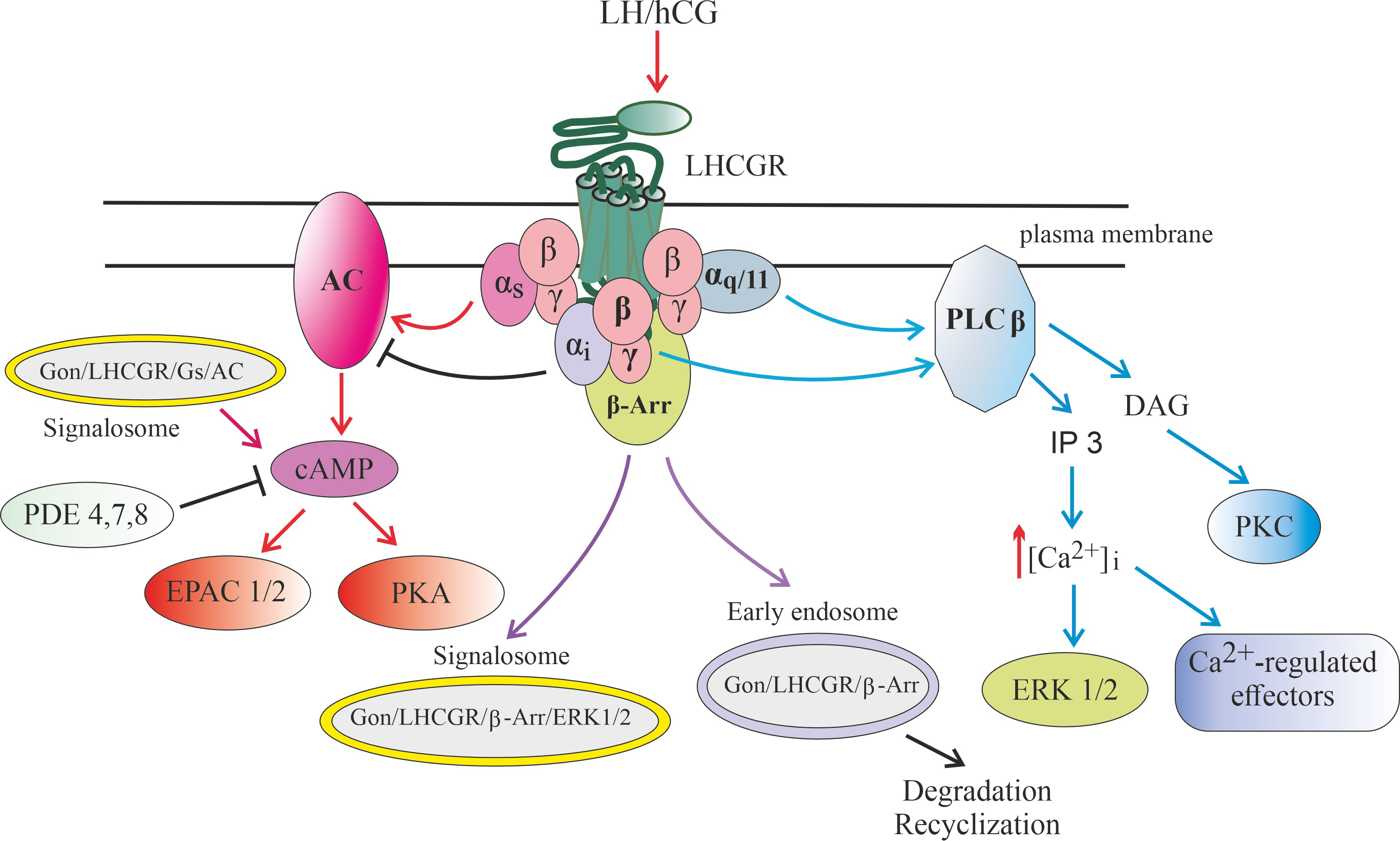 Fig. 3.
Fig. 3.
Signaling pathways realized through luteinizing
hormone/chorionic gonadotropin receptor (LHCGR), as well as endosomal signaling
carried out with the participation of gonadotropin-bound LHCGR. Abbreviation: AC, adenylyl cyclase; cAMP, 3-5-cyclic adenosine
monophoshate; -Arr, -arrestin; [Ca], intracellular
concentration of calcium ions; DAG, diacylglycerol; EPAC1/2, Exchange Protein
directly activated by Cyclic AMP, types 1 and 2; ERK1/2, extracellular signal-regulated kinases, types 1 and 2; , ,
, subunits of
-heterotrimeric G, G and G
proteins, respectively; IP3, inositol-3,4,5-triphosphate; LHCGR, receptor of
luteinizing hormone (LH) and chorionic gonadotropin (CG); PDE4,7,8,
cAMP-activated phosphodiesterases, types 4, 7 and 8; PKA, protein kinase A; PKC,
phorbol-sensitive protein kinase C, isoforms and ;
PLC, phosphoinositide-specific phospholipase C.
Binding of LH or hCG to the receptor leads to its transition to an active
conformation and triggering of several intracellular signaling cascades, which
can be carried out both through various classes of heterotrimeric G proteins and
through arrestins. After activation of the G protein, a free GTP-bound
subunit is formed, which mediates stimulation of the
membrane-bound AC, and this leads to an increase in the level of intracellular
cAMP and stimulation of PKA and/or factors of the EPAC family.
Along with this, after the formation of the gonadotropin-receptor-G
protein-AC complex, it is possible to form a signalosome that includes this
complex, which is translocated into the cell, providing a targeted increase in
the level of cAMP in its individual compartments. PKA phosphorylates a large
number of intracellular proteins that have specific sites for PKA
phosphorylation, including the transcription factor cAMP response element-binding
protein (CREB), and this largely ensures the regulatory effects of gonadotropins
on steroidogenesis, maturation of generative cells, as well as on metabolism,
growth, and survival of cells of the reproductive system.
Gonadotropin-induced dissociation of G protein leads to the generation
of a free subunit, which activates PLC, which
catalyzes the formation of second messengers, DAG and IP3. DAG causes stimulation
of DAG/phorbol-activated PKC isoforms and PKC-mediated phosphorylation of
intracellular effector proteins, while IP3 leads to activation of calcium
channels of the endoplasmic reticulum, causing the release of Ca from
intracellular stores and changes in the activity of many Ca-dependent
proteins, as well as activation of the MAPK cascade, primarily the extracellular signal-regulated kinases 1/2 (ERK1/2). Some PLC
isoforms, PLC2 and PLC3, can also be activated by the
dimer, the main donor of which is the G protein. In
this case, the subunit formed during the dissociation of this
protein can have an inhibitory effect on gonadotropin-induced AC activation,
preventing the overproduction of cAMP. In addition, a decrease in the level of
cAMP occurs due to its hydrolysis by cAMP-specific phosphodiesterases, such as
type 4 phosphodiesterase (PDE4), PDE6 and PDE7.
G protein-coupled receptor kinases (GRK)-mediated phosphorylation following hormonal activation of the LHCGR leads to
the recruitment of -arrestins, which not only inhibit G protein
signaling, but also lead to the formation of early endosomes into which the
ligand-receptor complex is included. Depending on the pattern of GRK
phosphorylation, the resulting endosomes can either provide endosomal trafficking
of such a complex with its subsequent lysosomal degradation or recycling of the
LHCGR into the plasma membrane, or form a signalosome responsible for the
activation of intracellular effectors, including ERK1/2, the effector components
of the MAPK cascade.
4.2 cAMP-Dependent Signaling Pathways
Hormones, including LH and hCG, stimulate cAMP-dependent effector proteins and
transcription factors through the “classical” AC signaling system, including as
the main components GPCR, a heterotrimeric G protein consisting of a
G subunit and a G dimer, and a membrane-bound
isoform of AC, the catalytic component of this system. AC catalyzes the reaction
of converting ATP into cAMP, which is a universal second messenger. After binding
to the hormone, the conformation of the G protein-binding surface of the receptor
changes, which includes segments of the ICLs and the intracellular vestibule of
the tunnel formed by the TMD. This ensures effective interaction of the receptor
with the G protein and its activation, including the replacement of GDP in
the guanine nucleotide-binding site of the G subunit with GTP and
the subsequent dissociation of the GTP-bound G subunit from the
G dimer. The monomeric Gs subunit in the active,
GTP-bound state is capable of maintaining weak bonds with the
G dimer, which allows it later, after hydrolysis of GTP and
the transition of the G subunit to the inactive, GDP-bound state,
to reassociate with G dimer at a high rate to form the
inactive complex. The GTP-bound G subunit interacts with
regulatory sites of AC, causing an increase in its catalytic activity and
stimulating the production of intracellular cAMP.
Synthesized cAMP interacts with effector proteins specific to it, primarily with
serine/threonine PKA and factors of the EPAC (Exchange Protein directly activated
by Cyclic AMP) family, which are also called cAMP-regulated guanine nucleotide
exchange factors exchange factor (cAMP-GEF) (Fig. 3). PKA in its inactive state
is a heterotetrameric complex consisting of two regulatory (inhibitory) and two
catalytic subunits. Each regulatory subunit includes two cAMP binding sites and
two sites located in the N-terminal region that interact with the
catalytic subunits [107]. When bound to cAMP, the regulatory subunits dissociate
from the complex, the released catalytic subunits are activated and catalyze the
transfer of the -phosphate group from ATP to the
serine/threonine-containing site of the phosphorylated protein, the target of
PKA. The main targets of the enzyme are cAMP-regulated transcription factors,
including the factor CREB (cAMP-responsive element binding protein), which
controls the expression of many genes.
The EPAC family factors, Epac-1 (cAMP-GEF-I) and Epac-2 (cAMP-GEF-II), have one
(Epac-1) or two (Epac-2) cAMP-binding sites in the N-terminal part, and
in their C-terminal part there is a catalytic GEF domain, the function
of which is to ensure GDP/GTP exchange and activation of small G proteins, such
as Rap1 and Rap2 [108]. As in the case of PKA, one of the targets of the EPAC1
and EPAC2 is the transcription factor CREB, through which they regulate gene
transcription, cell growth, apoptosis, cell migration, and mitochondrial dynamics
[109]. It should be noted that, in addition to the activation of small G
proteins, EPACs also activate a number of other effector proteins, including
PLC [110], Ca-calmodulin-dependent protein kinase II (CaMKII)
[111], phosphatidylinositol 3-kinase [112], and the components of the MAPK
cascade [113, 114]. All this significantly expands the range of their
physiological effects in response to cAMP stimulation. Since the dissociation
constants for the binding of cAMP to PKA and EPACs have similar values, in each
specific case the choice of effector is determined not by the intensity of cAMP
production, but by the availability of PKA and EPACs for activation by the cyclic
nucleotide [115].
In addition to the effector components of cAMP signaling, phosphodiesterases
(PDEs) play a significant and, in some cases, a decisive role in its regulation,
causing the degradation of cyclic nucleotides, including cAMP, and thereby
terminating the transduction of cAMP-dependent signals into the cell. The cells
of the male and female reproductive system contain PDE4, PDE7 and PDE8, which are
highly selective towards cAMP and hydrolyze it to inactive adenosine
5-monophosphate (Fig. 3). Importantly, the expression and activity of these
PDEs are characterized by cell and tissue specificity. In the ovaries, the PDE8A
and PDE8B isoforms are found in significant quantities in theca cells and
oocytes, the PDE4A isoform is found in oocytes and medullar stromal tissue, the
PDE4C and PDE4D isoforms are found in follicles, the PDE7A and PDE7B isoforms are
found in oocytes, while the PDE4B isoform is mainly in the outer layer of theca
cells [116]. Dual-specificity PDEs that hydrolyze both cAMP and cyclic guanosine
monophosphate (cGMP) may play a certain role in the control of cAMP signaling,
for example, the ovarian isoform PDE3B. The hydrolytic activity of PDE7 and PDE8
is detected even at low concentrations of cAMP inside the cell, while PDE4 is
activated only by relatively high concentrations of cAMP, under conditions of
intense AC stimulation with gonadotropins [116]. Leydig cells have demonstrated
high activity of the PDE8A and PDE8B isoforms and have been shown to be involved
in the regulation and modulation of the steroidogenic effects of gonadotropins
through cAMP-dependent pathways, and inhibition of these PDEs leads to increased
signal transduction, similar to hormonal stimulation [117, 118]. Thus, specific
PDE inhibitors mimic the stimulating effect of gonadotropins with LH activity on
target cells, which is of great practical importance for the pharmacology of
reproductive disorders [116, 118, 119, 120, 121, 122].
Phosphatases, which are targets for PKA and undergo phosphorylation, can also
play a certain role in the transduction of the LH-induced signal. It has been
shown that in ovarian granulosa cells the protein phosphatase 1 regulatory
subunit 12A (PPP1R12A) and serine/threonine-protein phosphatase 2A 56 kDa
regulatory subunit delta isoform (PPP2R5D) undergo cAMP-dependent
phosphorylation, which leads to changes in the functional activity of the enzymes
and causes dephosphorylation and inactivation of the receptor guanylate cyclase
natriuretic peptide receptor 2 (NPR2), which is involved in the control of oocyte meiosis [123]. However, there is
no reliable data on the direct influence of phosphatases on upstream components
of cAMP signaling.
Important for cAMP signaling are scaffold proteins, which ensure the integration
of GPCR, G proteins, AC, PKA, PDEs and other signaling and effector proteins into
multicomponent molecular assemblies. Among scaffold proteins, proteins of the
A-Kinase Anchoring Proteins (AKAP) family, which specifically interact with the
regulatory subunit of PKA, play an important role in the control of cAMP
signaling. This interaction includes the N-terminal helical domain of
the PKA regulatory subunit, responsible for its dimerization and docking
(N-terminal Dimerization/Docking domain), and the amphipathic
-helix of AKAP. These helical regions form a superhelical structure
responsible for the retention of PKA in a particular cellular compartment and for
the specificity of its interaction with target proteins [124]. Ezrin, radixin and
moesin (ezrin-radixin-moesin, ERM), which are responsible for the formation and
reorganization of the cytoskeleton and lipid rafts, participate in the regulation
of the activity of EPACs. By specifically interacting with the
N-terminal region of EPAC1, ERM proteins ensure the translocation of
EPAC1 to the membrane and its inclusion in the signalosome, thereby redirecting
cAMP signaling to the EPAC-dependent pathway [125].
4.3 Phospholipase Signaling Pathways
The result of hormonal activation of the G protein, mediated through
GPCRs, including LHCGR, is the exchange of guanine nucleotides in the
-subunit of the G protein, the dissociation of the GTP-bound
G subunit from the G dimer and its
functional interaction with phosphoinositide-specific PLC. Activated in
this way, PLC hydrolyzes the membrane-bound phosphatidylinositol
4,5-bisphosphate (PIP2), generating two important second messengers, such as
membrane-bound diacylglycerol (DAG) and water-soluble inositol-1,4,5-triphosphate
(IP3). DAG causes activation of phorbol-sensitive PKC isoforms, while IP3
mobilizes calcium ions from intracellular stores [126, 127, 128] (Fig. 3). The
regulatory effects of IP3 are realized through its binding to IP3-specific
receptors localized in the membrane of the endoplasmic reticulum [129]. This
leads to the leakage of calcium ions from intracellular stores, an increase in
their concentration in the near-membrane space of the endoplasmic reticulum and,
as a consequence, to the activation of Ca-activated ryanodine receptors,
which have a high conductivity for Ca [130]. A rapid increase in Ca
concentration inside the cell leads to the activation of a large number of
calcium-regulated proteins, primarily Ca-calmodulin-dependent, among which
the most important are various isoforms of Ca-calmodulin-dependent protein
kinase II [127]. The rapid increase in Ca concentration in certain cell
compartments caused by IP3 is subsequently quickly eliminated by pumping
Ca out of the cytosol using both plasma membrane calcium channels and
Ca-ATPases localized in the sarcoplasmic reticulum membrane. This is due
to the need to protect cells from hyperactivation of Ca-dependent effector
systems, which reduces cell survival. Termination of the G-mediated
signal is also carried out by the elimination of DAG and IP3, which are recycled
and, after phosphorylation, are converted back into PIP2, replenishing the
reserves of this phosphoinositide for the subsequent signal transduction cycle
[131, 132]. Within just a few minutes after hydrolysis induced by activation of
the G-mediated phospholipase pathway, rapid restoration of the PIP2 pool
begins, despite continued exposure of the cell to the hormonal stimulus.
Back in the early 1990s, it was found that the hormone-activated G
protein, or more precisely its G subunit, stimulates all four
known isoforms of PLC (PLC1, PLC2, PLC3 and
PLC4), while two of them, PLC2 and PLC3, can also be
activated by the G dimer, the main donor of which is the
pertussis toxin-sensitive G proteins [133, 134, 135]. Since the generation of
free G dimer occurs mainly due to the activation of
G-coupled GPCRs, their agonists, like agonists of G-coupled
GPCRs, can activate phospholipase pathways. Moreover, in the case of
PLC2 and PLC3, a synergistic calcium response can be observed
when agonists act simultaneously on G- and G-coupled GPCRs
[136]. Another possibility is a synergism between the G- and
G-mediated signaling pathways, which are activated by the hormone through
the same GPCR, characterized by coupling to both types of G proteins, which is
also true for LHCGR.
The target of the G subunit and G dimer is
the C-terminal domain of PLC, which, being in close interaction
with the catalytic domain, inhibits the hydrolytic activity of PLC. In
the case of the G subunit, both the distal and proximal
regions of the C-terminal domain are involved in binding to it, while in
the case of the G-dimer, only its distal regions, and the
interaction of PLC with the G dimer induces more
pronounced conformational changes in the C-terminal domain [137]. In
both cases, binding of the C-terminal domain to the G protein prevents
the inhibitory effect of this domain on the catalytic domain and ensures high
levels of PLC activity.
4.4 -Arrestin Signaling Pathways
Adapter proteins -arrestins, which, like G proteins, are capable of
specifically interacting with the membrane-proximal regions of the ICLs and the
cytosol-oriented vestibule of the GPCR transmembrane tunnel, are present in all
types of cells and tissues and are involved in the control of most physiological
processes [138, 139, 140, 141]. Of decisive importance for GPCR signaling, including that
realized through LHCGR, are two forms of -arrestins, such as
arr1 and arr2, which are also designated as
-arrestins-2 and -3 (78% homology of the primary structure).
arr1 and arr2 are cytosolic proteins, although arr1
can be localized within the nucleus. Their main function is the ability to
disrupt the interaction of ligand-activated GPCR with G protein and cause
internalization of the ligand-receptor complex within the early endosome into the
cell [142]. Along with this, -arrestins form an active complex with
GPCRs, which has its own signaling functions. This complex is responsible for the
activation of a number of intracellular effectors, including ERK1/2
(extracellular signal-regulated kinases, types 1 and 2), the effector
components of MAPK cascade [141]. Formation of an active complex with the
receptor requires dephosphorylation of Ser (arr1) or Thr
(arr2) located in the C-terminal part of -arrestins,
which are in a phosphorylated state before interacting with the ligand–GPCR–G
protein complex [141, 143]. It is important to note that dephosphorylation of
-arrestins is not necessary for -arrestin-mediated GPCR
desensitization that occurs upon receptor association with -arrestins,
since this process depends primarily on the pattern of -arrestin
isoforms and their relationship with receptors and other components of signal
transduction.
-Arrestin-mediated GPCR desensitization is due to the interaction of
-arrestin with receptor sites that, after hormonal activation, are
phosphorylated either by GPCR-specific kinases (GRK, homologous desensitization)
or by low-specific PKA and PKC (heterologous desensitization). However, in the
cytoplasmic regions of GPCRs, as a rule, there are several target sites for
phosphorylation by protein kinases, and the phosphorylation pattern depends on
many factors. Among them are the structural features of the active, hormone-bound
conformation of the receptor, the ability of the receptor to form homo- or
hetero-oligomeric complexes, the type and ratio of heterotrimeric G proteins
interacting with the receptor, as well as the type of protein kinases acting on
the receptor, including various isoforms of GRKs, the action of which on GPCR
sites, targets for phosphorylation, is characterized by high specificity. The
GPCR phosphorylation pattern is, in a certain sense, a “phosphocode” that
determines further signaling events involving -arrestins. Thus, the
“phosphocode” programs the internalization and subsequent degradation or
recycling of GPCRs, or favors the formation of a signalosome comprising the
GPCR--arrestin complex and the initiation of -arrestin-mediated
signaling cascades.
Among the effector components of the MAPK cascade, the main target of
-arrestins is protein kinases of the ERK-family [141, 144, 145] (Fig. 3), and various mechanisms of their activation are possible. According to one of
them, -arrestins act as scaffold proteins that ensure colocalization and
coordinated functioning of components of the MAPK cascade, such as protein
kinases Raf1, MEK1 and ERK1/2 [145, 146], and arr1 and arr2
differ in their influence on the activity of this cascade [141, 147]. Along with
this, there is evidence of the ability of -arrestins, mainly
arr1, through other mechanisms to directly activate Raf kinase, an
upstream component of the MAPK cascade [148], and a downstream effector protein,
the proto-oncogenic kinase Src [149]. The binding of arr1 to c-Raf
kinase occurs in its Ras-binding domain, since in the presence of the small
GTPase H-Ras, which also stimulates the activity of c-Raf kinase, the binding of
the enzyme to -arrestin is inhibited [148]. It has been established that
-arrestins can stimulate the activity of other members of the MAPK
cascade, such as p38-MAPK and c-Jun N-terminal kinase, but no direct interaction
between them has been detected [150, 151].
A study of the molecular mechanisms of -arrestin involvement in
GPCR-mediated signal transduction using genetically modified cells lacking
certain types of G proteins showed that -arrestin signaling, including
that mediating the activation of the MAPK cascade, requires the presence of
functionally active heterotrimeric G proteins, at least in small quantities
[152]. It is believed that the functions of -arrestins consist of fine
regulation and modulation of MAPK activity, which is based on their
allosterically determined influence on the functional interaction of the
hormone-activated receptor complex, including G proteins, with protein kinases
ERK1/2 [153, 154]. Thus, the question of the independence of -arrestin
signaling from G proteins has not been fully resolved, and the discussion here,
most likely, should be about the coordinated interaction of G proteins and
-arrestins within the signalosome at the post-receptor stages of signal
transduction.
