

 , Su Myat Phyu 1, Shisong Jiang 1
, Su Myat Phyu 1, Shisong Jiang 11 Department of Oncology, University of Oxford, OX3 7DQ Oxford, UK
Abstract
Survivin, an important inhibitor of apoptosis protein, contributes to cancer cells’ resistance to apoptosis, proliferation, and survival. It is a promising biomarker and therapeutic target due to being highly expressed in cancer cells relative to normal cells and universally expressed in almost all cancer types. Cancer cells release survivin to the tumour microenvironment (TME) not only as a free protein but also encapsulated in extracellular vesicles (EVs), especially small EVs (sEVs). The release of encapsulated survivin from cancer cells can be taken up by neighbouring cells, eliciting pathological responses such as tumorigenesis and metastasis. Consequently, EV survivin holds potential as a diagnostic, prognostic, and therapeutic biomarker for several types of cancer, including breast cancer, prostate cancer, pancreatic cancer, and glioblastoma. EV survivin expression is significantly elevated in cancer patients and correlates with unfavourable clinicopathologic parameters. Although no clinical studies have explored EV survivin as a therapeutic target, future research should explore survivin-based therapies in combination with EV-targeting therapies to effectively disrupt its roles in tumorigenesis and metastasis.
Keywords
- survivin
- extracellular vesicle
- cancer
- EV survivin
- apoptosis
The tumour microenvironment (TME) is a dynamic ecosystem comprising cancer cells, stromal cells, immune cells, and extracellular matrix components, playing a pivotal role in cancer biology by influencing tumour initiation, progression, metastasis, and response to therapy [1]. Within the TME, various cellular and molecular interactions occur, shaping the tumour’s behaviour and influencing its clinical outcomes. Among the key mediators of these interactions are extracellular vesicles (EVs) released by both cancer and stromal cells into the extracellular space. EVs carry a diverse cargo of biomolecules, including proteins, nucleic acids, lipids, and metabolites, which can reflect the features of the parent cells [2]. Importantly, EVs facilitate communication between different cell types within the TME by transferring their cargo to recipient cells via various mechanisms, such as receptor-mediated uptake or fusion with the cell membrane [3]. The impact of EVs on the TME is multifaceted—promoting tumour growth, angiogenesis, immune evasion, metastasis, and the establishment of a pre-metastatic niche [3]. Furthermore, emerging evidence suggests that exosomes derived from specific cell types within the TME exhibit distinct cargo profiles and functional properties, shaping the TME’s heterogeneity and driving tumour progression and therapeutic resistance [3]. Consequently, molecules and soluble factors implicated in the TME have emerged as attractive diagnostic markers or therapeutic targets of cancer [4].
Survivin, an important member of the inhibitor of apoptosis protein (IAP) family
with universal expression in cancer cells, is intricately involved in regulating
apoptosis, cell proliferation, metastasis, and resistance to therapy [5].