5. Regulatory Effects of hCG and LH on Intracellular Signaling
Cascades: Similarities and Differences
5.1 Stimulating Effects of LH and hCG on cAMP-Dependent Signaling
Pathways
Back in the early 1990s, it was shown that both hormones, LH and hCG, despite
their inherent structural differences, including different glycosylation
patterns, are capable of stimulating the activity of membrane-bound AC isoforms
and increasing the level of cAMP in frog oocytes and in cultured intestinal
L-cells that express mouse LHCGR [155, 156]. Gonadotropins also stimulated the
phospholipase cascade and caused the mobilization of calcium ions from
intracellular stores. Both processes were to a certain extent independent of each
other, which indicated the absence of significant interaction between signaling
cascades realized through heterotrimeric G and G proteins, which
are transducers between hormone-activated LHCGR and the effector proteins, AC and
PLC respectively [155, 156]. An increase in cAMP levels and a sharp rise
in intracellular Ca concentration after activation of LHCGR by
gonadotropins occurred quite quickly, on average one minute after exposure,
indicating similar kinetics of these processes [11, 157]. Back in 1996, it was
suggested that G proteins, with which ligand-activated LHCGR is also able
to interact, may be involved both in the negative regulation of AC activity after
its G-mediated stimulation by gonadotropin (short negative feedback) and
also act as donors G dimer involved in the activation of
PLC2 and PLC3, responsible for the regulation of calcium
signaling and stimulation of various isoforms of PKC [158] (Fig. 3).
The half-maximal concentration (EC) for stimulation of AC activity and
increase in intracellular cAMP levels by gonadotropins are in the picomolar range
of their concentrations, while nanomolar concentrations of LH and hCG are
required to trigger -arrestin (arr2)-mediated internalization
of LHCGR [11]. Along with this, it was established that to achieve the maximum
stimulating effect of gonadotropins on cAMP-dependent cascades, occupancy of no
more than 10% of LHCGR is sufficient, while for effective translocation of
-arrestins to the ligand-receptor complex and its further
internalization, the degree of occupancy of LHCGR should have exceeded 90% [11].
Activation of AC by gonadotropins with LH activity leads to an increase in the
level of phosphorylation of the transcription factor CREB and stimulation of
CREB-dependent expression of the gene encoding the cholesterol-transporting
Steroidogenic Acute Regulatory protein (StAR), which catalyzes the first,
rate-limiting stage of steroidogenesis, as demonstrated in Leydig cells [12].
Thus, the mechanisms of the influence of LH and hCG on AC activity and
cAMP-dependent intracellular targets are characterized by a certain similarity.
At the same time, the effectiveness of such influence varies quite significantly
[10, 12, 14].
Most of the data obtained using cell cultures demonstrate a higher potential of
hCG, compared to LH, regarding the activation of the AC system and downstream
effector units (PKA) (Table 2). In cultured COS-7 cells expressing LHCGR, the
ED value for the stimulatory effect of hCG on cAMP production was shown to
be 107 14 pmol/L, while the corresponding value for LH was five times
higher at 530 51 pmol/L [9]. When using hCG, the maximum stimulating
effect on intracellular cAMP levels was achieved one hour or more after
treatment, while when using LH this occurred faster, after 10 min [9]. The
efficiency of increasing cAMP levels in HEK-293 cells with LHCGR expressed in
them when exposed to hCG was significantly higher than when exposed to LH, and
the EC value for hCG was 213 pmol/L and was significantly lower than that
for LH (426 pmol/L), with the maximum response for both gonadotropins being
achieved at a concentration of about 10 nmol/L [17]. The differences in the
effectiveness of LH and hCG were greatest in ovarian granulosa cells [14].
Treatment of a primary culture of human granulosa cells (hGLC) for 36 hours with
hCG caused a more pronounced and time-sustained increase in cAMP levels compared
to treatment with recombinant LH taken at an equivalent dose. As a result, in the
culture treated with hCG, progesterone production was significantly higher than
in the control and in the culture treated with LH, indicating a more pronounced
stimulation of ovarian steroidogenesis caused by hCG [9].
Table 2.
Comparison of the effects of luteinizing hormone (LH) and human
chorionic gonadotropin (hCG) on adenylyl cyclase activity and cyclic adenosine
monophosphate (cAMP)-dependent pathways—similarities and differences.
| Similarities |
| Both gonadotropins in picomolar concentrations stimulate the gonadotropin–LHCGR–G protein–AC–cAMP–PKA/EPAC1/2 system, resulting in stimulation of the activity of cAMP-dependent transcription factors, including CREB, and stimulation of steroidogenesis and other cAMP-dependent processes. |
| Gonadotropins exert their effects with high efficiency both in ovarian cells (theca and granulosa cells) and in testicular Leydig cells. |
| Differences |
| (1) The stimulating effect of hCG on AC and cAMP production is achieved at lower concentrations and is more stable over time than that of LH, and this difference is specific to certain cell types and is most pronounced in granulosa cells, where the AC stimulating effect of LH is weakly expressed. The result is a more pronounced stimulatory effect of hCG at the early stages of steroidogenesis, despite the fact that at the later stages of steroidogenesis the effects of the hormones become comparable. |
| (2) The stimulatory effect of hCG on the activity of ERK1/2, an effector component of the MAPK cascade, realized via cAMP-dependent pathways and PKA is more pronounced than that of LH. This, along with differences in the influence of LH and hCG on the expression of proteins that regulate apoptosis, determines that hCG preferentially activates pro-apoptotic cascades, and LH anti-apoptotic cascades. |
| (3) LH and hCG have different effects on cAMP-dependent cascades during heterodimerization of LHCGR with FSHR. In the case of hCG, there is a potentiation of the AC stimulating effect, and in the case of LH this effect does not change significantly. |
Note: AC, adenylate cyclase; CREB, cAMP-dependent transcription factor (cAMP
Response Element-Binding protein); EPAC1/2, Exchange Protein directly activated
by Cyclic AMP, types 1 and 2; ERK1/2, extracellular signal-regulated kinases,
types 1 and 2, the effector components of mitogen-activated protein kinase
cascade; FSHR, follicle-stimulating hormone receptor; LHCGR, luteinizing
hormone/chorionic gonadotropin receptor; MAPKs, mitogen-activated protein
kinases; PKA, protein kinase A.
Similar results were obtained with long-term treatment of cultured goat ovarian
granulosa cells with gonadotropins, where hCG increased the intracellular level
of cAMP and activated PKA with significantly greater efficiency than LH [159].
The stimulating effect of hCG on cAMP levels and cAMP-dependent phosphorylation
of ERK1/2 during treatment of primary Leydig cell culture also significantly
exceeded the corresponding effects of LH, in the case of stimulation of
intracellular cAMP production by almost 10 times [12]. The higher potential of
recombinant hCG for AC activation, compared with recombinant LH, was demonstrated
in mouse Leydig tumor cells (mLTC-1), in which mouse LHCGR was expressed, as well
as in HEK273 cells expressing human LHCGR [13]. This indicates that the
peculiarities of obtaining recombinant forms of gonadotropins do not
significantly affect the higher potential for stimulation of the AC system
detected in the case of hCG, since both urinary and recombinant hCG were equally
superior to LH.
It should be noted, however, that cAMP level-dependent indicators such as the
degree of phosphorylation of the CREB factor and the expression of the gene for
the steroidogenic protein StAR in cultures of Leydig cells and HEK273 cells
treated with hCG and LH did not differ significantly [12, 14]. This may be due to
counter-regulatory influences that are triggered in the target cell with a
long-term and sustained increase in cAMP concentration, among which an increase
in the activity of cAMP-specific PDEs plays a significant role. It is also
interesting that progesterone production in different cell types treated with hCG
was significantly higher than with LH treatment, while testosterone levels varied
slightly [13]. Laura Riccetti et al . [13] explain this disappearance of
differences in the testosterone levels by the fact that in the case of exposure
to LH, the synthesis of testosterone is carried out to a greater extent by the
pathway that includes 17-hydroxyprogesterone as an intermediate instead of
progesterone (the so-called 5-pathway), while hCG ensures the synthesis
of large quantities of progesterone, which functions as a “parallel accumulation
product” and then largely enters the less efficient 4 pathway of
androgen synthesis. The result of this is a leveling off of testosterone
production induced by both gonadotropins during later stages of testicular
steroidogenesis, despite significantly greater amounts of progesterone
accumulating in hCG-treated cells [13].
A study of the effects of hCG and LH (100 pmol/L) in human granulosa-lutein
cells revealed that hCG not only stimulates phosphorylation of the factor CREB
and increases the expression of the StAR protein gene with greater efficiency
compared to LH, but also enhances proapoptotic cascades to a greater extent
[160]. The proapoptotic effect of hCG was strongly attenuated in the presence of
physiological doses of -estradiol (200 pg/mL), as illustrated by
increased activity of the anti-apoptotic enzyme Akt kinase, assessed by its
Ser phosphorylation, and inhibition of the cleavage of procaspase-3, a
key component of the apoptotic pathway, leading to increased cell survival. With
regard to the expression of the aromatase (cytochrome CYP19A1) gene, the
situation is the opposite, and the stimulating effect of LH 72 hours after
treatment exceeded that of hCG. This causes an increase in aromatase activity and
stimulates the conversion of androgens into estrogens, which are necessary to
maintain the growth of follicles and oocytes and increase the survival of
granulosa-luteal cells [160]. LH also more effectively, compared to hCG,
increased the expression of genes encoding the anti-apoptotic protein XIAP
(X-linked inhibitor of apoptosis protein) and cyclin D2, thereby exerting a
pronounced anti-apoptotic effect [160]. This indicates that LH-stimulated
cAMP-dependent pathways are characterized by a greater anti-apoptotic potential,
while the corresponding hCG-activated pathways are primarily triggers of
proapoptotic cascades, although the resulting effect of gonadotropins on
apoptosis is thought to depend on from many factors and counter-regulatory
influences (Table 2).
Since testing for LH and hCG is carried out in both human and rodent cell lines
and uses gonadotropins from different sources, it is important to evaluate the
possible species-specific effects of their effects on cAMP-dependent pathways.
With various combinations of gonadotropins and target cells, their primary
effect, which consists of activation of the AC signaling system, in the case of
hCG, as a rule, significantly exceeds that of LH, and this is due to differences
in the efficiency of interaction of the ligand (LH or hCG) with the LHCGR
ectodomain and the peculiarities of conformational changes caused in the TMD and
the LHCGR–G protein interface due to such interaction [12, 13, 14, 160, 161, 162].
However, at later, effector, stages of hormonal signal implementation, at the
stage of phosphorylation of the factor CREB and the final stages of
steroidogenesis, the differences in the effectiveness of LH and hCG begin to
weaken and, ultimately, may disappear altogether. It should be noted, however,
that this applies most to the stimulation of testicular steroidogenesis, while in
follicular cells the differences between LH and hCG tend to persist into the
later, effector stages of gonadotropin signaling [12]. To a certain extent, the
equivalence of LH and hCG in terms of stimulation of testicular steroidogenesis
is important for the treatment of androgen deficiency and hypogonadotropic
hypogonadism in men with gonadotropins. Thus, there is an observation that
treatment of a man with central hypogonadism sequentially with LH and hCG
normalized his blood testosterone level to the same extent [163].
An important point is that the AC-stimulating effects of hCG and LH may be
modulated differently in the presence of FSH. This is due to both the
heterodi(oligo)merization of LHCGR and the structurally similar FSHR, which
changes the efficiency and pattern of their activation by gonadotropins, and the
cross-interaction of intracellular signaling cascades activated by FSH and
gonadotropins with LH activity inside the target cell. Under the in
vitro conditions using human granulosa-luteal cells, it was shown that in the
presence of FSH, the ability of hCG to enhance the production of intracellular
cAMP increases on average five times, which entails increased phosphorylation of
the factor CREB and the steroidogenic response to hCG [164]. In this case, the AC
stimulating effect of recombinant LH in the presence of FSH does not change
significantly, as a result of which treatment of cells with a combination of
recombinant LH and FSH does not lead to a significant increase in the level of
CREB phosphorylation and the production of steroid hormones (Table 2).
Interestingly, FSH potentiates the stimulatory effects of LH (not hCG) on the
activity of ERK1/2, a key effector component of the MAPK cascade, and on the
activity of Akt kinase, an important mediator of anti-apoptotic processes, which
leads to increased cell growth and survival [164]. The effect of FSH on hCG- and
LH-induced signaling is in a good agreement with the powerful proliferative
potential of endogenous LH in the follicular phase and after trophoblast
formation, as well as with the strongly pronounced steroidogenic effect of
classical hCG, necessary to maintain pregnancy after leaving the luteal phase.
The participation of LH-stimulated cAMP-dependent mechanisms in the positive
control of proliferation was shown for a pool of primordial follicles in the
ovaries of prepubertal female mice treated with the anticancer drug cis-platinum,
which blocks cell growth [165]. Simultaneous administration of LH and
cis-platinum to mice preserved the activity of cAMP-dependent signaling pathways
and prevented a decrease in the reserve of primordial follicles, maintaining the
fertility of animals when they reached reproductive age [165]. At the same time,
it cannot be excluded that the different effectiveness and pattern of activation
of the effector components of the cAMP-dependent pathways caused by LH and hCG
may differently influence the proliferative response of follicular cells to these
gonadotropins.
It is a well-known fact that with long-term exposure of target cells to
gonadotropins, there is a significant decrease in the density of LHCGR on the
cell surface, which is based on the internalization of ligand-receptor complexes
as part of early endosomes into intracellular compartments. A factor that
accelerates the internalization of LHCGR is the formation of large
LHCGR-containing aggregates [166]. Using fluorescence microscopy in the early
2000s, it was shown that such aggregates are formed much faster and in larger
quantities when LHCGR binds to hCG than when they bind to LH, and this causes a
higher rate and intensity of desensitization and downregulation of LHCGR when
they are activated by hCG [167, 168]. Further studies showed that after
hCG-induced activation, LHCGR accumulate in the form of significant aggregates
inside the cell, both in detergent-resistant membrane microdomains with an
average diameter of about 170 µm, and in smaller membrane structures with
an average diameter of 80 to 160 µm. In cells without hCG treatment, a
significant portion of LHCGR was localized in larger membranous structures (with
an average diameter of about 200 µm), which were able to efficiently
integrate into the plasma membrane. As soon as hCG was removed from the medium,
large aggregates began to gradually dissociate and the released LHCGR moved into
larger membrane vesicles, thereby acquiring the ability to effectively
translocate into the plasma membrane and bind to the hormone [169]. LH with less
intensity induced the formation of large aggregates containing LHCGR, which led
to a less pronounced decrease in the sensitivity of target cells to gonadotropins
with LH activity and to a more rapid recovery after LHCGR desensitization caused
by prolonged exposure to gonadotropin (Table 2). The ability of hCG with greater
intensity, as compared to LH, to induce the formation of large receptor
aggregates is associated with a more effective interaction of the hCG–LHCGR
complex with -arrestins, which mediates the internalization of the
ligand-receptor complex with subsequent aggregation of LHCGR and their movement
into relatively small membrane microdomains [13].
5.2 Intracellular cAMP Signaling Activated by Gonadotropins with LH
Activity
In the recent years, the paradigm that the internalization of GPCRs into the
cell leads to the termination of hormonal signal transduction has undergone
changes, since a lot of evidence has accumulated that, being part of the early
endosome, ligand-receptor complexes continue to carry out their signaling
functions already inside the cell. This is in good agreement with the
localization of other components of cAMP signaling, primarily PKA and various PDE
isoforms, in intracellular compartments, including the nucleus [170, 171, 172].
Moreover, it is assumed that for many receptors the main contribution to the
regulation of the activity of effector proteins is made by “intracellular”
signaling [173]. This is also quite applicable for cAMP signaling mediated
through gonadotropin-stimulated LHCGR [174].
There is evidence that cAMP signaling for many types of GPCRs, including
gonadotropin receptors, involves two phases of activation of its components, and
these phases are separated both in space and time [175, 176, 177, 178]. The first, fast
phase includes signaling events that occur immediately after the hormone binds to
the receptor and occurs in the plasma membrane, where functional coupling occurs
between the ligand-activated GPCR, G protein and AC. It must be emphasized
that previously all ideas about the functioning of the hormone-sensitive AC
system were limited mainly to this phase, and further events were considered
exclusively in terms of down-regulation of the ligand-activated receptor complex
during endocytosis. However, following the fast phase, at least in some cases,
there is a constant, longer phase of cAMP signaling called the endosomal phase.
It starts after the internalization of the ligand-receptor complex as part of the
early endosome into intracellular compartments, where cAMP synthesis occurs, and
the formation of this second messenger can be spatially linked to a specific cell
compartment [175, 178, 179]. This makes it possible to influence cAMP-sensitive
effector proteins in specific compartments without inducing a total increase in
the level of cAMP in the cell, which is intended to ensure the specificity and
targeting of signal transduction.
Initially, it was believed that the implementation of the endosomal phase
requires the interaction of hormone-activated G-coupled GPCR with
-arrestin, which ensures both the internalization of the signaling
complex within the endosome and its functional activity in the second phase of
cAMP signaling. However, it was later shown that this process may be independent
of -arrestins [178]. The function of -arrestins in this case is
only to delimit the fast phase of signal transduction, interrupting signal
transduction through the G protein in the plasma membrane, from the late,
endosomal phase. In this case, in the endosome, -arrestins are no longer
involved in signaling events. It is assumed that, depending on the
phosphorylation pattern of the ligand-activated GPCR, -arrestins act as
“switchers”, redirecting some ligand–GPCR–G protein complexes along the
path of their degradation or recycling, and “packaging” other complexes into
the forming signalosomes, in which intracellular endosomal cAMP signaling occurs
[178].
For LHCGR, endosomal cAMP signaling was studied in detail using fluorescence
resonance energy transfer (FRET) microscopy in intact follicles isolated from the
ovaries of transgenic mice embedded with sensors to assess cAMP production [174].
Follicles were stimulated with LH (20 µg/mL) for 10 min, taking into
account the fact that this time was sufficient for the internalization of LHCGR,
after which the hormone was washed off. AC stimulation and increase in cAMP
levels continued for at least 20 min [174]. This is in good agreement with the
fact that lysosomal degradation of LHCGR occurs approximately 30–60 min after
the onset of their activation by the hormone [180]. It can be assumed that such
degradation is intended to stop endosomal cAMP signaling, which lasts
significantly longer than it should if only its first, fast phase was
implemented, which is inevitably followed by desensitization of LHCGR. In the
presence of dynasor, an inhibitor of clathrin-mediated endocytosis, disruption of
the second, endosomal phase of cAMP signaling was detected [174]. It is important
that a number of regulatory effects of LH are realized during long-term
stimulation of cAMP-dependent effector proteins, which may be due to endosomal
signaling.
5.3 Effects of Gonadotropins on cAMP Signaling in the Oocyte
Mediated through Components of the cGMP System
Along with the direct influence of gonadotropins with LH activity on the level
of cAMP in ovarian cells, their indirect, modulatory effects on cAMP signaling
are possible without triggering the LHCGR–G protein–AC–cAMP signaling
pathway, in which ligand-activated LHCGR and changes in intracellular levels cAMP
is realized in different cellular compartments and in different cells, as well as
at different time points. This mechanism is realized in the case of LH-dependent
control of oocyte maturation. In this case, LH, acting on ovarian granulosa
cells, regulates the activity of PDE3 in the oocyte, changing the ratio of the
concentrations of cGMP and cAMP in it and, thereby, controls the functional
activity of transcription factors responsible for the entry of oocytes into
meiosis [181, 182, 183].
In the absence of an ovulatory LH surge, FSH, through activation of FSHR in
granulosa cells, stimulates AC activity and thereby increases cAMP levels in
these cells. Along with this, the expression of natriuretic peptide precursor C
(NPPC) increases in granulosa cells, which subsequently binds to the receptor
guanylate cyclase NPR2 on the surface of cumulus cells, causing the accumulation
of cGMP in them. One of the factors activating the synthesis of cGMP is
FSH-mediated stimulation of estrogen receptors by increasing the production of
estradiol. In this case, NPPC functions as an autocrine factor, dependent on
estrogen levels and controlling the ratio of cAMP- and cGMP-dependent pathways in
the cumulus-oocyte complex. cGMP, like cAMP, enters the oocyte from cumulus cells
through connexin-37 localized in gap junctions and, through a competitive
mechanism, inhibits the activity of PDE3 [184]. Since this isoform of PDE
hydrolyzes cAMP, cGMP-induced suppression of its hydrolytic activity causes an
increase in the level of cAMP in the oocyte. It should be noted that part of cAMP
enters the oocyte from granulosa cells through the connexin system, and part is
synthesized directly in the oocyte using the constitutively active G
Protein-Coupled Receptor 3 (GPR3) and G Protein-Coupled Receptor 12 (GPR12)
functionally coupled to AC [181, 185]. Elevated cAMP levels lead to activation of
PKA, which phosphorylates and inhibits maturation promoting factor (MPF) composed
of cyclin-dependent kinase 1 (CDK1) and cyclin b1. The result of this is the
arrest of phase I of oocyte meiosis, which prevents its maturation [183].
Knockout of the genes encoding PDE3 and the GPR3 in the oocyte leads to a
decrease in the level of cAMP and stimulates oocyte maturation [186], while PDE3
inhibitors [187, 188, 189], as well as positive modulators of cAMP signaling [190]
cause the opposite effect, preventing oocyte meiosis. The relationship between an
increase in the cAMP level in the oocyte and blocking phase I of meiosis is
observed in oocytes of humans and various mammalian species, indicating the
universality of this mechanism in the control of oocyte maturation.
The LH surge, through the activation of LHCGR in granulosa cells, leads to the
suppression of the activity of intracellular androgen and estrogen receptors in
them, thereby inhibiting the expression and secretion of NPPC and preventing its
activating effect on NPR2 receptors in cumulus cells [191]. Along with this,
activation of the LH cascade causes an increase in the expression of epidermal
growth factor (EGF), which leads to activation of EGF receptors and a resulting
increase in intracellular Ca levels, which also contributes to the
inhibition of NPR2 activity. It should be noted that stimulation of EGF-dependent
pathways mediated by LH and hCG has been demonstrated in humans and a large
number of experimental animals [181, 192, 193, 194]. In addition to the suppression of
cGMP synthesis, the LH surge leads to the activation of ERK1/2-family kinases,
which prevents the entry of cyclic nucleotides, cGMP and cAMP, into the oocyte
through the connexin gap junction system. Thus, the level of cGMP in the oocyte
sharply decreases, which leads to activation of PDE3, a decrease in the
intracellular concentration of cAMP and inhibition of PKA [181, 183]. The result
of a decrease in PKA activity is the abolition of the inhibitory effect of this
enzyme on the MPF factor, which leads to stimulation of the activity of
meiosis-associated proteins and ensures the resumption of oocyte meiosis.
Despite the fact that stimulation of EGF-dependent pathways by gonadotropins
with LH activity and cross-talk between the signaling pathways stimulated by
these gonadotropins and EGF is shown for both LH and hCG, differences in the
influence of LH and hCG on oocyte meiosis cannot be excluded, and this may have
great implications for ARTs and requires further research.
5.4 Effect of hCG and LH on Phospholipase Pathways and Intracellular
Calcium Signaling
Both in the testes and ovaries, phospholipase signaling pathways, realized
through the functional coupling of LHCGR with the G proteins, play no
less important role in LH-mediated signaling than cAMP-dependent pathways [15].
The G-mediated activation of PLC is required for normal
ovulation, ensuring proper activation of progesterone receptors and follicle
rupture [195, 196] (Table 3). Stimulation of PLC activity through other
G-coupled GPCRs may also contribute to the ovulatory response, as
demonstrated for the prostanoid receptor. Activation of this receptor by
prostaglandin F2 in luteal cells stimulates the expression of the
Akr1c18 gene, encoding 20-hydroxysteroid dehydrogenase, which
causes increased conversion of progesterone to 20-hydroxyprogesterone
and is necessary for normal parturition [196]. At the same time, LH-induced
G-mediated signaling is not involved in the formation of the corpus
luteum and maintenance of pregnancy, since deletion of the gene encoding the
G subunit in granulosa cells does not lead to disruption of
these processes. Processes important for ovulation, such as the resumption of
meiosis, proliferation of the cumulus cell layer and ovarian angiogenesis,
dependent on the activity of the VEGF signaling pathways, are mediated mainly
through LH-activated cAMP-dependent pathways and occur normally in granulosa
cells deficient in genes encoding G subunits, that is, they do
not directly depend on phospholipase signaling [195, 196].
Table 3.
Comparison of the effects of luteinizing hormone (LH) and human
chorionic gonadotropin (hCG) on phospholipase pathways – similarities and
differences.
| Similarities |
| Both gonadotropins stimulate the gonadotropin–LHCGR–G protein–PLC–IP3/DAG–increased [Ca]/phorbol-sensitive PKCs system, and this requires higher concentrations of gonadotropins than in the case of AC activation. LH/hCG-stimulated phospholipase pathways are involved both in the steroidogenic response and in the regulation of growth and survival of target cells. |
| Differences |
| (1) The effects of hCG on phospholipase pathways and calcium signaling are less pronounced compared to LH and have differences in the pattern of regulation. In addition, the steroidogenic effect of hCG is highly dependent on extracellular Ca concentration, indicating a significant contribution of plasma membrane calcium channels to the hCG-induced increase in intracellular calcium levels, in addition to that of PLC activation. |
| (2) LH activates ERK1/2 through G-mediated stimulation of phorbol-sensitive PKC, and thereby has a pronounced proliferative effect on ovarian granulosa cells, while hCG is ineffective in this regard. |
Note: AC, adenylate cyclase; DAG, diacylglycerol; ERK1/2, extracellular
signal-regulated kinases, types 1 and 2; IP3, inositol-3,4,5-triphosphate; LHCGR,
luteinizing hormone/chorionic gonadotropin receptor; PKC, protein kinase C;
PLC, phosphoinositide-specific phospholipase C.
In contrast to cAMP signaling, activation of phospholipase pathways requires
significantly higher concentrations of gonadotropins, since, for example, the
EC for hCG, which ensures activation of PLC and IP3-mediated
mobilization of calcium ions from intracellular stores, is 20 times higher than
the corresponding EC values for the same gonadotropin required for
half-maximal activation of AC and an increase in the intracellular cAMP
concentration [8]. The stimulating effect of hCG on the accumulation of IP3 in
the target cell is more pronounced at high density of LHCGR and decreases at low
density, which indirectly indicates the preference for the interaction of
hormone-activated LHCGR with G rather than with G protein.
However, the EC value for hCG-induced stimulation of IP3 production at
both low and high receptor densities remains unchanged [8]. The effectiveness of
the stimulating effect of hCG on the mobilization of intracellular Ca 60
seconds after treatment with gonadotropin was significantly higher than that for
LH, but over time the differences disappeared, which was due to the activation of
the processes of pumping Ca from the cytosol. As a consequence, the
integrated values for the increase in cytosolic Ca concentration over a
three-minute interval varied to a significantly lesser extent [17].
It is necessary to distinguish mechanisms caused by activation of PLC
signaling pathways from mechanisms involving activation of L-type plasma membrane
calcium channels, which make a significant contribution to the steroidogenic
effects of gonadotropins. In this case, the entry of calcium ions into the cell
is necessary to replenish their reserves in intracellular depots. The first data
on the participation of plasma membrane calcium channels in the stimulatory
effect of gonadotropins with LH activity on the expression of steroidogenic
proteins and the production of steroid hormones were obtained in experiments on
mouse Leydig tumor cells in 1999 [197]. It was shown that in a Ca-enriched
environment (Ca concentration was 1.5 mM), the stimulating effect of hCG
on the expression of the gene encoding the StAR protein is enhanced by 1.7 times,
and this is associated with a significant increase in progesterone production. At
the same time, when complexing agents that specifically bind calcium ions or the
calcium channel blocker veropamil were added to the extracellular environment, a
sharp decrease in hCG-induced stimulation of steroidogenesis occurred. The
transcription steroidogenic factor-1 (SF-1) plays a positive role in the
Ca-induced potentiation of the steroidogenic effect of hCG, while the
nuclear receptor DAX-1 (dosage-sensitive sex reversal, adrenal hypoplasia
critical region, on chromosome X, gene 1) plays a negative role [197].
Subsequently, the role of L-type calcium channels in the modulation of
gonadotropin-stimulated steroidogenesis was demonstrated in rat granulosa cells,
where the inhibitory effect of amphetamine, which reduces the concentration of
intracellular Ca, both basal and increased under the influence of various
agents, on gonadotropin-stimulated expression of steroidogenic genes was
demonstrated [198].
Intracellular calcium signaling is involved not only in the steroidogenic
effects of LH and hCG, but also in the implementation of their proliferative
effects (Table 3), as demonstrated for LH in its action on the epithelial ovarian
cancer cells OV207 and OVCAR-3 [199]. The basis of the proliferative effects of
LH is the activation of the MAPK cascade, including its effector component
ERK1/2, and LH-induced stimulation of ERK1/2 occurs both through activation of
L-type plasma membrane calcium channels and through stimulation of the
phospholipase pathway, the effector component of which is a DAG-sensitive protein
kinase C. LH-induced migration and proliferation of cultured ovarian
cancer cells was suppressed by inhibiting the ERK1/2 stimulating effect of
gonadotropin, which can be achieved by reducing the extracellular Ca
concentration using chelating agents, as well as inhibiting calcium current
across the plasma membrane using the calcium channel blocker verapamil and by
suppressing release of Ca from intracellular stores by dantrolene. In
addition, the stimulatory effects of LH on ERK1/2 were suppressed in the presence
of protein kinase C inhibitors, the nonselective GF 109203X and the
highly selective rottlerin, as well as small interfering RNA specific for this
enzyme [199]. It was found that cAMP-dependent signaling pathways, through which
phosphorylation and activation of ERK1/2 can also occur, are not involved in this
process [199].
Since hCG activates cAMP-dependent pathways with much higher efficiency, it is
not surprising that it affects phospholipase pathways and ERK1/2 activity with
significantly less efficiency and, accordingly, has a low proliferative potential
and, moreover, even has antiproliferative activity. This is supported by the
results of a comparative study of long-term exposure of a primary culture of goat
granulosa cells to LH and hCG [159]. It was found that LH significantly
stimulates the activity of DAG-sensitive protein kinase C and significantly
increases the ratio of phosphorylated and non-phosphorylated forms of ERK1/2,
which is associated with activation of ERK1/2-mediated proliferation of
follicular cells, while hCG does not affect these indicators and, on the
contrary, reduces the proliferation of granulosa cells, acting through
cAMP-dependent mechanisms [159]. These data indicate a different pattern of
effects of LH and hCG on phospholipase pathways and intracellular calcium
signaling involved in the control of cell growth and differentiation (Table 3).
5.5 hCG- and LH-Induced Interaction of the LH/hCG Receptor with
-Arrestins
After activation of LHCGR by gonadotropin, which induces its functional
interaction with G proteins, the process of recruitment of -arrestins is
triggered, resulting in the internalization of the ligand-receptor complex as
part of early endosomes into the cell, and this leads either to the degradation
of LHCGR or to the induction of intracellular signaling [200]. It is important
that the mechanisms of interaction with -arrestins for LHCGR differ
significantly from those in the case of FSHR. Recruitment of -arrestins
and the formation of their complex with hormone-activated FSHR requires
site-specific phosphorylation of the cytoplasmic C-terminal domain of
the receptor at serine and threonine residues by various kinases of the
GRK-family. Moreover, depending on the set of phosphorylation sites in the
C-terminal domain, -arrestins can cause either desensitization
of FSHR or its inclusion in signaling complexes in early endosomes [201, 202, 203].
In the nonphosphorylated state, FSHR is unable to bind effectively to
-arrestins. This distinguishes it from LHCGR, which is able to recruit
-arrestins while in a non-phosphorylated state, making LH-regulated
-arrestin pathways independent of the pattern and activity of GRKs
[204]. The interaction of LHCGR with -arrestins involves the receptor
ICL3 and ADP-ribosylating factor 6 (ARF6) [22, 204, 205, 206]. In the inactive,
GDP-bound state, ARF6 binds to -arrestin, and the resulting complex is
anchored in the plasma membrane, being available for interaction with cytoplasmic
regions of LHCGR. After activation of LHCGR by gonadotropin, a transient complex
is formed between the receptor and the ARF6 protein, which leads to the exchange
of GDP for GTP at the nucleotide-binding site of ARF6. In this case, a
reorganization of the complex occurs between the active, GTP-bound form of ARF6
and -arrestin, as a result of which the latter is released from the
complex with ARF6 and binds to ICL3 of LHCGR, which ensures down-regulation and
internalization of the ligand-receptor complex [203, 206].
A comparative analysis of the effects of LH and hCG on the recruitment of
-arrestins demonstrates that hCG is more active in this regard, causing
-arrestin-mediated effects much faster and at lower concentrations
(Table 4). This is indicated by the EC value for hCG-induced arr2
recruitment in mouse Leydig tumor cells (about 10 nM), which is 13 times lower
than that for LH (130 nM). Moreover, based on the magnitude of the stimulatory
effect on -arrestin recruitment, LH can be classified as a partial
agonist of LHCGR, in contrast to hCG, which exhibits agonistic activity for
-arrestin-specific signaling [13]. As noted above, cAMP-dependent
pathways play a key role in stimulating steroidogenesis, and hCG is a more
effective activator of steroidogenesis compared to LH up to the stage including
progesterone synthesis. In this regard, it is of interest that
-arrestins positively regulate the synthesis of progesterone by
gonadotropins, since the reduction of their expression by microRNAs leads to a
significant inhibition of the production of this steroid hormone, a precursor of
androgens and estrogens [13]. hCG, in comparison with LH, more effectively
stimulates the recruitment of -arrestins, as a result of which this
effect, along with more pronounced stimulation of AC, can provide higher
steroidogenic activity of hCG, which, however, is limited to the stage of
progesterone synthesis. At later stages of testicular and ovarian
steroidogenesis, the steroidogenic activity of hCG becomes comparable to that of
LH, resulting in similar final steroidogenic responses to LH and hCG drugs
observed in the clinic [163].
Table 4.
Comparison of the effects of luteinizing hormone (LH) and human
chorionic gonadotropin (hCG) on -arrestin pathways – similarities and
differences.
| Similarities |
| Following activation of LHCGR, both gonadotropins, LH and hCG, induce the recruitment of -arrestins, thereby internalizing and down-regulating the activated receptor. Along with this, activation of LHCGR by gonadotropins can induce intracellular (endosomal) signaling, which involves -arrestins. |
| Differences |
| (1) The stimulating effects of hCG on the recruitment of -arrestins are more pronounced in comparison with those of LH and are realized at lower concentrations, and therefore hCG is considered as a full agonist in relation to the recruitment of -arrestins, while LH is a partial agonist. |
| (2) hCG strongly stimulates -arrestin-mediated internalization and down-regulation of LHCGR, while the corresponding effects of LH are much less pronounced, including even with prolonged exposure to LH at relatively high concentrations. The result of this is multiple aggregation of receptors in endosomes, which leads to a weakening of cAMP signaling during long-term hCG-induced activation of LHCGR. |
Note: cAMP, cyclic adenosine monophosphate; LHCGR, luteinizing
hormone/chorionic gonadotropin receptor.
The lower activity of LH in relation to -arrestins makes it possible to
prevent or at least reduce the internalization and down-regulation of LHCGR
during prolonged exposure to even relatively high concentrations of LH, thereby
maintaining the sensitivity of target cells to endogenous gonadotropins, and
preventing depletion of the pool of functionally active LHCGRs both in the plasma
membrane and in intracellular compartments (Table 4). Activating mutations in
LHCGR, including the replacement of the Asp located in ICL3 with glycine
or tyrosine, cause a significant (in the case of AspGly fivefold)
increase in the degree of internalization of constitutively activated LHCGR into
the cell [207]. It is important to note that activating mutations induce the same
conformations of LHCGR as hCG, as indicated by the lack of significant effect of
hCG on the redistribution of mutant LHCGR in membrane microdomains, which is due
to their interaction with -arrestins [208].
The localization of the -arrestin-competent region in ICL3 of LHCGR and
the key role of the Asp residue in -arrestin binding are also
indicated by the results of studying synthetic peptides corresponding to various
regions of ICL3 [204, 209]. Using surface plasmon resonance, it was shown that
the peptide corresponding to the central part of ICL3 binds specifically to
arr2 in picomolar concentrations, while its binding to arr3
occurs with significantly lower affinity, in millimolar concentrations.
Substitutions of the Asp with glycine, asparagine, glutamic acid, and
leucine suppressed the specific binding of the “mutant” peptide to
arr2, indicating its functional importance for the formation of the
arr2-binding surface of LHCGR [204].
6. N- and O-Glycosylation of Gonadotropins, Their
Role in Signal Transduction and Possible Allosteric Effects
6.1 Common Features of Glycosylation of Gonadotropin Subunits
Of decisive importance for the functional activity of gonadotropins and their
signaling cascades is the process of N-glycosylation at asparagine
residues, and in the case of hCG and other hCGs, also the process of
O-glycosylation at serine-containing sites localized in the
C-terminal segment of their -subunits. As noted above,
-GPH, common to LH and hCG, undergoes N-glycosylation at two
sites, where the targets of glycosyltransferases are residues Asn and
Asn, while -LH and -hCG have one and two sites for
N-glycosylation (Fig. 2). Along with differences in the degree of
glycosylation and localization of glycans in the - and
-subunits, the nature and charge of oligosaccharide chains play an
equally important role in gonadotropin-induced signal transduction. For example,
in the - and -subunits of LH and the sulphated hCG, a
significant part of the oligosaccharide chains contain terminal residues of
sulphated GalNAc, which have a significant negative charge, while at the ends of
N-glycans in classical hCG and in the - and -subunits
of FSH, mainly sialic acid residues are localized, the negative charge of which
is not so pronounced in comparison with sulphated GalNAc [76]. The total charge
of the N-glycan is determined by the number and ratio of terminal
glycosyl residues, which, in turn, depends on the degree of branching of the
N-glycan. The N-glycans in the - and
-subunits of LH and hCG are significantly less branched than those in
the FSH, and this not only makes a significant contribution to the resulting
charge of these subunits, but also determines their spatial configuration and
ability to bind to the orthosteric site LHCGR.
It should, however, be taken into account that the structure of
N-glycans in the - and -subunits that form dimeric
gonadotropin can vary greatly, which is due to differences in their
post-translational processing, including in the machinery of their
N-glycosylation, as well as in the possibility of exchange and
reassociation of these subunits. Thus, recent studies of the pattern of
N-glycans in FSH using mass spectrometry have shown that -FSH
have fucose-modified N-glycans, of which approximately one third are
weakly branched bi-antennary glycans, and another third are highly branched
tri-antennary glycans and tetra-antennary glycans [23]. In turn,
N-glycans in -FSH do not contain fucose residues and are bi-
and triantennary, and the Asn residue in the subunits may have a massive
N-glycan corresponding in mass to tetra-antennary structures, but in
reality is tri-antennary, additionally containing many lactosamine repeats [23].
The glycosylation pattern and chemical structure of N-glycans influence
the folding of gonadotropins, their maturation and intracellular transport, the
stability and lifetime of gonadotropins in the bloodstream, and are also the most
important regulatory mechanisms that control their binding to the receptor and
specific biological activity [18, 19, 20, 22, 23, 210, 211, 212, 213, 214].
Taking into account the high variability of glycosylation of the - and
-subunits of gonadotropins [19, 23, 86, 211, 213, 215, 216, 217], the total
number of glycoforms of -heterodimer complexes of LH and hCG
can be very significant even in one organism and amount to tens, hundreds and
even thousands of variants [19, 218]. The interaction of gonadotropins with LHCGR
is highly dependent on their glycosylation, as a result of which the resulting
effect of a gonadotropin is directly dependent on the ratio and pattern of its
glycoforms. Taking into account the peculiarities of the location of
N-glycans in gonadotropins, as well as O-glycans in the
C-terminal part of hCG and equine gonadotropins, there is every reason
to believe that glycosyl components are able to interact with receptor sites
other than its orthosteric site, thereby exerting an allosteric effect on the
affinity of gonadotropin for LHCGR, on the bias of intracellular signaling and
the efficiency of the cellular response [18, 20, 22, 23, 219]. This is of great
importance for assessing the specific biological activity of recombinant human LH
and hCG, as well as hCG isolated from urine when used in ARTs [19]. In addition,
the hCG glycosylation pattern is important for monitoring the course of the first
trimester of pregnancy, assessing possible risks for embryo development and
pregnancy complications [74, 220, 221, 222, 223, 224, 225, 226, 227]. The need to match recombinant forms of LH
and hCG [19], as well as chemically synthesized hormones with LH activity [228],
with natural glycoforms is one of the most difficult problems of modern
gonadotropin-based pharmacology, being the main obstacle to create
physiologically relevant LH and hCG drugs.
Most of the data on the effect of N-glycosylation on the binding
characteristics and activity of gonadotropins were obtained in relation to FSH,
which is largely predetermined by the intensive development and characterization
of pharmacological preparations of FSH, primarily recombinant FSH, for use in
ARTs [23]. And these data indicate that N-glycans may play the role of
the most important allosteric modulators of FSH signaling, controlling its
efficiency and bias, and this is based both on the study of FSH isoforms that
differ in the degree of N-glycosylation and the structure of
N-glycans, and on the study interactions of highly and weakly
glycosylated FSH dimers with the FSHR ectodomain [23, 229, 230].
Despite some differences in the structural organization of FSH and its receptor,
as well as N-glycans that modify - and -FSH, from
those of LH, hCG and LHCGR, the main patterns of the influence of
N-glycosylation on functional activity are also valid for gonadotropins
with LH activity. Normally or weakly glycosylated forms of hCG stimulate
cAMP-dependent signaling pathways with greater efficiency and, as a result, they
are effective stimulators of the synthesis of steroid hormones, while
hyperglycosylated forms of hCG are less active in this regard [21], and their
regulatory effects are realized largely in relation to the stimulation of
mitogenic pathways, which may involve receptors other than LHCGR [231]. In this
case, the stability of various forms of hCG also plays an important role, since
normally or weakly glycosylated forms of the hormone have a significantly shorter
half-life in the bloodstream, in contrast to hyperglycosylated forms, in which
sites for proteolytic cleavage are protected by many glycosyl groups [74, 232].
Moreover, important information is provided by studying the interaction of
glycosylated forms of TSH with TSHR, which is structurally similar to LHCGR
[233]. It is quite interesting that hyperglycosylated forms of hCG, but not
normally glycosylated forms, are able to stimulate TSHR, increasing the level of
thyroid hormones, which may cause an increased risk of miscarriages and the
development of autoimmune thyroid diseases [234], especially in the case of
expression of the TSHR variant with the replacement of Val with an
isoleucine in the TMD, leading to hypersensitivity to hCG [235].
The following facts and theoretical principles support the allosteric nature of
the influence of N- and O-glycosylation of gonadotropins on
their activity and regulatory properties.
6.2 Effect of Gonadotropin Glycosylation on Its Binding to the
Receptor
A modulating role of N-glycans is postulated in the process of hormone
binding to the receptor, without direct interaction with the orthosteric site. It
is assumed that high-affinity binding of a glycoprotein hormone (GPH) to the
orthosteric site of a glycoprotein hormone receptor (GPHR) should involve almost
exclusively specific interactions between their specific amino acid residues or
their clusters, that is, based on the principles of protein-protein interaction
[236, 237, 238]. The interactions between the N-glycan and the polypeptide
chain in this case cannot be highly specific and high-affinity, since the
structure of N-glycans is very variable, and therefore they can be
involved in the interaction of the hormone with the orthosteric site only
indirectly, modulating its binding through allosteric mechanisms. Indeed,
deglycosylated forms of gonadotropins retain the ability to bind to the receptor,
and in some cases their affinity even exceeds that of glycosylated forms [239, 240].