Physiologically, survivin plays essential roles in mitosis regulation, apoptosis
inhibition, and angiogenesis [6]. In cancer, survivin is involved in
tumourigenesis through various mechanisms: inhibition of apoptosis pathways,
regulation of cytokinesis and cell cycle progression, and participation in
numerous pathways, including p53, Wnt, hypoxia, transforming growth factor
(TGF)-
Survivin has been shown to localise intracellularly in mitochondria, cytosol,
and nuclei, where it regulates cellular apoptosis and mitosis [18, 19, 20]. Several
studies have shown that cancer cells release survivin to the extracellular space
(TME) packaged in EVs, especially small EVs (size
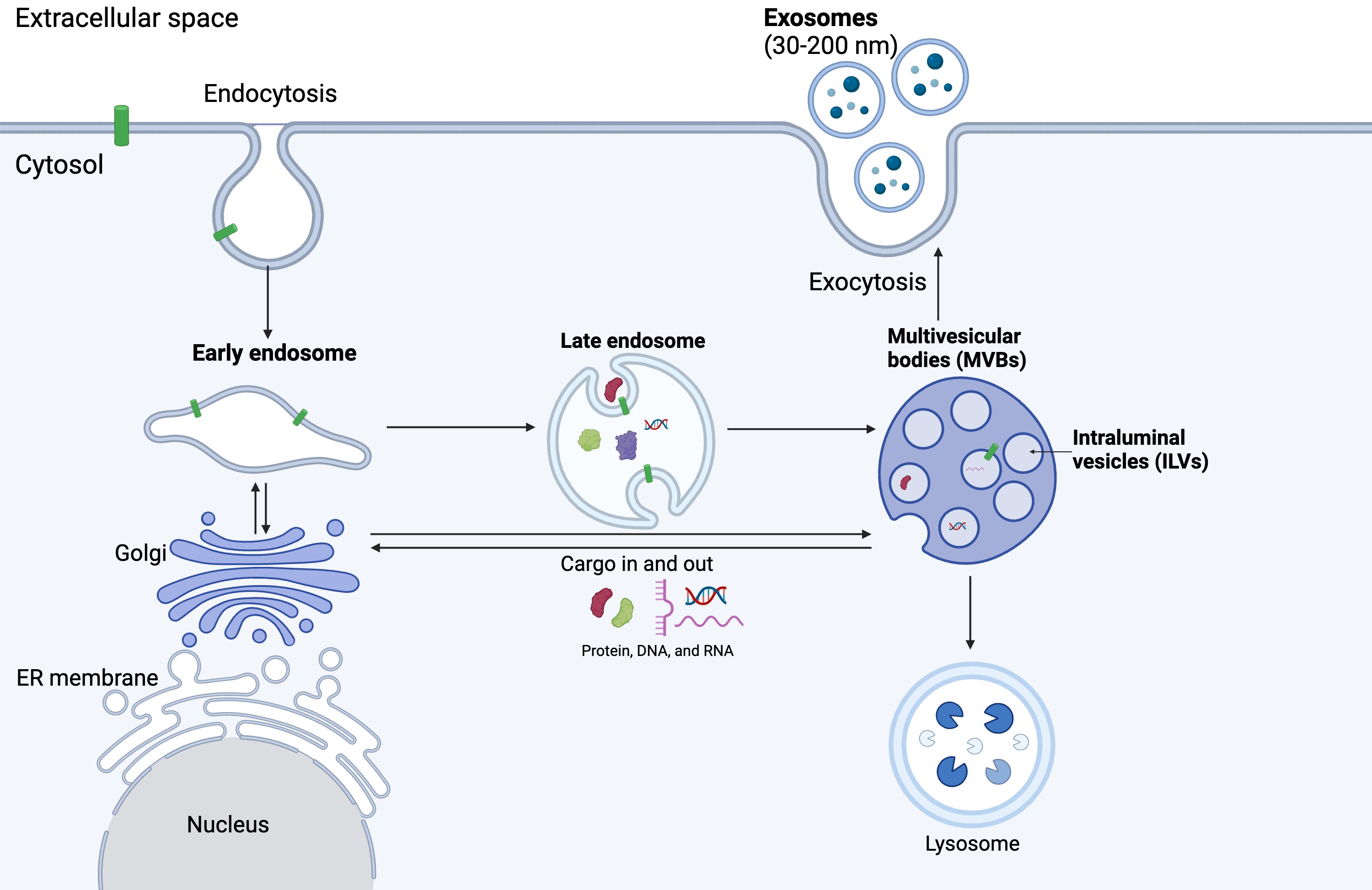
Exosomes are the smallest subset of EVs, ranging from 30 to 200 nm in size. They are secreted by various cell types and have garnered significant attention in recent years due to their diverse biological functions and potential clinical applications. The biogenesis of exosomes (Fig. 1) is a complex and tightly regulated process involving multiple cellular pathways [27]. Exosomes are formed through the endosomal pathway, which begins with the internalisation of cargoes by the invagination of the plasma membrane (endocytosis). These cargoes are sorted to form early endosomes, which consequently mature into late endosomes, also known as multivesicular bodies (MVBs), through the inward budding of the endosomal membrane. This process leads to the accumulation of intraluminal vesicles (ILVs) – small vesicles within the lumen of MVBs [28]. The cargo packaged into ILVs includes proteins, lipids, nucleic acids, and other biomolecules, reflecting the composition of the parent cell. Cargoes are delivered from the trans-Golgi network and cytosol [29, 30, 31].
 Fig. 1.
Fig. 1.
Exosome biogenesis. Exosome biogenesis begins with the endocytosis of cargos, forming early endosomes. Early endosomes mature into late endosomes and later multivesicular bodies consisting of multiple intraluminal vesicles (ILVs). ILVs are released as exosomes into the extracellular space through the process of exocytosis (Created with BioRender.com).
Several molecular mechanisms regulate the sorting of cargo into ILVs during exosome biogenesis. The endosomal sorting complexes required for transport (ESCRT) machinery play the primary role in this process [32]. The ESCRT machinery consists of four protein complexes (ESCRT-0, ESCRT-I, ESCRT-II, and ESCRT-III) that work sequentially to recognise and sort ubiquitinated cargo proteins into ILVs. Initially, ubiquitinated proteins destined for packaging in exosomes are recognised and gathered into endosomal membranes. This process is facilitated by ESCRT-0 (hepatocyte growth factor-regulated tyrosine kinase substrate [Hrs] and signal transducing adaptor molecule [STAM]), which recognises and clusters ubiquitinated cargo. ESCRT-I and ESCRT-II complexes are recruited to the endosomal membrane, where they further concentrate the ubiquitinated cargo and start the budding process to form vesicle precursors inside the endosome. Next, ESCRT-III is recruited to the budding site, where it forms filaments that constrict the neck of the emerging vesicle, leading to its separation from the endosomal membrane [33, 34].