Extracellular regions of the receptor can also be targets for
oligosaccharyltransferases, which suggests the possibility of glycan-glycan
interactions between glycosylated forms of the hormone and glycosylated sites of
the receptor ectodomain. Taking into account the dominance of the negative charge
in N-glycans that modify gonadotropins and their receptors, the glycosyl
components of the hormone and the receptor will be subject to mutual repulsion,
as a result of which their role in stabilizing the ligand-receptor complex seems
unlikely, being limited, most likely, to the negative control of the formation
such a complex. In agreement with this are the results obtained using molecular
modeling and structural-phylogenetic analysis for the GPHs and GPHRs, including
for TSH and its receptor, about the absence of N-glycans in the hormone
binding sites of GPHRs. Thus, in all studied vertebrate species in all types of
GPHRs, including LHCGR, there are four regions free of N-glycans,
including two on the concave faces of the LRR subdomain, responsible for
high-affinity binding of the hormone [238]. At the same time, N-glycans
can influence the relative position and efficiency of interaction between the
binding sites of the hormone and the receptor, including by changing the location
of the receptor ectodomain in relation to its TMD. All this will influence the
pattern of active conformations of the ligand-receptor complex, thereby
determining the activation of intracellular cascades and the predominance of
signal transduction [23, 241].
6.3 The Influence of Gonadotropin Glycosylation on the Stability of
Active Conformations of the LHCHR and on the Conadotropin-Regulated Intracellular
Cascades
The N-glycosylation directly affects the effectiveness of the
regulatory influence of gonadotropins on intracellular effector systems,
including cAMP-dependent signaling cascades and calcium signaling, and, as a
consequence, the magnitude and nature of the physiological response. Moreover,
the presence or absence of N- and (or) O-glycans can change the
pharmacological profile of gonadotropin, which was first demonstrated more than
thirty years ago. It was shown that deglycosylation of gonadotropins leads to a
decrease or complete loss of their specific activity [242], and in some cases,
deglycosylated forms of gonadotropins even acquired the properties of LHCGR
antagonists [210, 243]. It should be emphasized that the binding of
deglycosylated gonadotropins to receptors was preserved in most cases, although
its affinity, as a rule, changed significantly, and this again indicates the
allosteric nature of the influence of glycans on the activity of GPHRs.
One of the key molecular mechanisms that may mediate the influence of the degree
and pattern of N-glycosylation of gonadotropins on their ability to
activate the receptor is the influence of N-glycans on the relative
position of the ectodomain and TMD of the receptor. N-glycosylation of
gonadotropin may stabilize the active conformation of gonadotropin-bound
receptor, allowing efficient interaction between the hinge region of the
ectodomain and the ECL1 and the outer vestibule segments of the TMD. In this
case, N-glycosylation contributes to the activation of the signaling
cascade, and the gonadotropin functions as a full or partial agonist. Otherwise,
when a gonadotropin has an N-glycosylation pattern that prevents this
interaction, it functions as an antagonist or inverse agonist.
Deciphering the possible mechanisms of the influence of N-glycans on
the pharmacological profile of gonadotropins showed that the N-glycan,
which modifies the Asn residue in -GPH, can play a decisive role
here. This is supported by the fact that a similar mechanism is implemented for
different pairs of GPHs and GPHRs, which have identical -subunits in
their primary structure and different -subunits. In 2022, the role of
the N-glycan at position 56 of the TSH -subunit in TSHR was
established [244]. It has been shown that TSH, which includes the Asn in
-TSH modified by a large N-glycan (up to 16 monosaccharide
units), when interacting with TSHR, stabilizes its active conformation by moving
the LRR subdomain from the outer vestibule to the TMD, facilitating the
interaction of the latter with hinge region. The reason for this is the strong
steric and possibly electrostatic repulsion between the bulky N-glycan
of -TSH, directed orthogonally to the membrane surface, and membrane
phospholipids. The absence of this N-glycan does not allow stabilization
of the active conformation or leads to a minor effect on the equilibrium of
active and inactive conformations, which prevents TSHR activation and TSH-induced
stimulation of AC and other effector proteins [244]. To prove the exceptional
importance of the glycosyl component, experiments were carried out with the Fab
fragment of antibodies K1-70 with the activity of weak TSHR agonists [244],
which, when combined with TSH and stimulating antibodies, for example, with K1
-18, exhibited antagonistic properties [245, 246]. N-Glycosylation of
antibodies K1-70, which in terms of exposure of N-glycan and its
structural organization had similarities with glycosylation of the Asn in
-GPH (-TSH), led to a sharp increase in the ability of such
modified antibodies to stimulate the AC signaling system, due to whereby they
acted as full agonists. Thus, the hypothesis about the role of N-glycan
at position 56 of -GPH in stabilizing the active form of receptors of
the GPHR family, and possibly other receptors including ectodomains with multiple
LRRs, was confirmed [244].
In relation to FSHR, the role of interactions of -GPH
N-glycans (-FSH) with the membrane phase is also considered as
one of the mechanisms for stabilizing the active state of FSHR, and here the
structure and charge of the N-glycan modifying Asn in
-GPH (-FSH) is important [23]. It was established that in
this position in various glycoforms of FSH a bulky three-antennary
N-glycan is localized, which is critical for the biological activity of
the hormone, which, as in TSHR, is orthogonal to the surface of the plasma
membrane. Interestingly, both N-glycans localized in -FSH, as
well as Asn of -FSH, are located in the part of the FSH dimer
that faces the membrane. They are at an angle to the surface of the plasma
membrane, and not orthogonal to it, and therefore are only partially localized in
the cavity formed by the ectodomain and the outer vestibule of the TMD. In
accordance with this, they do not create significant steric tension in the
ligand-receptor complex. Despite this, deglycosylation of -FSH at
N-glycosylation sites has a significant impact on FSH activity,
including through changes in the localization and rigidity of the
Asn-coupled glycan and subtle reorganization of the entire ligand-receptor
complex. This is illustrated by the studies on the preservation or even
significant increase in biological activity of FSH dimers deglycosylated at one
or two sites of -FSH, and it has been shown that the increase in dimer
activity is positively correlated with an increase in the affinity of
hypoglycosylated forms of FSH for the receptor [239, 247, 248, 249].
It has been established that hypoglycosylated forms of FSH with a
-subunit having a mass of 18 or 21 kDa and, accordingly, deglycosylated
at positions Asn and Asn, stimulate FSHR and the cascades dependent
on it with significantly higher activity in comparison with the highly
glycosylated form of FSH with a mass of 24 kDa, which directly affects their
physiological effects, as well as the pharmacological profile in ARTs [23, 240, 249, 250, 251, 252, 253]. It can be assumed that the loss of one of the N-glycans in
-FSH increases the flexibility of the molecule and ensures its more
efficient anchorage in the cavity between the extracellular and transmembrane
domains with subsequent activation of FSHR. In turn, the N-glycan at
position 78 of -FSH, facing away from the membrane and located outside
the cavity formed by the ectodomain and TMD, does not affect the activity of the
FSH dimer [247, 248].
With regard to LHCGR, it has also been established that when it binds to hCG,
the ectodomain rotates by a mechanism similar to that of FSHR and TSHR. Cryogenic
electron microscopy data for LHCGR and TSHR have demonstrated similar rotation
angles of the main core of the ectodomain during the transition from the active
to the inactive conformation, at 45° and 38°, respectively [28, 244]. The similarity in the size of N-glycans at position Asn of
-GPHs in gonadotropins and TSH shows that they perform a similar
function in the process of receptor activation. Numerous studies on the effect of
deglycosylation of -GPHs in the hCG and LH dimers on LHCGR activity, as
well as the results of studies of mutant and hybrid forms of hCG and LH largely
confirm this mechanism of participation of glycans of the -subunit in
the binding and activation of LHCGR.
The functional importance of the Asn, the target of
N-glycosylation, in -GPH included in the hCG dimer is
supported by data on the activity of hCG lacking this site [254, 255]. More than
thirty years ago, Irwing Boime’s group [254, 255] found that this site for
N-glycosylation plays an extremely important role both in the formation
of the heterodimeric hCG complex and for the activation of cAMP-dependent
pathways and the regulation of steroidogenesis. Heterodimers containing a
normally glycosylated -subunit and an -subunit lacking an
N-glycan at position Asn were characterized by a significantly
reduced ability to increase intracellular cAMP levels and activate
steroidogenesis. In the same case, when the -subunit was deglycosylated,
hCG not only completely lost the ability to stimulate AC and induce
steroidogenesis, but also acquired the properties of a competitive antagonist,
interfering with the regulatory effects of normally glycosylated hCG [255]. The
authors noted that the absence of a glycan at position Asn of the
-subunit, as well as in both sites for N-glycosylation in the
-subunit (Asn, Asn) was less critical for the
steroidogenic activity of hCG, but only if the site Asn remained
glycosylated (Fig. 2). It was also found that of the two sites for
N-glycosylation present in the -subunit, the Asn site is
more important for activation of the AC signaling system [255].
A significant decrease in LH-like activity upon deglycosylation of Asn
was also shown for the -heterodimer of equine CG, which
contained a normally glycosylated -subunit
(eCG/56), both when assessing the ability of the
mutant dimer to stimulate testosterone production in a primary culture of Leydig
cells, and when assessing its ability to stimulate aromatase in cultured ovarian
granulosa cells [90, 256]. At the same time, the mutant dimer also had reduced
FSH-like activity, characteristic of eCG [90]. Treatment of gonadotropins with
peptide-N-glycanase F, which cleaves off N-glycan, including at
position 56 of -CG, also led to the loss of gonadotropin biological
activity [90]. Deletion of the C-terminal region of equine -CG,
which contains numerous sites for O-glycosylation
(eCG-D/), led to impaired secretion of the mutant
gonadotropin, but had a lesser effect on the functional activity, while the
double mutant (eCG-D/56), which lacked the
O-glycosylation site in -CG and the Asn site in
-CG, was completely devoid of both LH- and FSH-like activities [90].
The EC values for the eCG/56 and
eCG-D/56 mutants were reduced by 2.1 and 3.4 times,
and the number of binding sites on the cell surface was 56 and 65% of those for
wild-type CG [257].
It was noted above that equine LH, like equine CG, has both LH- and FSH-like
activity. At the same time, the same changes in the glycosylation status of
equine LH can have different effects on these LH- and FSH-like activities, as has
been demonstrated using the example of hybrid gonadotropin molecules. Hybrids
that included equine -LH and -FSH deglycosylated at
Asn and C-terminally truncated equine -LH and
-hCG that were unable to undergo O-glycosylation, as well as
hybrids that were different combinations of mutant subunits with wild-type
subunits were studied [92]. Hybrids N(56)dg-eLHalpha:eLHbeta(t) and
N(56)dg-eFSHalpha:eLHbeta(t) bound to LHCGR with an affinity similar to that of
wild-type LH, but had a very low ability to stimulate testosterone synthesis in
Leydig cells and stimulate progesterone production and aromatase activity in
ovarian granulosa cells. The hybrid N(56)dg-eLHalpha:eLHbeta(t) demonstrated an
inhibitory effect on the stimulation of ovarian steroidogenesis caused by FSH,
and was significantly superior in this regard to N(56)dg-eLHalpha:eLHbeta and
N(56)dg-eCGalpha:eLHbeta(t). Interestingly, N(56)dg-eLHalpha:eCGbeta did not have
an inhibitory effect on the effects of FSH and, at relatively high
concentrations, retained the ability to increase the expression and activity of
aromatase [92]. It is possible that in the latter case, a certain role
compensating for the absence of N-glycan at the Asn residue of
-eLH is played by the tail of equine -CG, glycosylated at
serine residues. Thus, the glycosyl components in hCG dimers can, to a certain
extent, compensate or modulate each other’s functions.
6.4 The Influence of Glycosylation on the Heterodimerization,
Receptor Specificity and Stability of Gonadotropins
The important role of N-glycans in the formation and stability of
heterodimeric gonadotropin complexes has been demonstrated. This determines the
affinity of gonadotropins for the receptor, affects the bias of signal
transduction, being one of the allosteric mechanisms of gonadotropin signaling,
and is also essential for the bioavailability and pharmacokinetic characteristics
of gonadotropins. Long-term incubation (72 hours) of cell cultures with the
N(56)dg-eLHalpha:eFSHbeta hybrid, in which equine -LH was
deglycosylated at the Asn residue, led to an unstable dimeric complex with
-FSH and a complete loss of activity of the hybrid gonadotropin [92].
At the same time, for hCG, the -subunit of which was
O-glycosylated, N-glycosylation of -hCG at the
Asn residue did not significantly affect the stability of the hCG
heterodimer, although it was critical for its activity. The dissociation
constants for -hCG complexes with normal and Asn-deglycosylated
-hCG were similar, and CD spectroscopy and NMR data showed only slight
differences for these complexes, mainly associated with local changes in the
spatial organization of the AAR cluster of -hCG, potentially contacting
the N-glycan at the Asn site [258]. At the same time, the
biological activity of the hCG heterodimer with Asn-deglycosylated
-hCG was sharply reduced, which confirms the importance of the
N-glycan at this position for interaction with LHCGR, in accordance with
the model proposed for FSHR and TSHR [23, 244]. Crosslinking of -CG with
a mutant -subunit with the replacement of the Thr with alanine,
which prevents normal glycosylation at Asn, resulted in a single-chain
phCGalphabeta molecule. This construct was characterized by normal binding of
phCG to LHCGR and was active, while the dimer with such a
mutation was slightly active [259]. Thus, the covalent crosslink of mutant
subunits with reduced resistance to the formation of active dimeric complexes can
ensure their preservation of functional activity. It should also be noted that
during the formation of such a single-chain gonadotropin, a number of other
mutations critical for the activity of hCG did not cause a significant decrease
in its specific activity. This suggests that changes in the localization and
accessibility of molecular determinants, including those represented by the
glycosyl moiety, mediating allosteric interactions of gonadotropin with the
receptor, can critically affect the affinity and effectiveness of hCG.
Hyperglycosylation makes an even more significant contribution than
deglycosylation to the ability of hCG to form heterodimeric complexes and plays a
critical role in the pattern of their specific biological activity [260],
especially given the very large set of glycans involved in the modification of
the hCG molecule [213]. In the case of the -hCG subunit, it has been
shown that excessive glycosylation leads to disruption of its association with
-hCG, which was first shown back in 1990s [261]. Monomeric
-hCG, containing various bulky N-glycans that prevent complex
formation, is secreted by the placenta, pituitary gland, and some tumors [260, 262], and have also been identified in significant quantities in seminal fluid
[263]. It has been shown that N-glycans in hyperglycosylated hCG
subunits, which are enriched in seminal fluid, contain a large number of fucose
residues, which significantly changes the conformational mobility of both the
glycans themselves and the hCG subunits [264].
Hyperglycosylated -GPH (-CG) can function in the form of
both monomers and homodimers of -CG–-CG, which is also true
for hyperglycosylated -CG. It has been shown that hyperglycosylation of
-CG not only disrupts the stability of their complex with the
-subunit, but also promotes the generation of free forms of
-CG and their homodimers -CG–-CG. At the same time,
hyperglycosylated hCG, including a population of monomeric -hCG, not
only interacts with LHCGR with lower affinity [21], but also acquires the ability
to effectively stimulate transforming growth factor- (TGF)
receptors [75, 232, 265]. Stimulation of these receptors, along with
LHCGR-mediated stimulation of cAMP-dependent pathways and various isoforms of
PKC, leads to the activation of post-receptor effector proteins such as ERK1/2,
Akt kinase and SMAD (Similar to Drosophila Mothers Against Decapentaplegic)
family proteins [266]. This determines the powerful mitogenic and anti-apoptotic
potential of hyperglycosylated forms of -CG, which is realized
independently of signaling pathways including LHCGR and makes a decisive
contribution to the growth and differentiation of the embryo in the early stages
of its development. By acting on TGF receptors, hyperglycosylated hCG
functions to a large extent as an auto- and paracrine factor [231, 232, 265]. In
an unfavorable scenario, hyperglycosylated forms of -CG may be involved
in tumorigenesis and provoke the development of gonadotropin-dependent tumors
[267, 268, 269].
The different receptor specificities of the weakly and highly glycosylated hCG
isoforms are believed to be due to differences in the accessibility of certain
regions of the molecule to interact with LHCGR, which allows the
hyperglycosylated form of hCG to activate the TGF receptor. Thus, the
B-152 antibody, which specifically recognizes O-glycans bound to residue
132 in the C-terminal part of -hCG, is unable to precipitate
with hCG heterodimers [270, 271]. This indicates the release of the
C-terminal region in hyperglycosylated forms of hCG, either due to the
dissociation of -hCG from the dimeric complex, or due to a significant
change in the conformation of the latter, ensuring the presentation of this
region for interaction with antibodies. Along with this, hyperglycosylation leads
to a decrease in the affinity of hCG for LHCGR and a weakening of its stimulating
effects on the effector systems of the target cell, which can be considered as an
allosteric effect of N- and O-glycans on receptor binding and
the specific activity of the hormone. The EC for LHCGR activation in the
case of classical hCG with normal glycosylation status was about 3.3 pM, while
for hyperglycosylated forms of the hormone it was from 7.1 to 14.0 pM [21]. Thus,
the excess amount of glycans in the hCG functions as a tethered NAM in relation
to the activation of LHCGR.
It is important that under the in vivo conditions, a significant
contribution to the activity of hCG can be made by the influence of the
N- and O-glycosylation pattern on post-translational
processing, secretion, stability and bioavailability of gonadotropin. As noted
above, in equine -CG, the C-terminal O-glycosylation
sites are critical for normal gonadotropin processing and secretion, as
demonstrated by studies of the eCG-D/ dimer mutant [90, 256, 257]. Based on this, chimeras were constructed in which the C-termini of
the - and -subunits forming heterodimers of eel LH or FSH were
linked to each other using the C-terminal sequence of equine
-CG, containing functionally active sites for O-glycosylation
[272]. This led to a mutant gonadotropin (LH-M), which, on the one hand, was able
to be secreted efficiently and quickly, and on the other, had the ability to bind
to the LHCGR and stimulate the AC signaling pathway, increasing the intracellular
level of cAMP. Moreover, the AC stimulating effect and EC value for LH-M
were not inferior to those obtained for wild-type LH [272]. However, further
studies are needed to assess the effectiveness of such chimeric gonadotropins,
including taking into account their effect on other LH-dependent signaling
cascades.
6.5 The Influence of Glycosylation on the Receptor Complex Formation
It should be noted that N-glycosylation and other modifications of
gonadotropin receptors can also affect the stability of homo- and
heterodi(oligo)meric receptor complexes. Despite the fact that gonadotropin
receptors can function as monomers, their formation of homo- and
heterodi(oligo)meric receptor complexes largely determines their functional
activity, and the ratio and structural organization of such complexes plays an
important role in the pattern of signal transduction, especially in the case of
heterodimers, formed by LHCGR and FSHR (see Section 7). Various types of
glycan-mediated interactions may be involved in the stabilization of receptor
complexes, including glycan-protein and glycan-glycan interactions, which can
stabilize or, conversely, destabilize the association of protomers within the
receptor complex or in higher-order complexes.
For the FSH–FSHR system, it has been shown that the process of formation of
receptor complexes may also depend on the degree of glycosylation of the hormone.
The FSHR binding to high concentrations (30 nM) of hypoglycosylated forms of FSH
containing 18 and 21 kDa -FSH leads to an increase in the proportion of
monomeric forms of FSHR, from 70% in the ligand-free state to 80% in FSH-bound
state, and this effect is realized after only 2 min and positively correlates
with the FSH-induced activation of cAMP production [214]. Highly glycosylated
FSH, including 24 kDa -FSH, is less effective in this regard, and an
increase in the proportion of the monomeric form of FSHR and activation of cAMP
signaling are achieved much later. The deglycosylated form of equine LH,
including eLH deglycosylated at Asn and eLH lacking the
C-terminal segment 121–149 containing sites for
O-glycosylation, on the contrary, increases the proportion of the
oligomeric form of the receptor to 50% and higher. Exposure of FSHR to low
concentrations (1 nM) of weakly glycosylated forms of FSH leads to the same
result [214]. All this indicates that the degree of FSHR oligomerization depends
not only on the degree of N-glycosylation of gonadotropin, but also on
its concentration, and demonstrates a relationship between
N-glycosylation and the kinetic parameters of FSHR complex formation.
All of the above may also be true for LHCGR, especially in terms of its
heterodimerization with FSHR, and this requires further research.
6.6 Impact of Gonadotropin Glycosylation on Signal Transduction
Bias
The degree and pattern of N-glycosylation of gonadotropins can have a
significant impact on signal transduction bias, as well as determine the fate of
the ligand-receptor complex in the cell, which largely depends on the nature of
the interaction of ligand-activated LHCGR and other GPHRs with
-arrestins. Most of the evidence on the influence of
N-glycosylation on intracellular signaling bias and cellular response
selectivity, and more broadly on folliculogenesis and oogenesis, relates to FSH
and its signaling cascades [214, 230, 253, 273, 274]. At the same time, there is,
although indirect, data regarding gonadotropins with LH activity. Thus, in the
middle of the menstrual cycle in women, at the stage of ovulation induction,
there is not only a significant increase in the total concentration of LH
glycoforms, but also a predominance of the proportion of the weakly glycosylated
form of the hormone, containing two N-glycans in the -subunit
(on average 62–65%). This is synchronized with a powerful activation of
LH-dependent signaling pathways in ovarian cells, leading to follicle rupture
[20]. At the same time, at the early follicular and luteal phases, both the total
concentration of LH in the blood and the proportion of its weakly glycosylated
form (LHdi) sharply decrease, which coincides with a weakening of LH-dependent
signaling in the ovaries. It is important to note that by the 14th day of the
cycle, the ratio of terminal sialic acids and sulphated GalNAc in
N-glycans, which determine the total negative charge of LH molecules,
significantly changes, and this also makes a significant contribution to their
interaction with LHCGR [20].
When studying equine LH with LH/FSH-like activity, evidence was obtained of
signaling bias of its glycosylated and Asn-deglycosylated forms in
relation to the cAMP-dependent and -arrestin pathways, which are
mediated through activation of FSHR [275]. At the same time, deglycosylation led
to a significant decrease, compared with the native hormone, in the stimulating
effect of equine LH on the AC activity and downstream cAMP-dependent cascades,
and at the same time maintained and even potentiated the activation of the
-arrestin pathway, resulting in activation of ERK1/2 and increased
phosphorylation of the ribosomal protein rpS6. Phosphorylation of rpS6 was
carried out independently of PKA through a signaling pathway including
mTOR-mediated phosphorylation and activation of ribosomal kinase p70S6K [275].
6.7 Some Comments on the Role of Gonadotropin Glycosylation in LHCGR
Signal Transduction
Thus, glycosylation has a significant impact on the stability of heterodimeric
gonadotropin complexes, on their binding characteristics and on the interaction
with LHCGR. Moreover, in the case of hyperglycosylated forms of -CG, the
receptor specificity of gonadotropins changes, with an increase in the degree of
glycosylation, shifting from LHCGR to TGF receptors. It is also possible
that N-glycosylation of extracellular regions of LHCGR influences the
formation and activity of the ligand-receptor complex. However, it is not always
possible to separate the allosteric effects of glycosylation from steric effects
affecting the accessibility of interaction sites of gonadotropin and its
receptor, and from the influence of glycosylation on post-translational
processing and secretory activity of gonadotropins, as well as on the processing
and translocation of the receptor into the plasma membrane. In addition, the
effect of glycosyl residues largely depends not only on their quantity and
location, but also on the type of glycosylation and the chemical structure of
glycans (the ratio of sialic acid and sulphated GalNAc, which determines the
charge of N-glycans, and the presence of fucose residues, which
determines them conformational rigidity, etc.). Attempts have been made to
separate the functions of N- and O-glycosylation for the
activity and structural organization of gonadotropins [210], but still, due to
objective circumstances, this remains a very difficult task. This is due both to
the fact that the results obtained in the in vitro cannot always be
relevantly translated into the bioactivity of gonadotropins and their signaling
systems in the in vivo, and to the fact that the structural diversity of
N- and O-glycans and the multiplicity of enzymatic systems
responsible for the modification and processing of their oligosaccharide backbone
make a significant, and in certain cases decisive, contribution to the stability
and pattern of specific activity of gonadotropins.
It is also important that glycosylation, as noted above, critically affects the
pharmacokinetics of gonadotropins and their resistance to proteolytic
degradation, which also affects the effectiveness of their regulatory effect on
intracellular signaling. In addition, during complicated pregnancy and in
conditions of reproductive dysfunction, as well as when taking various drugs,
including contraceptives, significant changes in the glycosylation pattern are
observed, which inevitably affects the pharmacokinetics, bioavailability and
pattern of gonadotropins with LH activity, primarily various forms of hCG, and
mediates the various effects of LH and hCG glycoforms on steroidogenesis,
folliculogenesis, oogenesis and embryogenesis [276, 277, 278, 279].
7. Formation of Receptor Complexes as an Allosteric Mechanism for LHCGR
Regulation
7.1 Common Principles of the Formation of Homo- and Heterodimeric
Receptor Complexes
The formation of homo- and heterodi(oligo)meric GPCR complexes, as well as their
complexes with other signal proteins (G proteins, -arrestins, RAMPs,
etc.) plays an important role in signal transduction, and a significant
contribution is made here by allosteric influences [42, 280, 281, 282, 283]. Thus, there is
numerous data on the effect of complex formation between class C GPCRs on their
functional activity and binding characteristics [282, 284, 285, 286]. Complex
formation has been less studied for class A GPCRs, which include LHCGR and
receptors for other pituitary glycoprotein hormones. The prevailing view here is
that class A GPCRs are active predominantly in monomeric form, while their
formation of complexes is due to their transition to an inactive state and/or is
necessary to modulate the binding characteristics and effectiveness of the
orthosteric agonist [42, 280, 281, 283], as demonstrated for rhodopsin, one of
the most structurally simple representatives of class A GPCRs [287, 288].
Homodi(oligo)merization may also be involved in the desensitization, processing,
and translocation of class A GPCRs.
At the same time, there are good reasons to believe that, depending on the type
of class A GPCRs, their formation of homo- and heterodi(oligo)meric complexes may
play a more significant role in signal transduction than signal modulation alone.
It may provide a mechanism for trans-activation of receptors, make a significant
contribution to biased activation of intracellular cascades and, especially in
the case of heterocomplex formation, mediate multidirectional regulation of these
cascades [289, 290]. Moreover, the molecular mechanisms of the influence of
protomers on the functional activity of complexes also include allosteric
interactions [280, 283, 291]. The formation of receptor complexes itself is under
the control of various allosteric regulators, specifically interacting with sites
in protomers that form contacts between them, including sites interacting with
membrane lipids and adapter proteins [288, 292].
Homodimeric complexes have been characterized for many members of class A GPCRs,
including rhodopsin [287, 288] and histamine H3 receptors [293]. Homo- and
heterodimeric complexes form different subtypes of -adrenergic
receptors, and this is of great importance for the adrenergic regulation of the
functions of the cardiovascular and other systems [294, 295]. Using various
approaches, heterocomplexes have been identified between the D2-dopamine receptor
and the protomers of A2A-adenosine [296, 297], CB1-cannabinoid [298] and oxytocin
receptors [299], between the 5-HT2A-serotonin receptor and the protomers of
5-HT1A-serotonin [300], 5-HT4-serotonin [283], µ-opioid [301] and oxytocin
receptors [302], between neuropeptide Y receptor and galanin receptor type 2
[303]. There are heterocomplexes between protomers of GPCRs belonging to classes
A and C, for example, between the 5-HT2A-serotonin and metabotropic
GluR2-glutamate receptors [304, 305], as well as between protomers of class A
GPCRs and receptors with tyrosine kinase activity, for example, between 5-HT2A
serotonin receptor and fibroblast growth factor receptor-1 (FGFR1) [300].
In the case of gonadotropins, there are various models describing the possible
role and assessing the contribution of complex formation in the regulation of the
functional activity of class A GPCRs and biased agonism. This is illustrated by
the dynamic pattern of monomeric and oligomeric forms of LHCGR, their mutual
transitions during receptor binding to ligands and during activation [24, 26, 28, 306, 307], as well as the existence and specific functional activity of
heterodimeric complexes formed by LHCGR and FSHR [14, 25, 27, 29, 30, 308].
Indirectly, the importance of complex formation for LHCGR is supported by
numerous data on the dimerization capacity of FSHR [214, 308, 309] and TSHR [24, 310, 311, 312, 313].
7.2 Homodi(oligo)meric Complexes of the LHCGR
As noted above, in the basal state, the main population of LHCGR (about 60%),
like FSHR (about 70%), is presented in a monomeric form, which is typical for
most GPCRs of class A [26]. Upon activation by an agonist, the proportion of
monomeric forms of the receptor increases, although to a small extent, and this
is due to the formation of an activated complex in which LHCGR is represented as
a monomer. Thus, using cryoelectron microscopy, it was demonstrated that in the
activated complex, including hCG-bound LHCGR and the associated G protein,
which mediates the activation of the AC and cAMP-dependent pathway, the receptor
molecule is preferably in the monomeric form [28]. It is important that when
LHCGR is activated by a small molecule allosteric agonist that binds to an
allosteric site located within the transmembrane channel, the receptor is most
likely also in the form of a monomer [28].
Using dual-color photoactivatable dyes and localization microscopy (PD-PALM), it
was shown that during the association process, LHCGRs form predominantly
homooligomeric, tri- and tetrameric complexes, while the proportion of
homodimeric complexes is significantly lower [26]. The authors carried out the
association of mutant forms of LHCGR, which were either deprived of the ability
to bind a ligand (LHR), or lost the ability to couple with the G
protein and activate AC (LHR). This made it possible to assess the role of
dimerization and oligomerization in the binding of gonadotropin and the
implementation of its regulatory effects to the cAMP level. It was found that
with the co-expression of mutant LHCGR protomers, both the processes of hCG
binding and activation of cAMP production are realized, which was evidence of the
formation of functionally active receptor complexes and, in addition, indicated
in favor of trans-activation of LHCGR [26]. It is important that other
authors, long before this, demonstrated the trans-activation mechanism
for the mutant form of LHCGR. This form of LHCGR lacked the ability to bind
gonadotropin but retained all the molecular determinants required for G protein
activation (LHR). It formed a complex with an isolated ectodomain of the
same receptor, fixed on the outer surface of the membrane using the transmembrane
glycoprotein CD8, and the result of this was not only high-affinity binding of
hCG to the resulting construct, but also activation of AC [314]. It should be
emphasized that in the monomeric state, ligand-bound LHCGR was also able to
activate AC, and with high efficiency, but in this case this can only occur
through the cis-activation mechanism.
The study of the mechanisms of formation of di- and oligomeric LHCGR complexes
showed a key role in their formation of interhelical contacts formed by the TM of
each protomer, and in the case of different complexes the set of such
interactions differed, although to a small extent. In all three pairs of
protomers studied, such as the associated protomers LHR, the
homodi(oligo)mer LHR-LHR, and the heterodimer
LHR-LHR, the complexes were stabilized by contacts between helices
TM4 and TM1, as well as by contacts TM3-TM3 and TM5-TM5. At the same time, in the
case of the formation of the LHR-LHR homodi(oligo)mer, an
additional contribution was made by the TM4-TM4 and TM1-TM5 contacts; in the case
of the LHR-LHR heterodimer, the TM4-TM4, TM6-TM1 and TM2-TM2
contacts were also formed, and in the case of the LHR-LHR
homodimer, the TM6-TM7 contact [26]. The involvement of TMs in the stabilization
of homodi(oligo)meric complexes is supported by the results of studying chimeric
LHCGRs, including those lacking the ectodomain or its interface with the TMD,
which showed the important role of TMs in complex formation [315]. It is
important to note that the TMD is involved in the di(oligo)merization of LHCGR
and under conditions of its activation by gonadotropin, which binds to LHCGR
ectodomain. Conformational changes occurring in the LRR subdomain and hinge
region extend to the TMD, which changes the pattern and efficiency of
interactions between TM protomers, thereby affecting the stability of the
receptor complex [316]. In this case, the influence of the gonadotropin-bound
ectodomain of one protomer on the ligand-free ectodomain of another protomer
occurs not directly, but through changes induced by hormone binding in the TMD
[316].
The involvement of the TMD in the formation of homodi(oligo)meric complexes
entails modulation of the binding of G proteins by the receptor, which, in turn,
affects the efficiency of activation of G protein-dependent intracellular
effectors. An assessment of the role of complex formation in the binding and
activation of the G protein showed that the formation of
homodi(oligo)meric complexes of LHCGR provides considerable stimulation of the
G protein when using hCG, but is not sufficient for its effective
stimulation when LHCGR binds to LH [26]. At the same time, gonadotropin-mediated
activation of the G protein, the effectiveness of which differs for hCG and
LH, is carried out mainly through the monomer LHCGR [28]. The formation of a
dimeric complex between wild-type LHCGR or constitutively activated mutant LHCGR
with mutant LHCGR having the AspAsn and TyrPhe substitutions,
which, despite maintaining binding to the hormone, is not able to mediate its
stimulation of the G protein and AC, leads to a decrease in the ability of
the receptor to activate cAMP-dependent signaling [317]. In this case, AC
stimulation, mediated either through the gonadotropin-bound constitutively active
or through the wild-type LHCGRs, is reduced to the same extent. In this case, the
mutant protomer lacking AC activity functioned as an inverse agonist, stabilizing
the inactive state of the receptor. It should be noted, however, that the
decrease in the stimulatory effect of gonadotropin on the intracellular cAMP
level during dimerization with the inactive mutant LHCGR protomer was expressed
to a small extent, and the EC value for this effect was reduced by half,
while the R did not change at all [317]. An attempt to heterodimerize
wild-type LHCGR with the type 3 melanocortin receptor, also functionally coupled
to G proteins, did not lead to a change in gonadotropin-induced stimulation
of AC, which indicates the specificity of the formation of the complex between
the mutant LHCGR and wild-type LHCGR protomers [317]. Significant changes in
signal transduction also occur in the case of the formation of heterodimeric
complexes between LHCGR and the structurally similar FSHR (see below for more
details). In this case, both G- and G-mediated signaling responses
change, and the first of them is enhanced during the formation of a heterodimer
complex, while the second is enhanced under conditions of its dissociation [25, 27]. This suggests a close relationship between the structure of mono- and
oligomeric forms of LHCGR and, to a certain extent, the nature of gonadotropin
(LH, CG), on the one hand, and the selectivity and efficiency of stimulation of
various types of G proteins coupled to LHCGR, on the other.
7.3 Heterodimerization of the LHCGR with the FSHR
With regard to assessing the allosteric effect of complex formation on the
activity of LHCGR, studies of heterodimeric complexes between LHCGR and FSHR are
of greatest interest, especially since the formation of such heterodi(oligo)meric
complexes leads to a significant change in LHCGR activity, including biased
activation of intracellular cascades. There is extensive evidence indicating the
formation of heterodimers formed by LHCGR and FSHR and their role in the control
of steroidogenesis and folliculogenesis [318], although there is still debate on
the formation of stable GPCR heterocomplexes for class A receptors under the
in vivo conditions [319], including for FSHR/LHCGR heterodimerization
[200].
In LHCGR/FSHR heterocomplexes, the binding of FSH to FSHR through a
trans-activation mechanism causes stimulation of the activity of
unliganded LHCGR, thereby triggering LH-dependent cascades even in the absence of
significant amounts of LH or hCG. The opposite situation may also occur. The
functioning of such complexes is critical for the stages of folliculogenesis,
including the early stages of antral follicle maturation, which are
FSH-dependent. At these stages, both the number of LHCGR and the concentration of
LH in the blood are very low and insufficient for efficient androgen synthesis.
At the same time, androgens are precursors for estrogens, significant amounts of
which are necessary for the growth and development of the antral follicle
[320, 321, 322, 323]. It is important that at the early stages of antral follicle
development, theca cells, which express LHCGR and produce androgens, are not yet
differentiated from granulosa cells, which are not LH-competent. It has been
suggested that during the preantral stage of folliculogenesis, there are cells
that exhibit both theca and granulosa cell properties and are enriched in FSHR,
but express only small amounts of LHCGR [324, 325, 326, 327]. Since the expression of LHCGR
and the level of LH at the preantral stage are not sufficient to ensure the
synthesis of such an amount of androgens that is necessary to maintain the high
level of estrogens characteristic of this stage, two models of regulatory
influences can be realized here. The first model, proposed almost 40 years ago,
suggests the development of LHCGR hypersensitivity to hormonal stimulation in the
preantral period [328], but no experimental evidence for this has been obtained
[200]. In accordance with the second model, confirmed in recent years by many
facts, FSHR/LHCGR heterocomplexes function according to the principle of
FSH-induced trans-activation of LHCGR [25, 30, 308]. A factor
contributing to the formation of such heterocomplexes is the significant
predominance of FSHR on the surface of follicular cells at the early antral stage
of folliculogenesis, since the ratio of FSHR to LHCGR is approximately 100:1
[329]. This makes FSHR easily accessible to interact with LHCGR and promotes the
formation of hetero-oligomeric complexes with a predominance of FSHR.
The following facts support the functional importance of FSHR/LHCGR
heterodi(oligo)mers. In women with inactivating mutations in the gene encoding
-LH, folliculogenesis up to the antral follicle stage proceeds normally,
and this is accompanied by normal levels of androgen and estrogen production
[330, 331]. In addition, back in the 1990s, numerous evidence was obtained that
FSH preparations are capable of causing the growth and maturation of follicles
and the formation of corpus luteum, and also contribute to an increase in
ovulatory potential, including in hypophysectomized rats and mice [332, 333, 334, 335], and
knockout of the gene encoding LHCGR completely prevented these effects of FSH,
indicating the requirement of LHCGR for FSH-mediated regulation of certain stages
of folliculogenesis [336]. Thus, at the early stages of the antral follicle,
LHCGR, to a greater extent than gonadotropins with LH-like activity, is necessary
to maintain the androgenic status, ensuring the normal course of these stages of
folliculogenesis [29, 337].
It should be noted, however, that in mice knockout of the gene for LHCGR,
expression of the constitutively active FSHR ensured follicle maturation from
antral to preovulatory and provided a phenotype associated with estrogen
production, although it did not support ovulation [338]. In addition, in
transgenic male mice knockout for the LHCGR gene, the expression of
constitutively active FSHR ensured the synthesis of androgen-dependent genes in
Sertoli cells and thereby restored, at least partially, spermatogenesis impaired
in Lhr mice [339]. All this indicates that high doses of FSH or
hyperactivated forms of FSHR are able to partially replace LHCGR-dependent
signaling in both the ovaries and testes, preventing a number of disorders of
folliculogenesis and spermatogenesis. This should be taken into account when
assessing the possible contribution of FSHR/LHCGR heterodimerization to the
control of follicle growth and maturation, as well as to the maturation of
spermatogenic cells [338].
Clusters combining FSHR and LHCGR on the surface of theca and granulosa cells
were first visualized back in 1980, which suggested the possibility of their
physical contact or even the formation of FSHR/LHCGR complexes [340].
Subsequently, using BRET, fluorescence correlation spectroscopy, and other
approaches, not only the formation of FSHR/LHCGR heterocomplexes was confirmed,
but also molecular determinants that may be involved in their stabilization were
identified [15, 25, 30, 308]. It was shown that the FSHR/LHCGR heterodimer is
stabilized primarily through the interaction of the outer surfaces of TM5, TM6
and TM7, which include a number of allosteric sites in contact with the lipid
phase of the membrane. A similar mechanism provides the stabilization of the
homodimeric LHCGR complex [26]. At the same time, the formation of
homodi(oligo)meric FSHR complexes, along with the TMD, involves regions of the
ectodomain [309], which is similar to the mechanisms of the formation of
complexes between type C GPCR protomers, where the formation of homo- and
heterodimeric complexes occurs involving both extracellular and transmembrane
domains [341, 342, 343]. In this regard, it cannot be excluded that extracellular
regions, including segments of the hinge region of the LHCGR and FSHR protomers,
are also involved in the formation of the FSHR/LHCGR complexes.
It should be noted that endogenous regulators of allosteric sites in contact
with the lipid phase of the membrane can be membrane lipids, primarily
cholesterol and phospholipids, which allows, through changes in the lipid
composition of the plasma membrane, to influence the functional activity of
GPCRs, as well as their formation of di(oligo)dimensional complexes. On the other
hand, the involvement of allosteric sites located on the lateral surface of the
TMD in the interaction between protomers in the receptor complex not only changes
the conformation and mobility of the TMD and its interfaces with the ectodomain
and ICLs, but also shields sites of interaction with membrane lipids and other
hydrophobic molecules, capable of specifically binding to such sites. This may
provide cause-and-effect relationships between the physicochemical and structural
features of the membrane lipid matrix and the functional state of GPCRs,
including their ability to form complexes.
The formation of FSHR/LHCGR heterocomplexes influences signal transduction bias
by weakening cAMP-dependent signaling pathways and, conversely, enhancing
phospholipase pathways leading to activation of intracellular calcium signaling
[25, 27]. It has been shown that stimulation of heterodimeric human FSHR/LHCGR
complexes with both gonadotropins with LH-like activity (LH or hCG) and FSH leads
to a significant attenuation of the stimulating AC signal [25]. On the other
hand, the formation of heterocomplexes between LHCGR and FSHR leads to increased
signals through G proteins that mediate the activation of PLC [27]. Ligand-free FSHR, when associated with gonadotropin-bound LHCGR,
significantly increases the latter’s ability to activate G protein,
releasing the G subunit, which stimulates PLC,
generating inositol 1,4,5-triphosphate and, as a consequence, stimulating release
of calcium ions from intracellular stores. When ligand-activated LHCGR associates
with FSHR, a reorganization of the LH–LHCGR–G protein complex occurs,
ensuring more efficient signal transduction along this pathway [27].
Depletion of intracellular calcium reserves by thapsigargin weakens the
potentiating effect of heterodimerization on LH-dependent activation of calcium
signaling. Along with the G-subunit, during heterodimerization
of FSHR/LHCGR, the G-dimer is also involved in LH-induced
stimulation of calcium signaling, and its action is most likely realized by
opening calcium channels of the plasma membrane, although other mechanisms of
their activation are not excluded, which are independent of the
G dimer. A significant contribution of extracellular Ca
to the effects of LH under conditions of association of LHCGR with FSHR is
supported by the weakening of the effect of gonadotropin on calcium signaling
with a decrease in the concentration of Ca in the extracellular space, as
well as the inhibition of this effect by inhibitors of calcium channels of
different types. In this regard, it is interesting that in the absence of FSHR,
the stimulating effect of LH on calcium signaling is realized mainly through the
release of Ca from intracellular stores, and not due to its pumping from
the extracellular space. This is, in particular, demonstrated by the lack of
influence on it by calcium channel blockers, nifedipine and 2-aminoethoxydiphenyl
borate (2-APB) [27]. Thus, ligand-free FSHR is a PAM for LH-induced stimulation
of G protein, and simultaneously a NAM for hormone activation of the AC
signaling system, demonstrating the properties of a biased allosteric modulator.
One of the mechanisms of the modulating effect of FSHR on LH signaling may be a
change in the subunit composition of hetero-oligomeric FSHR/LHCGR complexes.
There is evidence that in a ligand-free state such complexes can be tetrameric
and include three LHCGR molecules and one FSHR molecule, and when bound to LH
they are transformed into complexes with a smaller number of LHCGR protomers or
even with one such protomer [26, 27]. Thus, by replacing LHCGR protomers with
FSHR protomers, the effect of negative cooperativity shown for homodimeric LHCGR
complexes is blocked [24], which may be the root cause of increased
G-mediated signaling [27]. The complexes with a FSHR:LHCGR ratio of 3:1
are interesting because in the case of active FSHR complexes, homotrimeric
structures are shown, including three FSHR protomers [239]. In other words, at
the level of formation of FSHR/LHCGR complexes, another mechanism for the
regulation of signal transduction can be demonstrated, based on the different
ratio and number of protomers in such complexes.
Currently, heterodimerization of LHCGR with other GPCRs has not been proven,
although the formation of such complexes with the membrane estrogen receptor GPER
(GPR30), which is known to form heterodimeric complexes with FSHR, cannot be
ruled out [318, 344, 345]. There is evidence of a relationship between the
expression of FSHR and LHCGR and the efficiency of GPER-mediated signaling,
which, by analogy with FSHR/GPER heterodimers, may indicate the potential for the
formation of LHCGR/GPER heterodimers [346, 347], but this requires further study.
Interestingly, the expression and activity of GPER changes significantly in
diabetes mellitus (DM) and cancer [348]. This may indirectly affect gonadotropin
signaling or, on the contrary, be one of the consequences of changes in the
activity of FSHR- and LHCGR-competent signaling systems.