Additionally, ESCRT-independent mechanisms involving lipids, tetraspanins, and other proteins have been suggested in cargo sorting and ILV formation. Complex lipids, such as ceramide, can accumulate in certain areas of the membrane, leading to the formation of lipid rafts. These rafts are more ordered and tightly packed than the surrounding membrane, leading to inward budding and vesicle formation [35]. Clustering of tetraspanins (CD63, CD81, CD9) also induces membrane curvature and vesicle formation [36]. Other ESCRT-independent mechanisms include the syndecan-syntenin-ALG2 interacting protein X (ALIX) pathway [37] and the accumulation of sphingolipids [38].
Once formed, MVBs can follow two distinct fates: they can either fuse with lysosomes for degradation or with the plasma membrane for exosome release. The regulation of MVB trafficking and fusion is tightly controlled by various molecular mechanisms, including Rab GTPases (such as Rab27a and Rab27b), soluble N-ethylmaleimide-sensitive factor attachment protein receptors (SNAREs), and lipid signalling pathways. Upon fusion with the plasma membrane, MVBs release their ILVs into the extracellular space as exosomes [39].
Exosome release from cells is affected by a variety of physiological and pathological conditions. This regulation is essential to maintaining cellular homeostasis and facilitating intercellular communication. However, it can also contribute to the progression of diseases, including cancer, neurodegenerative diseases, and inflammatory conditions. Exosome release has been shown to increase under stress conditions, hypoxia [40], acidic microenvironmental pH [41], and cellular senescence [42]. In cancer, exosomes play important roles in intercellular communication.
For example, cellular stress, inflammation, and oncogenic transformation can
modulate exosome secretion and cargo composition. Furthermore, recent studies
have highlighted the role of cell-to-cell communication in regulating exosome
release, suggesting that exosomes may serve as important mediators of
intercellular communication in both physiological and pathological contexts. In
cancer, tumour cells use exosome release for communication, promoting tumour
growth, angiogenesis, and metastasis. The altered microenvironment and oncogenic
signalling in tumour cells significantly increase exosome secretion [43]. Details
of exosomes’ roles in cancer will be explored further in the next section. In
neurodegenerative diseases, such as Alzheimer’s and Parkinson’s, the dysregulated
release of exosomes contributed to the spread of neurotoxic proteins, such as
amyloid-
Microvesicles (MVs) typically range from 100 nm up to 1000 nm in diameter, with the average size being 250–400 nm [46]. MVs are EVs released by direct outward budding of the cell membrane through adenosine diphosphate (ADP)-ribosylation factor 6 (ARF6) [47] and small GTPase Ras homolog gene family member A (RhoA)-dependent rearrangement of the actin cytoskeleton [48]. MV biogenesis comprises several steps, including plasma membrane reorganisation, redistribution of phospholipids, outward repositioning of phosphatidylserine, disassembly of the cytoskeleton network, and actomyosin basal abscission via the activation of ESCRT-I, myosin light chain kinase (MLCK) and ADP ribosylation factor 6 [49, 50, 51]. The budding of the membrane occurs at particular locations on the plasma membrane and is influenced by phospholipid redistribution, together with Rho-kinase-mediated myosin light chain phosphorylation and contractile machinery to allow for vesicle pinching and detachment [52, 53]. MV cargo comprises cytosolic proteins, plasma membrane-associated proteins such as tetraspanins, lipids and fragmented nucleic acids (DNA and/or RNA) [54, 55, 56]. Despite the distinct mechanism for biogenesis and membrane origin, both endosome-origin EVs and MVs can work similarly, and specific markers are still lacking to distinguish MVs from exosomes [57].
The size of apoptotic bodies ranges from 50 to 5000 nm in diameter [58, 59]. They are released by dying cells through the characteristic blebbing and fragmentation of the cell membrane during cell death into the extracellular space [60, 61, 62, 63]. Apoptotic bodies contain whole cellular organelles [64], nuclear genomic DNA [65], fragmented nucleic acids and randomly enclosed cargo [66]. Apoptotic bodies have been demonstrated to present CX3 C-chemokine ligand 1 (CX3 CL1) and intercellular adhesion molecule 3 (ICAM3) to attract phagocytic cells for engulfment [67] and contain an abundant amount of 18S and 28S rRNA [55, 56]. In contrast to exosomes and MVs, apoptotic bodies contain intact organelles, chromatin, and small amounts of glycosylated proteins [68, 69]. Therefore, higher levels of proteins associated with the nucleus (i.e., histones), mitochondria (i.e., HSP60), Golgi apparatus, and endoplasmic reticulum (i.e., GRP78) were reported to be observed inside apoptotic bodies. In recent years, it has been hypothesised that apoptotic cells communicate with other cells via apoptotic bodies to propagate tumorigenicity and horizontal DNA transfer [65] and promote inflammation [70].