8. Antibodies to Gonadotropins and LHCGR as Potential Allosteric
Modulators of Their Activity
Allosteric effects on LHCGR can be exerted by autoantibodies produced against
its antigenic determinants located in extracellular sites. Recently, stimulating
autoantibodies to LHCGR were detected in the blood of women with polycystic ovary
syndrome and hyperandrogenemia [47]. In this regard, it must be emphasized that
inhibitory autoantibodies to FSHR have been found in women with ovarian failure
[349, 350]. Since LHCGR and FSHR can heterodimerize, autoantibodies to FSHR, like
autoantibodies to LHCGR, are characterized by cross-reactivity with respect to
the LH- and FSH-dependent cascades. It cannot be excluded that, as in the case of
autoantibodies to -adrenergic receptors [351, 352], antibodies to LHCGR
create a “regulatory buffer” that allows modulation of powerful activation
signals caused by significant changes in the level of gonadotropins during
reproductive cycles, or function as a compensatory mechanism that prevents the
development of reproductive pathology [42]. But this issue requires additional
study.
Synthetic antibodies produced to various epitopes of LHCGR are not only used to
assess the functional significance of various regions of the receptor, but are
also characterized by a wide range of biological activities, affecting the
receptor, including through allosteric mechanisms [45]. Thus, scFv 13B1
antibodies produced against the hinge region of LHCGR were characterized by
agonistic activity, and their effect was independent of the hormone and persisted
in the case of constitutively active LHCGR forms [45]. Epitope 313–349 was
identified in the C-terminal part of the LHCGR hinge region, which was
responsible for the ability of antibodies to stimulate cAMP production via LHCGR,
and the key role of the sulfated Tyr residue in the agonistic effect of
these antibodies was established [353].
Along with antibodies to receptors, antibodies to LH and hCG, which have
different profiles of pharmacological activity, but, as a rule, neutralize the
effect of gonadotropins, can have a significant role in the control of
LH-dependent cascades [46]. The hCG treatment of men with hypogonadotropic
hypogonadism resulted in the production of anti-hCG antibodies, which neutralized
the effects of hCG by interfering with LHCGR activation, rendering such therapy
ineffective [354, 355]. In women, antibodies to endogenous hCG led to decreased
fertility and pregnancy loss in the first trimester, when hCG production plays a
determining role in fetal development [356, 357]. The negative effects of
anti-hCG antibodies on reproductive function have been reported in experiments
with animals, including monkeys [43, 358].
Antibodies developed against equine CG, which has mixed LH- and FSH-like
activity, were characterized by a wide range of effects on FSHR and LHCGR, which
was due to different epitopes on the - and -subunits of the
hormone [88, 359]. Some of them suppressed the effects of equine hCG on the
activity of FSHR and LHCGR, while others, on the contrary, potentiated the
FSH-like effects of the hormone. It was extremely interesting that two antibodies
that potentiated the FSH-like activity of equine hCG either did not significantly
affect or inhibited the LH-like activity of the hormone. This indicates that the
same antibodies have different modulatory effects on the LH- and FSH-like effects
of equine hCG, functioning as NAMs, PAMs or silent allosteric modulators [88].
Since antibodies to gonadotropins are present in humans in normal and
pathological conditions, as shown for antibodies to hCG in humans [360, 361],
their ability to specifically influence gonadotropin signaling is one of the
factors controlling the functions of the male and female reproductive system [44, 46]. It must be emphasized that a complex spectrum of biological activity and the
ability to modulate the basal and hormone-stimulated activity of FSHR is
characteristic of antibodies to FSH, which gives strong grounds to consider
antibodies to gonadotropins as one of the endogenous allosteric regulators of
LHCGR, FSHR and their heterodi(oligo)mers [30, 46].
A study of the role of glycosylation in the production of autoantibodies to
equine hCG showed that the deglycosylated form of the hormone did not cross-react
with antibodies, while the glycosylated forms, on the contrary, intensively
immunoprecipitated with antibodies of different pharmacological activity, which
had stimulating, inhibitory or mild modulating effects on FSH-like activity of
equine hCG, assessed by progesterone production by Y1 cells derived from a mouse
adrenal tumor and stably expressing FSHR, as well as on LH-like activity,
assessed by testosterone production by rat Leydig cells [362]. Thus,
glycosylation status directly affects the production of antibodies and their
ability to biasly regulate gonadotropin signaling.
9. Low-Molecular-Weight Allosteric Regulators of LHCGR
In addition to the orthosteric site involved in the high-affinity binding of
gonadotropin molecules, LHCGR contains allosteric sites localized in various loci
of the molecule, including in the upper half of the transmembrane tunnel, which
remain free when gonadotropin binds [30, 42]. It has been established that the
transmembrane tunnel of LHCGR contains at least two allosteric sites, the main
one and the one modulating its activity [219, 363, 364]. The first of them, as in
a number of other GPCRs, is formed by the internal surfaces of helices TM4, TM5,
TM6 and TM7 [363, 365, 366], while the second is formed by the helices TM1, TM2,
TM3 and TM7 [349]. Both sites, the main and modulatory ones, physically overlap
each other. The consequence of this overlap, as well as interaction, direct or
indirect, with other allosteric sites, is to provide a wide pharmacological range
of allosteric regulation of LHCGR. A similar arrangement of allosteric sites in
the upper half of the transmembrane tunnel has been postulated for other
receptors of pituitary glycoprotein hormones [30, 42, 367]. In the process of
developing LMW ligands of the transmembrane allosteric site of LHCGR, both full
and inverse allosteric agonists with intrinsic activity and allosteric modulators
were developed, and allosteric modulators, based on their pharmacological
activity, can be classified as PAMs and NAMs [30, 42]. Along with this,
allosteric regulators with ago-PAM activity have been identified [42]. A wide
range of LMW allosteric regulators, the targets of which are allosteric sites
located in the transmembrane channel, have also been developed for FSHR and TSHR
[42, 368, 369].
9.1 Thienopyrimidine Derivatives as Allosteric Full Agonists and
PAMs of LHCGR
The first LMW ligands of LHCGR were developed by Dutch scientists in 2002, who
synthesized thieno[2,3-d]pyrimidine derivatives (TPDs) with agonist activity,
including the most active compound Org41841
(N-tert-butyl-5-amino-4-(3-methoxyphenyl)-2-(methylthio)thieno[2,3-D]pyrimidine-6-carboxamide)
and its analog Org43553. Both of these compounds activated LHCGR at nanomolar
concentrations, and Org43553 showed higher activity [370]. Subsequently, these
compounds became prototypes for a large number of TPDs with the activity of
allosteric regulators of both LHCGR and TSHR [42, 371, 372, 373, 374, 375, 376, 377, 378, 379, 380, 381, 382, 383, 384]. We, based on
Org43553 as a prototype of LMW ligands of LHCGR, have developed a series of TPDs
with LHCGR agonist/ago-PAM activity, including the in vivo active
compounds
5-amino-N-tert-butyl-4-(3-(isonicotinamido)phenyl)-2-(methylthio)thieno[2,3-d]pyrimidine-6-carboxamide
(TP01),
5-amino-N-(tert-butyl)-2-(methylthio)-4-(3-(thiophene-3-carboxamido)phenyl)thieno[2,3-d]pyrimidine-6-carboxamide
(TP02),
5-amino-N-tert-butyl-2-(methylsulfanyl)-4-(3-(nicotinamido)phenyl)thieno[2,3-d]pyrimidine-6-carboxamide
(TP03),
5-amino-N-tert-butyl-4-(3-(1-methyl-1H-pyrazole-4-carboxamido)phenyl)-2-(methylsulfanyl)thieno[2,3-d]pyrimidine-6-carboxamide
(TP4/2),
5-amino-N-(tert-butyl)-4-(3-(2-methoxynicotinamido)phenyl)-2-(methylthio)thieno[2,3-d]pyrimidine-6-carboxamide
(TP21), and
5-amino-N-tert-butyl-4-(3-(2-chloronicotinamido)phenyl)-2-(methylthio)thieno[2,3-d]pyrimidine-6-carboxamide
(TP23) [379, 380, 383, 384, 385, 386] (Table 5, Ref. [363, 370, 373, 375, 376, 379, 380, 383, 384, 385, 386, 387, 388, 389, 390, 391, 392, 393, 394, 395, 396, 397, 398, 399, 400, 401, 402, 403]).
Table 5.
Low-molecular-weight allosteric regulators of luteinizing
hormone/chorionic gonadotropin receptor (LHCGR): chemical structure,
pharmacological profile and efficacy.
| Compound |
Efficiency |
Structure |
| Full allosteric LHCGR agonists and/or ago-PAMs |
| Thieno[2,3-d]-pyrimidine derivatives (Org41841, Org43553) [370, 373, 375, 376, 387, 388, 389] |
Stimulate G proteins and AC in cells with expressed LHCGR; when administered to male rats, they increase testosterone levels; when administered to female rats and female volunteers, they induce ovulation. There is no competition with gonadotropins for binding to LHCGR |
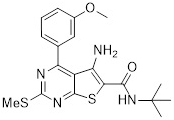 |
| Org41841 |
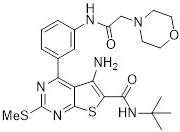 |
| Org43553 |
| Thieno[2,3-d]-pyrimidine derivatives (TP01, TP02, TP21, TP23) [379, 380, 385] |
Stimulate the activity of G proteins and AC in testicular and ovarian membranes, increase testosterone production when administered intraperitoneally and orally to male rats |
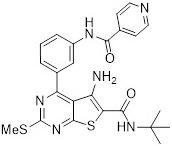 |
| TP01 |
 |
| TP02 |
 |
| TP21 |
 |
| TP23 |
| Thieno[2,3-d]-pyrimidine derivatives (TP03, TP4/2) [383, 384, 386, 390, 391, 392, 393] |
Stimulate the AC system in testicular membranes; activate steroidogenesis and testosterone production in cultured Leydig cells; stimulate testicular steroidogenesis when administered intraperitoneally, subcutaneously and orally to male rats, including those with diabetes mellitus (DM), and aging animals. Activate ovarian steroidogenesis and cause induction of ovulation in mature and immature female rats. Potentiate the steroidogenic effects of hCG in the in vitro and in vivo conditions |
 |
| TP03 |
 |
| TP4/2 |
| Pyrazole derivative, Compound 1 [394] |
Stimulates AC activity and testosterone production in Leydig cells |
 |
| Pepducin 562–572-K(Palm)A, and its dimeric analogue, including cross-linked regions 562–572 [395, 396] |
Stimulate AC and G proteins in the testicular and ovarian membranes of rats. Peptide 562–572-K(Palm)A increases testosterone production in male rats when administered intratesticularly |
Asn-Lys-Asp-Thr-Lys-Ile-Ala-Lys-Lys-Nle-Ala-Lys(Palm)-Ala-amide; |
| (Asn-Lys-Asp-Thr-Lys-Ile-Ala-Lys-Lys-Nle-Ala)-Lys-amide |
| Allosteric LHCGR antagonists and/or NAMs |
| Terphenyl derivative, LUF5771 [363, 397] |
Inhibits the stimulatory effects of hCG and allosteric agonists on LH-dependent intracellular cascades |
 |
| Benzamide derivatives (ADX68692, ADX68693) [398] |
They inhibit the activity of LH-dependent signaling cascades stimulated by gonadotropins. The specificity of the effects in different cell types and with different ratios of LHCGR and FSHR expression has been shown |
 |
| ADX68692 |
 |
| ADX68693 |
| Tetrahydro-1,6-naphthyridine derivatives (BAY-298, BAY-899) [399] |
Reduce the production of steroid hormones stimulated by gonadotropins and allosteric agonists; inhibit testicular and ovarian steroidogenesis; suppress folliculogenesis when administered to female rats |
 |
| BAY-298 |
 |
| BAY-899 |
| Dichlorodiphenyl trichloroethane [400, 401] |
Reduces the stimulating effect of hCG on AC, and suppresses LH/hCG-induced recruitment of -arrestins. Does not significantly affect gonadotropin-stimulated testosterone production, indicating that it selectively acts on different signaling cascades |
 |
| NAMs and/or inverse allosteric LHCGR agonists |
| Thieno[2,3-d]-pyrimidine derivative TP31 [402, 403] |
Reduces the stimulating effects of hCG and TP03 on AC activity in testicular membranes (to a greater extent in the case of TP03 stimulation), and inhibits hCG-induced stimulation of testosterone production in male rats. Induces a slight decrease in basal testosterone levels |
 |
| Pyrido[3,4-d]pyrimidine derivative (PP17) and pyrimido[4,5,6-de][1,6]naphthyridine (PP10) derivatives [402] |
Inhibits the stimulatory effects of hCG and TP03 on the AC system in testicular membranes (more so in the case of hCG), and inhibits hCG-stimulated and basal testosterone levels in the blood of male rats |
 |
| PP17 |
 |
| PP10 |
When studying the compound Org43553, it was shown that it specifically binds to
LHCGR (K, 2.4 nM), and in its presence, the binding of gonadotropins to the
orthosteric site of LHCGR and their stimulating effect on AC activity is
preserved, which indicates a mismatch in the localization of the allosteric and
orthosteric sites [404]. The result of binding of Org43553 to LHCGR in cells
where LHCGR was expressed was stimulation of AC and cAMP-dependent transcription
factor CREB with EC values of 28 and 4.7 nM, respectively, although the
effectiveness of Org43553 was lower compared to LH [387]. The compounds we
developed, TP01, TP02, TP03, TP4/2, TP21 and TP23 stimulated AC activity in
fractions of plasma membranes isolated from the testes and ovaries with moderate
efficiency, and did not significantly affect the AC-stimulating effects of hCG,
and their effects were suppressed by cholera toxin, the substrate of which are
G proteins [379, 380, 381, 383, 385, 405, 406] (Fig. 4). The most active in the
in vivo conditions were TP03 and TP4/2, which contained pyridin-3-yl and
1-methyl-1H-pyrazol-4-yl groups in a variable part, and according to the
results of molecular docking, they most effectively interacted with the
transmembrane allosteric site of LHCGR [383, 384, 386]. Both compound Org43553
and compounds TP03 and TP4/2, when exposed to primary cultures of rodent Leydig
cells, increased the intracellular level of cAMP and enhanced steroidogenesis,
which led to a dose-dependent increase in testosterone production [375, 384].
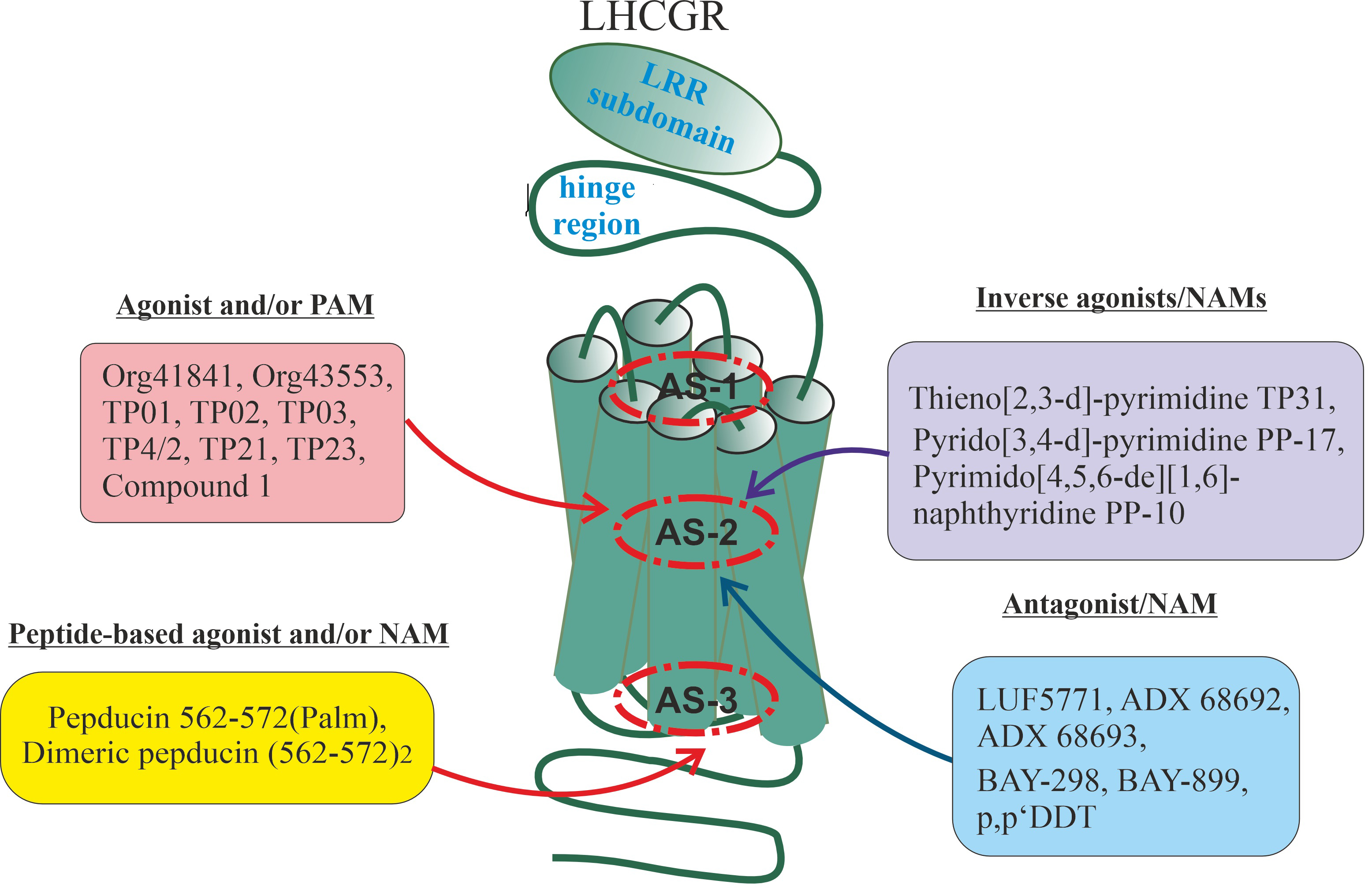 Fig. 4.
Fig. 4.
Synthetic allosteric regulators of luteinizing hormone/chorionic
gonadotropin receptor (LHCGR) with different pharmacological activity profiles.
The localization of allosteric sites in LHCGR and their low-molecular-weight
ligands with different pharmacological activity profiles, including pepducins,
which are palmitoylated fragments of the third cytoplasmic loop of LHCGR, are
shown. For designations and chemical formulas for the compounds shown in the
figure, see Table 5. AS-1, AS-2 and AS-3: allosteric sites in the loci 1, 2 and 3.
Despite the fact that Org43553 and the TPDs we studied were significantly
inferior to LH and hCG in terms of binding affinity and AC-stimulating effect,
their lower affinity and moderate AC stimulation are more a positive factor than
their disadvantage, because it does not lead to the hyperactivation of
cAMP-dependent cascades and prevents the resulting side effects of gonadotropins.
The Org43553, TP01, TP03, TP4/2 and TP23 had little or no effect on FSHR and TSHR
activity, suggesting their receptor specificity for LHCGR [381, 383, 385, 387, 407]. This distinguishes them from the compound Org41841, which, although to a
small extent, interacted with the transmembrane allosteric site of TSHR, which
subsequently made it possible to create on its basis a wide range of allosteric
TSHR regulators with different profiles of pharmacological activity [371, 372, 374, 377, 378].
As mentioned above, LH, through G proteins, stimulates the activity of
PLC, and activation of this enzyme requires higher concentrations of
gonadotropin than activation of AC [8, 15, 408]. Compound Org43553 and the TPDs
we developed in the micromolar concentration range stimulated AC both in cell
cultures and in plasma membrane fractions of LH-competent tissues, while weakly
affecting the activity of G proteins, PLC and phosphoinositide
metabolism [380, 385, 387] (Fig. 5). It was shown that Org43553, even at
relatively high concentrations (10–10 M), stimulated PLC
activity by only 33–37%, which is less than 5% of the corresponding effect of
LH [387]. There is reason to believe that Org43553 also weakly affects the
activity of -arrestins, which are key targets of gonadotropins, as
illustrated by the lack of a pronounced effect on components of the MAPK cascade
and the low intensity of LHCGR endocytosis [387]. It is also very important that
TPDs, unlike gonadotropins, even when applied in a course in the in vivo
conditions, have a relatively weak effect on the expression of the gene encoding
LHCGR and on the density of LHCGR in target cells, which prevents resistance to
the action of endogenous gonadotropins with LH-activity [383, 384, 409].
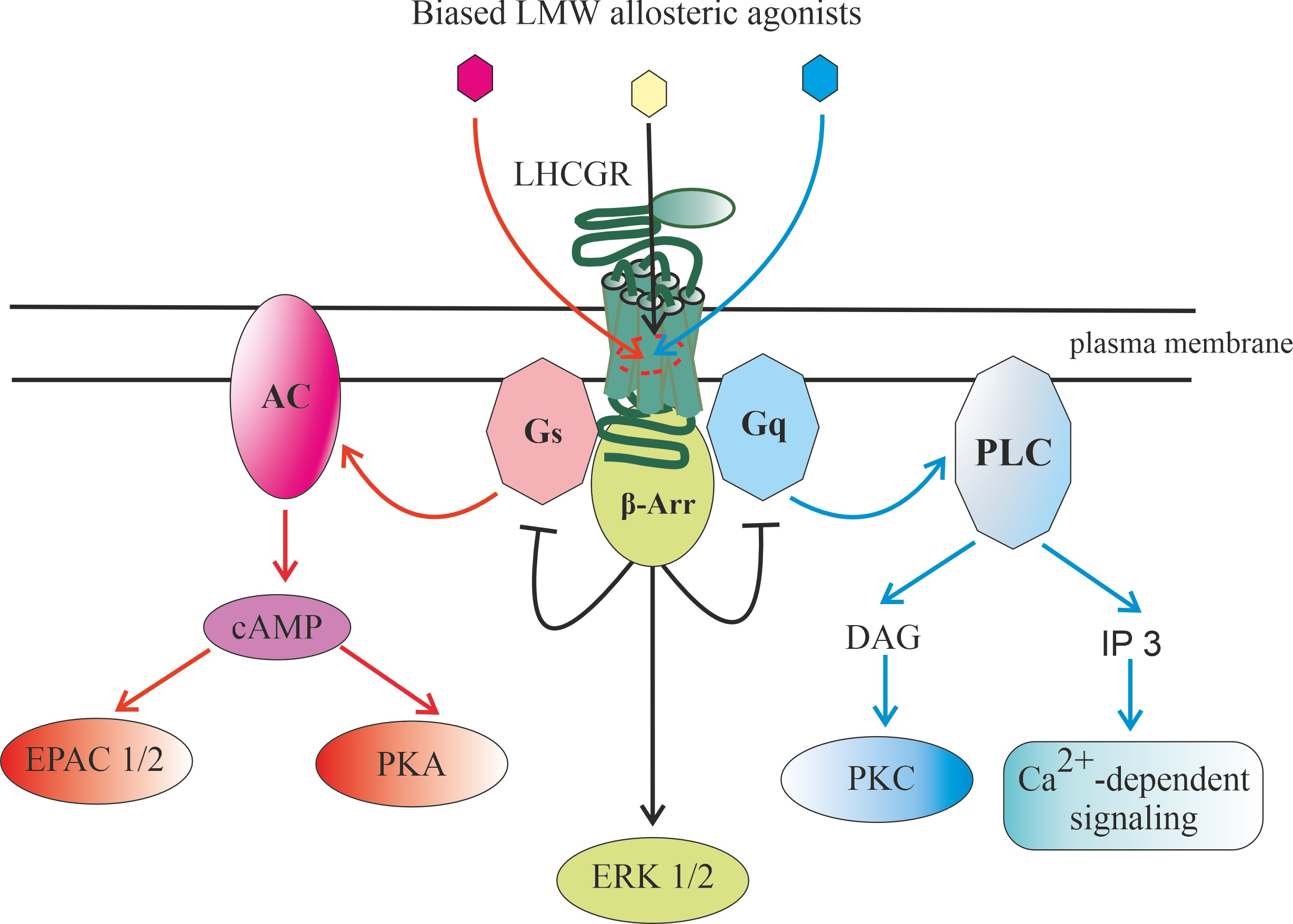 Fig. 5.
Fig. 5.
Regulation of luteinizing hormone/chorionic
gonadotropin receptor (LHCGR) activity by biased low-molecular-weight allosteric
agonists. A characteristic feature of allosteric agonists is a bias in the
activation of intracellular signaling cascades, which is clearly shown for thienopyrimidine derivatives (TPDs)
such as Org43553, TP03 and TP4/2, which activate cAMP-dependent signaling
pathways with high selectivity. The figure shows various variants of biased
signaling, when an allosteric agonist preferentially activates either the
adenylyl cyclase signaling cascade (red arrows), or phospholipase C and
calcium signaling (blue arrows), or -arrestins and processes
dependent on them (desensitization and down-regulation of the receptor,
-arrestin endosomal signaling) (black arrows, including those
blocking G protein-mediated signaling). The bias of allosteric agonists may be
due to the characteristics of their chemical structure when binding to the same
allosteric site, or the ability to bind to topologically different allosteric
sites. Abbreviations: AC, adenylyl cyclase; G, heterotrimeric G
proteins that stimulate adenylyl cyclase activity; PKA, protein kinase A; EPAC
1/2, Exchange Protein directly activated by Cyclic AMP, types 1 and 2; PLC,
phosphoinositide-specific phospholipase C; G, heterotrimeric G
proteins that and stimulate PLC and calcium signaling;
DAG, diacylglycerol; IP3, inositol-3,4,5-triphosphate; PKC, protein kinase C; ERK
1/2, extracellular signal-regulated kinases, types 1 and 2, the effector
components of mitogen-activated protein kinase cascade; LMW agonist, low-molecular-weight agonist.
Site-directed mutagenesis and molecular docking of Org41841, Org43553 and their
analogues were used to reconstruct the transmembrane allosteric site of LHCGR.
For this purpose, the AARs forming the allosteric site TSHR, located in the
transmembrane tunnel of the receptor, were replaced with LHCGR residues
corresponding in localization, as a result of which this TSHR site was
structurally close to that of LHCGR [371]. The LeuPhe substitution in
ECL2 TSHR resulted in Org41841 binding to the mutant receptor with an EC
value of 800 nM, while the double substitutions LeuPhe/PheThr and
LeuPhe/TyrPhe resulted in Org41841 binding with an EC
value of 1000 nM. Simultaneous replacement of nine residues (IleVal and
LeuPhe in ECL2, PrpThr, AlaSer, LeuGln,
AlaVal and PheThr in TM5, TyrPhe and IleAla in
TM6) with the corresponding LHCGR residues resulted in a mutant receptor, upon
binding to which Org41841 had the activity of full agonist. Its maximum AC
stimulating effect was comparable to that of TSH. These data indicate the
important role of the LeuPhe, PheThr, and TyrPhe residues
in the formation of the hydrophobic surface of the LHCGR allosteric site located
in the transmembrane channel [371]. The key for interaction with Org41841 is the
negatively charged residue Glu, localized in TM3 and highly conserved in
GPCRs. Its replacement with alanine blocked the binding of Org41841 to mutant
LHCGR and prevented its AC activation. The side carboxylate of Glu is proposed to
form a salt bridge with the positively charged amino group of the low molecular
weight ligand, ensuring its efficient binding to the receptor [371, 372].
In 2021, Duan et al. [28] examined the complex of Org43553 with the
transmembrane allosteric site LHCGR using cryo-electron microscopy and showed
that this small molecule agonist occupies the upper part of the pocket formed by
this site. In this case, the morpholine ring of Org43553 is directed outward, to
the boundary between the hinge region and the extracellular part of the TMD,
while the tert-butylamine group is directed deep into the transmembrane tunnel,
to the place where the orthosteric site is located in most class A GPCRs. The
allosteric site includes amino acid residues belonging to helices TM3, TM5, TM6
and TM7, loops ECL2 and ECL3 and the segment of the hinge region bordering TM1,
and substitutions of two of them, AlaTrp and IlePhe,
significantly reduce the agonistic activity of Org43553, despite maintaining the
affinity of this TPD to the receptor. The importance of hydrophobic interactions
between Org43553 and the transmembrane allosteric site of LHCGR has also been
shown [28].
Previously, using homology modeling methods and molecular dynamics, we assessed
the binding parameters of TP4/2 and other TPDs developed by us with LHCGR and
also showed that hydrophobic interactions play a decisive role in stabilizing the
complex of a LMW agonist with a transmembrane allosteric site, while Coulomb
interactions and hydrogen bonds were less significant [383]. As in the case of
Org43553, the 1-methyl-1H-pyrazol-4-yl group was directed to the extracellular
entrance of the transmembrane tunnel, and the tert-butylamine group to
the central part of this tunnel. A reduced ability of TP4/2 to interact with the
transmembrane allosteric site of TSHR was also shown, which is in agreement with
the data on the absence of a significant effect of this compound and its
analogues on TSH-stimulated AC activity in the membranes of the thyroid gland,
and in vivo on the levels of thyroid hormones and the expression of
genes responsible for their synthesis [383, 385, 407].
9.2 The In Vivo and Clinical Effects of Thienopyrimidins Derivatives
with Agonistic Activity
It is important that Org43553 and the TPDs we developed are active not only
in vitro, but also in vivo, and they are effective both in
parenteral (intraperitoneal, subcutaneous) and oral routes of administration
[375, 376, 379, 381, 383, 384, 385, 386, 388, 389]. Retention of activity upon oral
administration indicates good absorption of TPDs in the gastrointestinal tract
and their high bioavailability with this method of delivery. The oral
bioavailability of Org43553 was 79% in rats and 44% in dogs compared to
parenteral bioavailability [375]. We demonstrated comparable bioavailability and
effectiveness in stimulating steroidogenesis with intraperitoneal and oral
delivery methods for compounds TP03 and TP4/2 [379, 383, 384]. A single oral
administration of Org43553 at a dose of 50 mg/kg caused ovulation in immature
mice and mature rats, and the resulting oocytes were of good quality,
characterized by high fertility, normal implantation of fertilized eggs into the
uterus, and a high yield of viable embryos [375]. A single oral administration of
Org43553 at the same dose to male rats stimulated testicular steroidogenesis and
increased blood testosterone levels [375]. Both oral and intraperitoneal
administration of TP03 and TP4/2 increased testosterone production in male rats,
and this effect was maintained during a course of drug administration for up to 5
days or more [383, 384]. Various routes of administration of TP03 and TP4/2,
including oral administration, induced ovulation in immature female rats [386, 392]. Since oral administration is preferred and is especially valuable when
using LHCGR agonists in assisted reproductive technologies, the comparability of
bioavailability and physiological response between oral and parenteral delivery
routes of TPDs is one of their advantages over gonadotropins, which can only be
administered parenterally.
When TP4/2 and TP03 were administered to male rats for five days, they showed a
moderate, sustained increase in blood testosterone levels [383, 384]. In contrast
to hCG, the steroidogenic effect of TPDs did not change significantly during the
treatment period, while in the case of gonadotropin it reached a maximum in the
first two days, decreased significantly on the third day (below the level of
treatment with low molecular weight agonists) and on days 4–5 was comparable to
that of TPDs [383, 384]. This is due to the fact that TPDs do not suppress the
expression of the gene encoding LHCGR and have little effect on its distribution
in the testes, while gonadotropins, especially hCG, sharply reduce both the
expression of this gene and the density of LHCGR in the testes, inducing Leydig
cells resistance to LH [383, 384, 409]. Despite the fact that TP03 and TP4/2 more
mildly, in comparison with hCG, stimulated the expression of the main genes of
testicular steroidogenesis (StAR, P450-17), these effects persisted
with long-term administration of the drugs, while in the case of hCG they were
weakened [383, 384]. In TP03-treated rats, there was an increase in the
proportion of sperm with progressive movement, as well as an increase in the
number of spermatogonia and pachytene spermatocytes in the seminiferous
epithelium, indicating stimulation of spermatogenesis by LMW agonists of LHCGR
[384].
The testicular steroidogenesis- and spermatogenesis-stimulating effects of TPDs
have been demonstrated not only in healthy male rats, but also in aging animals,
in rats with the types 1 and 2 DM, and with diet-induced obesity, all of which
had varying degrees of severe androgen deficiency and impaired spermatogenic
function, leading to decreased fertility [383, 384, 410].
When administered to male rats with streptozotocin type 1 diabetes and aging (18
months) animals, TP4/2 restored their androgenic status, normalized the thickness
of the germinal epithelium, the number of spermatogonia and spermatocytes,
thereby restoring impaired spermatogenesis [383]. TP03 compensated for androgen
deficiency and restored attenuated spermatogenesis in male rats with type 2 DM
induced by a high-fat diet and low dose of streptozotocin [384]. Despite some
weakening of the steroidogenic effects of single-administered TPDs in the types 1
and 2 DM and aging compared with those in control animals, it was less pronounced
than in the case of hCG and was absent with long-term administration of TPDs.
This may be due to the preservation of the amount of functionally active LHCGR in
the testes, which we assessed immunohistochemically [383, 384]. Five-week
treatment of male type 2 DM rats with metformin partially restored testicular
steroidogenesis and spermatogenesis and enhanced the steroidogenic effects of
TP03 and hCG, indicating improved testicular sensitivity to both allosteric and
orthosteric LHCGR agonists in metformin-treated animals. However, with long-term
administration of TP03 and hCG, their steroidogenic effects during metformin
therapy, on the contrary, were weakened, which may be a compensatory reaction to
hyperactivation of the testicular steroidogenesis system under the combined
influence of metformin and LHCGR agonists [384, 411]. This opens up prospects for
the development and optimization of approaches to correct androgen deficiency,
which is based on metabolic disorders, using combination therapy, including
allosteric LHCGR agonists.
Pharmacokinetic studies have shown that Org43553 is degraded faster than
gonadotropins [375]. Thus, in rats, the half-life of Org43553 was about 3 hours,
while for hCG it reached 6–7 hours. Our assessment of the half-life of TP03 and
TP4/2 showed similar values for both oral and intraperitoneal routes of
administration (Shpakov, Derkach, unpublished data). Reducing the
half-life is of great practical importance because it reduces the activation time
of LHCGR, and this prevents hyperactivation of LH-dependent cascades and prevents
the development of resistance to endogenous gonadotropins. As a result, the risk
of developing ovarian hyperstimulation syndrome, one of the most serious
complications during controlled ovulation induction using hCG and LH, is reduced,
and the potential oncogenic risks associated with the use of gonadotropins in the
correction of androgen deficiency in men are also reduced.
With a single treatment of mature rats with hCG, a significant increase in the
size of the ovaries, an increase in vascular permeability, and hypersecretion of
vascular endothelial growth factor (VEGF) by granulosa cells are observed, which
are characteristic signs of ovarian hyperstimulation syndrome. At the same time,
when Org43553 was administered to female rats, the size of the ovaries and
vascular permeability in them changed to a small extent, and even repeated
treatment of animals with this agonist did not cause ovarian hyperstimulation
syndrome [376]. One reason for this is the weak effect of Org43553 on the
production of VEGF, a potent angiogenic factor and inducer of increased vascular
permeability [376]. When studying orally administered TP4/2 (40 mg/kg) to
immature female rats stimulated two days earlier with Follimag, it was shown that
TP4/2 time-dependently stimulates the production of progesterone and the
formation of the corpus luteum, increases the expression ovarian genes
responsible for steroidogenesis and involved in the control of ovulation, and
also increases the gene expression and amount of the ADAMTS metalloproteinase,
the most important marker of ovulation [386, 392]. All these effects had some
similarity to those of hCG, but were less pronounced, indicating a moderate
stimulation of ovulation by the LMW agonist of LHCGR. There were significant
differences between hCG and TP03 regarding the stimulation of gene expression of
the proangiogenic factor VEGF type A (VEGF-A). If, after treatment with hCG, the
level of expression of the VEGF-A gene in the ovaries remained elevated
throughout 24 hours after treatment with gonadotropin in comparison with the
group treated only with Follimag, while in the case of TP03 it was increased only
after 4 hours, and then decreased to the control level [386]. It should be noted
that increased levels of VEGF in the tissues of the reproductive system are a
trigger for the development of malignant neoplasms and contribute to the
metastasis of existing tumors. As a result, LMW agonists of LHCGR are believed to
have low pro-tumorigenic potential, which, however, requires further study.
Successful animal experiments allowed for clinical trials of Org43553 as an
ovulation inducer in women [389]. After oral administration of Org43553 at the
doses from 25 to 900 mg, its peak concentration was reached after 0.5–1 hour,
and the half-life was 30–47 hours. At a dose of 300 mg, Org43553 induced
ovulation in 83% of women of reproductive age, without causing significant side
effects. effects. There were no signs of ovarian hyperstimulation syndrome when
taking Org43553 in female volunteers [389]. Compound Org43553 is patented by
Organon/Merck & Co/Merck Sharpe & Dohme (MSD) (Kenilworth, USA), its main
developer [373]. The ability of this compound and other TPDs with LHCGR agonist
activity to increase testosterone levels in men can be used to compensate for
androgen deficiency, correct hypogonadotropic conditions, and also to increase
muscle size and strength in athletes when using TPDs as anabolic steroids. In
this regard, Org43553 and its analogues were recommended for inclusion in the
list of controlled substances in doping tests already at an early stage of their
development [412].
9.3 Additivity of Steroidogenic Effects of Thienopyrimidines and hCG
and Possible Chaperone-Like Effects of Thienopyrimidines with LHCGR Agonistic
Activity
One of the approaches to prevent complications of gonadotropin therapy is to
reduce their dose, but this, as a rule, leads to a significant weakening of the
effectiveness of the drugs and failure to achieve the required effect, including
assisted reproductive technologies. We and other authors have demonstrated the
partial additivity of the effects of low doses of gonadotropins and allosteric
LHCGR agonists on AC activity and testosterone production in the in
vitro conditions [385, 387]. This made it possible to formulate a hypothesis
about their additivity and potentiation under the in vivo conditions.
Such effects are due to both different localization of orthosteric and allosteric
sites in the LHCGR (additivity) and the mutual influence of these sites
(potentiation). In the first case, TPDs can act as a full allosteric agonist; and
in the second case, they can function as PAM or ago-PAM. In support of this, we
showed that pretreatment of male rats with TP03 in doses from 7.5 to 25 mg/kg
(i.p.) almost doubled the steroidogenic effect of hCG and reduced its effective
dose, and also significantly changed the hCG-stimulated expression pattern
steroidogenic genes [413]. There is evidence that gonadotropin is able to
facilitate the binding of LHCGR to a LMW agonist. This is based on a change in
the interaction of the LRR subdomain and hinge region with the extracellular
loops and the outer vestibule of the TMD after receptor binding to gonadotropin.
The result is a change in the superposition of TM6 and TM7 and an increase in the
volume of the transmembrane tunnel cavity in which the allosteric site of LHCGR
is localized, and this changes its accessibility and affinity for the LMW ligand
[95].
Along with the mutual influence of the orthosteric and allosteric sites, an
important contribution to the potentiation effect can be made by the chaperone
properties inherent in allosteric LHCGR agonists, which can be most pronounced in
the case of mutant forms of the receptor with a reduced ability to translocate
into the plasma membrane (Fig. 6). LHCGR with AlaPro and SerTyr
mutations in the TMDs are not capable of translocation into the plasma membrane
and remain in the endoplasmic reticulum, remaining in an inactive state, despite
retaining the ability to bind to gonadotropins. Mutant receptors have been
identified in patients with reproductive dysfunctions caused by hypoplasia of
testosterone-producing Leydig cells [414, 415, 416, 417]. The compound Org42599 with LHCGR
agonist activity, which is a trifluoroacetate salt of Org43553, restores the
activity of mutant LHCGR with AlaPro and SerTyr substitutions
[418]. Incubation of cells expressing mutant LHCGR with Org42599 increased
expression of the mutant receptor, the proportion of LHCGRs with normal folding
of the polypeptide chain and suitable topology in the membrane, and also
increased the density of LHCGRs on the cell surface. This effect was associated
with the ability of Org42599 to penetrate the plasma membrane of Leydig cells and
specifically bind to the allosteric site of the receptor located in the reticular
membrane, which ensured the correct folding of LHCGR and its translocation into
the membrane [418]. The transition of LHCGR to the active state prevented its
degradation in proteasomes, which was due to a change in the interaction of the
mutant receptor in the Org42599-bound state with enzymes and chaperone proteins
responsible for the maturation, translocation and degradation of LHCGR. The
mutant LHCGR in complex with Org42599, like the normal receptor, was able to be
modified by protein disulfide isomerase, which catalyzes the formation of
disulfide bonds in proteins and, thereby, ensuring their appropriate folding.
Along with this, mutant LHCGR in complex with Org42599 lost the ability to form
complexes with 94 kDa glucose-regulated protein (Grp94) and Ig-binding protein
(BiP), which transport misfolded proteins to the site of their degradation in
proteasomes [418].
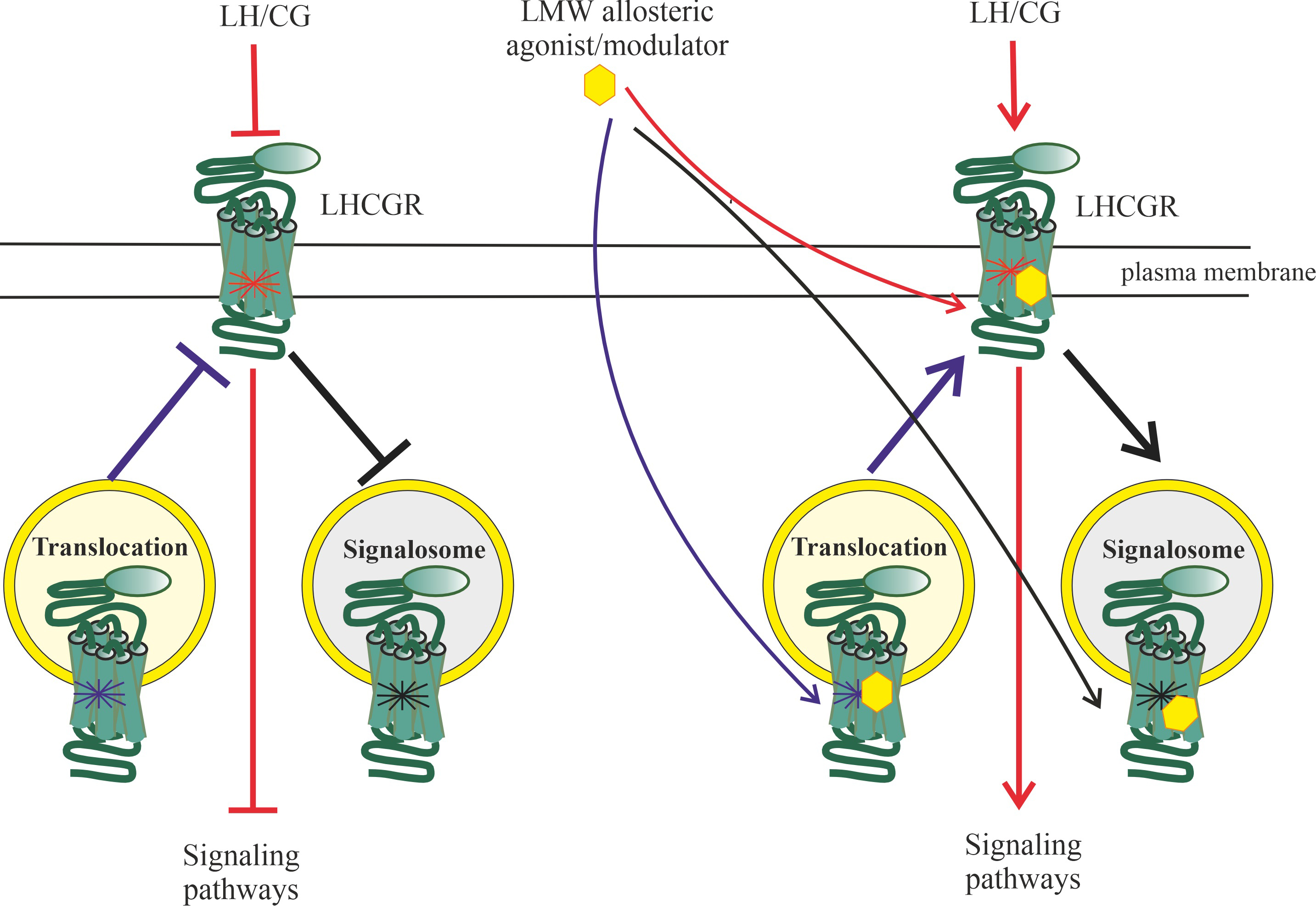 Fig. 6.
Fig. 6.
Chaperone-like activity of low-molecular-weight (LMW)
ligands of luteinizing hormone/chorionic gonadotropin receptor (LHCGR) allosteric
sites. Mutations in LHCGR can lead to a decrease in the affinity of the receptor
for gonadotropin and weakening of gonadotropin-induced receptor activation (red
blocking arrow), deterioration of post-translational processing of the receptor
and its translocation into the plasma membrane (purple blocking arrow), as well
as disruption of interaction with -arrestins and prevention of the
endosomal signaling (black blocking arrow). An allosteric LMW agonist or
modulator is capable of binding to allosteric sites of LHCGR located on the cell
surface (direct interaction of receptor with an extracellular allosteric ligand),
as well as to LHCGR located in intracellular compartments, which is due to the
ability of a hydrophobic allosteric ligand to penetrate through the plasma
membrane into the target cell. Binding of the mutant LHCGR to a suitable
allosteric ligand leads to stabilization of the receptor conformation, similar to
that of the wild-type LHCGR, which can ensure normal binding of the allosteric
ligand-bound mutant LHCGR to gonadotropin and its activation (red arrow),
normalize translocation of the mutant receptor into the membrane (purple arrow),
as well as ensure the formation of the signalosome responsible for intracellular
LHCGR-mediated signal transduction (black arrow). Thus, a suitable allosteric
ligand can act as an extracellular and/or intracellular LMW chaperone for the
mutant receptor, which is in both a free and gonadotropin-bound state.
Potentially, such chaperones can be either hydrophobic allosteric ligands of
allosteric sites located inside the receptor transmembrane channel, or pepducins
(peptides modified by fatty acid radicals that are derivatives of the receptor
intracellular loops), which are able to penetrate the membrane into the cell and
specifically interact with the cytoplasmic allosteric sites of the receptor.
The chaperone-like effect of TPDs may be mediated not only by the potentiating
effect of these compounds on the effects of gonadotropins in vitro and
in vivo, but also by the high efficiency of TPDs, including TP03, in
stimulating steroidogenesis in rats with the types 1 and 2 DM. Moderate and
severe forms of type 1 DM in humans, as well as in animals with experimental
models of type 1 DM, are characterized by severe androgen deficiency and impaired
sensitivity to gonadotropins, as demonstrated by us and other authors [33, 419, 420, 421, 422]. The reasons for this are an increase in the content of reactive oxygen
species, a weakening of the antioxidant defense system and the intensification of
inflammatory and apoptotic processes in the testes, as well as structural changes
in testicular tissue [423, 424, 425, 426]. Based on the above, it can be assumed that when
diabetic rats are treated with TPDs, these compounds, being hydrophobic
substances, penetrate the plasma membrane and stabilize intracellularly localized
LHCGR in a conformation resistant to degradation, allowing their translocation
into the membrane. Thus, the chaperone-like activity inherent in TPDs ensures
their restorative effect on androgen deficiency in conditions of metabolic
disorders that negatively affect the activity of LHCGR, including due to
disturbances in its post-translational modifications, and also in the case of
mutant LHCGR with reduced sensitivity to gonadotropins.
9.4 The Other Low-Molecular-Weight Allosteric Regulators of LHCGR
Along with TPDs, other allosteric LHCGR ligands with heterocyclic structures
have been developed that have full agonist or PAM activity. Of greatest interest
among them are pyrazole derivatives, including Compound 1. It stimulated AC
activity (EC, 20 nM) and increased testosterone synthesis by Leydig cells
(ED, 1.31 µM) in the in vitro conditions, and its effectiveness was
comparable to that of hCG [394]. Under the in vivo conditions, when
administered intraperitoneally to male rats, Compound 1 (32 mg/kg) increased the
level of testosterone in the blood of animals. Like Org43553, the pyrazole
derivative did not compete with the labeled hormone [I]-hCG for binding
sites with LHCGR. This indicates the interaction of pyrazole derivatives with the
allosteric site of the receptor, which does not overlap with the orthosteric site
of LHCGR and is probably localized in its transmembrane tunnel [394].
Along with full agonists and/or PAMs, LMW allosteric ligands for LHCGR with the
activity of NAMs, neutral antagonists or inverse agonists have been developed,
including the derivatives of terphenyl [363, 397], tetrahydro-1,6-naphthyridine
[399], pyrimido[4,5,6-de][1,6]naphthyridine, pyrido[3,4-d]pyrimidine [403],
benzamide [398] and thieno[2,3-d]-pyrimidine [402, 403], as well as
dichlorodiphenyltrichloroethane [400, 401].
Among terphenyl derivatives, the compound LUF5771 suppressed LHCGR stimulation
by both gonadotropins and LMW agonists. LUF5771 at a concentration of 10 µM
accelerated the rate of dissociation of [H]Org43553 from LHCGR by 3.3
times, and also reduced the stimulating effects of hCG and Org43553 on
LH-dependent signaling cascades by 2–3 times [363, 397]. The study of the
molecular determinants responsible for the binding of LUF5771 to the receptor
showed that the binding site for LUF5771, as in the case of Org43553 and other
TPDs, is localized in the transmembrane tunnel, and partially overlaps with the
TPDs-binding site. At the same time, LUF5771-binding site differs from the
TPDs-binding site in its set of AARs, including amino acids localized in helices
TM1, TM2, TM3, TM6 and TM7, and also in ECL2 [363]. Another terphenyl derivative,
compound LUF5419, in the presence of which the inhibitory effects of LUF5771 were
reduced by 2–10 times, bound to the same site as LUF5771, with the only
difference that the LUF5419-binding site did not include AARs localized in
helices TM1 and TM2 [363, 397]. There is reason to believe that it is these AARs
that may make a decisive contribution to the inhibitory effect of LUF5771 on
LHCGR activity.
In 2011, German scientists developed the compounds BAY-298 and BAY-899 based on
tetrahydro-1,6-naphthyridine, which have NAMs activity for LHCGR [399]. BAY-298
inhibited testicular and ovarian steroidogenesis stimulated by LH and the
allosteric full agonist Org43553, and also impaired follicular maturation when
administered to female rats. Importantly, BAY-298 bound to a site distinct not
only from the extracellular orthosteric site, but also from the transmembrane
allosteric site responsible for binding of Org43553. Like BAY-298, BAY-899 also
dose-dependently suppressed the production of steroid hormones when administered
to female rats and arrested the cycle in the diestrus and metestrus phases [399].
We have developed two allosteric inverse LHCGR agonists,
8-amino-5-methyl-2-(methylsulfanyl)-6-(methoxycarbonyl)-7H-pyrimido[4,5,6-de][1,6]naphthyridine-9-carboxylic
acid ethyl ester (PP10), derivative pyrimido[4,5,6-de][1,6]naphthyridine, and
7-amino-4-(3-(ethoxycarbonyl)piperidin-1-yl)-2-(methylsulfanyl)pyrido[4,3-d]pyrimidine-8-carboxylic
acid (PP17), a pyrido[3,4-d]pyrimidine derivative [403]. At a concentration of
100 µM, they weakened the AC-stimulating effects of hCG and TP03 in
testicular membranes, and when intratesticularly injected into male rats (10
mg/kg), they inhibited testosterone production. However, when administered
intraperitoneally, only PP17 (45 mg/kg) retained the ability to suppress both
basal and hCG-induced testicular steroidogenesis, indicating low bioavailability
or instability in the bloodstream of PP10 [403].
Additionally, we have developed TPDs with allosteric LHCGR antagonist activity.
The most active among them was
5-amino-N-(tert-butyl)-2-(methylthio)-4-[3-(2-(ethylamino)nicotinamido)-phenyl]thieno[2,3-d]pyrimidine-6-carboxamide
(TP31) [402, 403]. In the in vitro experiments, TP31 (100 µM)
effectively inhibited the AC-stimulating effects of hCG and TP03, and in the
in vivo experiments, when administered intratesticularly to male rats,
TP31 (10 mg/kg) reduced the basal level of testosterone in the blood, and its
inhibitory effect increased during time, reaching a maximum 5 hours after
injection. Intraperitoneal administration of TP31 (45 mg/kg) resulted in a
decrease in both basal and hCG-stimulated testosterone levels and prevented the
hCG-induced increase in expression of testicular steroidogenec genes [403].
Thus, a series of inverse agonists or NAMs for LHCGR has been developed, both
based on TPDs (TP31) and on other heterocyclic compounds (LUF5771, PP17), which
can be used to create drugs that reduce the sensitivity of cells of the
reproductive system to gonadotropins, which is important for contraception and
treatment of gonadotropin- and steroid-dependent tumors.
More recently, it was found that dichlorodiphenyltrichloroethane (p,pDDT)
negatively affects the activity of LHCGR by preventing its activation by
gonadotropins [401]. In cultured CHO cells, p,pDDT reduced the stimulatory
effect of hCG on the production of intracellular cAMP and also inhibited the
recruitment of -arrestins induced by LH and hCG. At the same time,
p,pDDT had little effect on testosterone production stimulated by these
gonadotropins. Taken together, these data indicate that p,pDDT functions as
NAMs towards LHCGR, and its influence is characterized by a bias towards certain
intracellular cascades [401]. It is interesting to note that p,pDDT is not
specific for LHCGR and is capable of influencing FSHR activity, in which case it
potentiates its activation by FSHR [400]. Perhaps this dual specificity is due to
the ability of FSHR and LHCGR to heterodimerize these receptors.
Another class of compounds that have demonstrated activity against both FSHR and
LHCGR are the benzamide derivatives ADX68692 and ADX68693. They inhibited the
gonadotropin-stimulated activity of both of these receptors with varying
intensity [398]. Quite unexpectedly, the actions of ADX68692 and ADX68693 were
specific to both the specific cell types in which FSHR and LHCGR were expressed,
and also depended on the expression pattern and ratio of these receptors. This
gives grounds to believe that the target of benzamide derivatives was the
heterocomplexes of these receptors, which is responsible for their unusual
activity profile. Thus, in contrast to allosteric full LHCGR agonists, allosteric
neutral antagonists and NAMs are less specific towards LHCGR, and this may be due
to both the similarity in the configuration of the “inhibitory” allosteric site
of FSHR and LHCGR, and their ability to heterodi(oligo) merization [30, 398].
In addition to ligands of the transmembrane allosteric site, ligands of
cytoplasmic allosteric sites, including regions involved in interaction with G
proteins and -arrestins, are promising. Hydrophobic LMW heterocyclic
compounds can be used as such ligands, for example, parmodulins in the case of
proteinase-activated receptors [427, 428] and vercirnon and its derivatives for
chemokine receptors [429, 430]. Also of significant interest are synthetic
peptides corresponding to the ICL2, ICL3 regions and the proximal segment of the
C-tail cytoplasmic domain of GPCR, as well as structurally including the
interfaces of the cytoplasmic loops and TMD, important for signal transduction
[431, 432, 433, 434]. These peptides, interacting with their complementary regions within
the intracellular allosteric site, modulate signal transduction at the stage of
receptor coupling with G proteins and -arrestins, and their action can
be highly selective in relation to both the receptor and intracellular cascades.
Moreover, they themselves are capable of triggering signal transduction, in the
absence of an orthosteric agonist. In order to be available for interaction with
an intracellular allosteric site, such peptides must cross the plasma membrane,
as a result of which they are modified with a hydrophobic radical, usually a
palmitic acid residue (such peptides are called pepducins) [432, 433, 435, 436, 437].
Based on the fact that the C-terminal half of ICL3 is involved in
interaction with the G protein, which indirectly indicates its overlap
and/or interaction with the intracellular allosteric G-competent site, we
synthesized and studied the C-terminally palmitoylated peptide
NKDTKIAKK-Nle-A(562–572)-K(Pal)A, which corresponded to the region 562–572 of
LHCGR, and its analogs [395, 396, 438, 439]. Under the in vitro
conditions at micromolar concentrations, this peptide increased AC activity and
GTP binding in testicular membranes, and when administered intratesticularly to
male rats, it stimulated their testicular steroidogenesis and increased
testosterone production [395, 439]. At the same time, it significantly inhibited
the stimulating effects of hCG on the activity of the AC system in vitro
and on testosterone production in vivo, which indicates its
pharmacological profile as a PAM antagonist [395, 439]. A series of derivatives
of peptide 562–572 with acyl radicals of different localization and
hydrophobicity was synthesized and it was found that the highest activity was
characterized only by those derivatives that had a fatty acid residue at the
C-terminus of the peptide, which in native LHCGR borders the intracellular end of
the TM6 helix [438]. Thus, it was shown that the hydrophobic radical is necessary
not only for the efficient penetration of pepducin through the membrane, but also
for its anchoring on the cytoplasmic side of the membrane, near the site of its
interaction with the complementary regions of the receptor ICLs. This provides a
suitable position of pepducin for allosteric regulation of LHCGR-mediated signal
transduction [438]. It should be noted, however, that when administered
subcutaneously and intravenously, the peptide 562–572 LHCGR showed low stability
and little activity, which requires further research to improve its resistance to
proteolysis.
10. Conclusion and Perspectives
In the process of evolution of vertebrates, along with the complication and
improvement of the mechanisms regulating reproductive functions, including a more
subtle organization of hormonal control of the estrous cycle in females, there
was an increase in the complexity and flexibility of controlling these
mechanisms, including at the level of the LH-regulated signaling system. To
increase its information capacity and efficiency, the range of signaling cascades
and effector proteins activated by gonadotropins with LH activity was expanded,
and mechanisms were developed to ensure the targeting of LH signaling and its
fine tuning under specific physiological conditions. And to achieve this, various
strategies have been implemented.
Some of these strategies were implemented by changing the structure of
gonadotropins, endogenous ligands of the orthosteric site of LHCGR, without
significantly changing the structure of this site. To this end, at the genome
level, through gene duplication and alternative splicing, various forms of
gonadotropins were generated, which in humans and some primates are represented
by -LH and -CG, and, in addition to this, the glycosylation
status of gonadotropin subunits at asparagine residues (LH) and at serine or
threonine residues (CH, equine LH) also varied significantly. Thus, not only was
the efficiency and bias of gonadotropin-induced signal transduction required
under certain physiological conditions achieved, but also additional levels of
its regulation were provided. First, since LH and CG can be produced by different
tissues (pituitary and extrapituitary origin), their expression and secretion in
these tissues will be regulated by different factors and by different mechanisms.
Secondly, in different tissues and cell types there are significant differences
in the machinery of N- and O-glycosylation, due to different
patterns of expression and activity of glycosyltransferases and glycosidases,
which leads to an almost inexhaustible variety of glycoforms of LH and CG. The
multiplicity of glycoforms of dimeric gonadotropins is also ensured by the
combination of - and -subunits of gonadotropins, which have a
different set of glycans. In this case, not only the number of glycans plays an
important role, but also the degree of their branching and the content and
distribution of negatively charged glycosyl residues at the ends of the glycan
backbone. It should be noted that a significant, and possibly decisive,
contribution to the interaction of both various gonadotropins and their
glycoforms with LHCGR is made by allosteric mechanisms. They are due to the fact
that the binding of LHCGR to LH or hCG molecules and their various glycoforms can
directly or indirectly affect the structural characteristics of allosteric sites
localized both in the LRR subdomain, hinge region and ECLs, and in the TMD, as
well as on the accessibility these allosteric sites for ligands.
It is also important that this changes the superposition of “internal”
allosteric regulators, the functions of which are performed by fragments of the
hinge region and, possibly, other parts of the receptor molecule. Direct
interaction of N- and O-glycans of gonadotropins with
allosteric sites of LHCGR or glycan-glycan interactions between gonadotropins and
the receptor, changing the conformational characteristics of such sites, cannot
be excluded.
Other strategies involve “gonadotropin-independent” influence on the
conformational characteristics of the LHCGR and its ability to interact with
gonadotropin, and these strategies rely almost exclusively on allosteric
mechanisms. Firstly, this is the homodi(oligo)merization of LHCGR or the
formation of its heterocomplexes with other receptors, primarily with FSHR. The
formation of receptor complexes can lead to a weakening or even prevention of the
gonadotropin signal (LHCGR homodimerization), and to a certain extent determines
its bias towards intracellular targets. In the case of the LHCGR/FSHR
heterocomplex, due to trans-activation, LH-dependent intracellular
cascades can be triggered by FSH. Combining the allosteric effects of the
formation of LHCGR complexes on LH/CG-mediated transduction with those of various
combinations of gonadotropin subunits and their glycoforms leads, on the one
hand, to an even greater variety of potential effects of gonadotropins in the
target cell, and on the other hand, allows for finely regulated selectivity and
the intensity of the hormonal signal, which is of great importance for the
development of drugs with LH-like activity for use in reproductive medicine.
Little studied, but of considerable interest, is the possible impact of
autoantibodies developed against molecular determinants localized in the
ectodomain of LHCGR on its activity, since such autoantibodies, interacting with
LHCGR, are capable of both changing its basal (constitutive) LHCGR activity and
modulating the effects endogenous gonadotropins on LHCGR, as shown for anti-TSHR
antibodies.
The presence of allosteric sites in LHCGR, localized in the TMD and ICLs,
creates good opportunities for the development of artificial allosteric
regulators of different chemical nature with a wide range of pharmacological
activity. Currently, the main emphasis is on the creation of low-molecular
regulators of a heterocyclic nature, targeting allosteric sites localized inside
the transmembrane tunnel, the most effective of which are TDs. These regulators
are endowed with the intrinsic activity of full or inverse agonists and the
activity of PAMs and NAMs. They moderately affect LHCGR activity, biasly regulate
LH-dependent intracellular cascades, and also retain specific activity when
administered orally. Thus, they have a number of advantages over
gonadotropin-based drugs, and in the future can be used as prototype drugs for
correcting androgen deficiency and in ARTs.
Allosteric sites located in the extracellular and cytoplasmic regions of the
receptor can also become targets for the development of allosteric regulators. In
this case, peptide regulators corresponding in primary structure to the
membrane-proximal regions of ICL2, ICL3 and the C-terminal domain, as well as
peptide constructs similar to regions of the hinge region and ECLs of LHCGR, are
of particular interest. For ICL-derived peptides with the activity of allosteric
regulators of GPCRs, there are very successful developments based on the creation
of pepducins (ICL-derived peptides modified by hydrophobic radicals), including
for LHCGR, FSHR and TSHR, as well as for the structurally related relaxin
receptor [435, 437, 440, 441]. The main obstacle to the widespread use of such
peptide constructs is their low resistance to proteolytic degradation, especially
considering their enrichment in positively charged AARs. Along with pepducins,
allosteric regulators of LHCGR, there may also be LMW compounds that can
penetrate the plasma membrane and effectively interact with allosteric sites
localized in ICLs and in the intracellular vestibule of the transmembrane tunnel,
as shown for parmodulins, intracellular LMW allosteric regulators of
protease-activated receptors [428, 442].
Finally, it is of particular interest to assess the influence of cholesterol,
phospholipids and other membrane lipids on the functional activity of LHCGR
through their interaction with allosteric sites located on the lateral surface of
the TMD and at its interfaces with extracellular and cytoplasmic loops. In this
case, a relationship can be established between the composition of the diet,
metabolic disorders and other factors affecting the lipid composition of the
membrane, and the activity of LHCGR signaling. It is also impossible to exclude
the influence of steroid hormones on the activity of LHCGR, not according to the
classical scenario of negative feedback, but through direct interaction with
allosteric sites of the receptor.
All this stimulates further research into the allosteric mechanisms of LHCGR
functioning and regulation, as well as the search for new technologies to
specifically influence the functional activity of the LHCGR signaling system.
Abbreviations
AAR, amino acid residue; AC, adenylyl cyclase; ART, assisted reproductive technology; BAM, biased allosteric modulator; cAMP, 3-5-cyclic adenosine monophoshate; CG, chorionic gonadotropin; CREB, cAMP-dependent transcription factor (cAMP Response Element-Binding protein); DAG, diacylglycerol; DM, diabetes mellitus; eCG, equine chorionic gonadotropin; ECL1,2,3, extracellular loops 1, 2 and 3; EPAC1/2, Exchange Protein directly activated by Cyclic AMP, types 1 and 2; ERK1/2, extracellular signal-regulated kinases, types 1 and 2, the effector components of mitogen-activated protein kinase cascade; FSH, follicle-stimulating hormone; FSHR, follicle-stimulating hormone receptor; GalNAc, N-acetylgalactosamine; G, heterotrimeric G proteins that inhibit adenylyl cyclase activity; GnRH, gonadotropin-releasing hormone; GPCR, G protein-coupled receptor; GPH, glycoprotein hormone; GPHR, glycoprotein hormone receptor; G, heterotrimeric G proteins that and stimulate PLC and calcium signaling; GRK, G protein-coupled receptor-specific protein kinases; G, heterotrimeric G proteins that stimulate adenylyl cyclase activity; hCG, human chorionic gonadotropin; ICL1,2,3, intracellular loops 1, 2 and 3; IP3, inositol-3,4,5-triphosphate; LH, luteinizing hormone; LHCGR, luteinizing hormone/chorionic gonadotropin receptor; LMW, low-molecular-weight; LRR, leucine-rich repeat; MAPKs, mitogen-activated protein kinases; NAM, negative allosteric modulator; PAM, positive allosteric modulator; PDE, cyclic nuleotide-activated phosphodiesterase; PKA, protein kinase A; PKC, protein kinase C; PLC, phosphoinositide-specific phospholipase C; StAR, cholesterol-transporting protein (Steroidogenic Acute Regulatory protein); TGF, transforming growth factor-; TM, transmembrane helix; TPD, thieno[2,3-d]pyrimidine derivative; TSH, thyroid-stimulating hormone; TSHR, thyroid-stimulating hormone receptor; VEGF, vascular endothelial growth factor.
Author Contributions
AS was responsible for the entire preparation of this manuscript.
Ethics Approval and Consent to Participate
Not applicable.
Acknowledgment
Not applicable.
Funding
The work is supported by the Russian Science Foundation (№ 19-75-20122).
Conflict of Interest
The author declares no conflict of interest. Given his/her role as Guest Editor, Alexander O. Shpakov had no involvement in the peer-review of this article and has no access to information regarding its peer review. Full responsibility for the editorial process for this article was delegated to Marina Ivanišević and Jordi Sastre-Serra.