EVs play various roles in cancer progression and shaping the tumour microenvironment. EVs act as key mediators of intercellular communication by carrying a cargo of proteins, lipids, and nucleic acids, which contribute to tumorigenesis, metastasis, immune evasion, and therapeutic resistance.
EVs help create complex communication networks in the TME by carrying oncogenic signals (proteins, RNA/DNA) from cancer cells to other cells, which support tumour proliferation, angiogenesis, and immune evasion. For instance, small EVs derived from metastatic melanomas increased the metastatic behaviour of primary tumours by educating bone marrow progenitors through the receptor tyrosine kinase mesenchymal epithelial transition (MET) [71]. Tumour-derived EVs also carry angiogenic factors and microRNAs (miRNAs) that promote the formation of tumour blood vessels. A miRNA of colorectal cancer (CRC), called miR-2503p, can be transferred from CRC cells to endothelial cells via small EVs. EV miR-25-3p regulated the expression of vascular endothelial growth factor receptor 2 (VEGFR2), zonula occludens-1 (ZO-1), occluding, and Claudin5 in endothelial cells by targeting Kruppel-like Factor (KLF)2 and KLF4, promoting vascular permeability and angiogenesis. It also induces vascular leakiness and enhances CRC metastasis in the liver and lung of mice [72].
EVs have also been implicated in the transmission of genomic instability from cancer cells towards normal cells. EVs released from Harvey Rat sarcoma virus (HRAS)-driven rat intestinal epithelial cells were shown to be transferring genomic DNA, including oncogenic sequences, to endothelial cells. This transfer leads to abnormal cellular behaviours, such as the formation of aberrant micronuclei, increased cell migration, and proliferation, all of which are associated with cancer development and progression [73]. Tumour-derived EVs have also been suggested to contribute to immune evasion by transporting immunosuppressive molecules. Programmed death-ligand 1 (PD-L1) has been demonstrated to be secreted in tumour-derived EVs, suppressing T cell activation in the draining lymph node. In addition, EV PD-L1 appears to be resistant to anti-PD-L1 antibody blockade. Meanwhile, suppression of EV PD-L1 inhibits tumour growth, even in models resistant to anti-PD-L1 antibodies [74].
EVs also contribute to therapeutic resistance in cancer by transferring drug resistance genes, proteins, and microRNAs (miRNAs) between cancer cells, promoting the spread of resistance mechanisms throughout the tumour. Recipient cancer cells are then able to evade the effects of chemotherapy and targeted therapies despite being initially sensitive. As an example, exosomes carry and transfer P-glycoprotein, an efflux pump associated with multidrug resistance, and miRNAs that can suppress the expression of drug targets or activate survival pathways. EVs can also gather and remove anticancer drugs from cells, reducing the intracellular concentrations of these drugs and thus decreasing their efficacy [27, 75]. EVs not only speed up the spread of resistance among cancer cells but also significantly hinder the success of cancer therapeutics.
Aberrant expression of IAP proteins is one of the mechanisms contributing to the
resistance to apoptosis in human cancers. The IAP family is a group of
apoptosis-negative regulators characterised by the presence of at least one copy
of the baculovirus IAP repeats (BIR) domain (containing 70 amino acids) at their
N-terminus [76]. Survivin is the smallest member of the IAP family, containing
only 1 BIR domain [8, 9]. Survivin physiological functions include mitosis
regulation, apoptosis inhibition, angiogenesis, and cell motility [6]. In cancer,
survivin is involved in tumorigenesis through a variety of mechanisms, including
inhibition of apoptosis pathways, regulation of cytokinesis and cell cycle
progression, and participation in numerous pathways, including p53, Wnt, hypoxia,
TGF-
Traditionally, survivin has been shown to localise intracellularly in mitochondria, cytosol, and nuclei, where it performs its functions [18, 19, 20]. Recently, survivin has also been found in the extracellular space, contained in EVs secreted by various cancer cell lines [77, 78], especially small EVs (Fig. 2 (Ref. [18]), Table 1 (Ref. [22, 23, 78, 79, 80, 81, 82, 83, 84, 85])) [21, 22, 79, 80, 81, 82]. Mechanisms regarding survivin release via EVs have not been fully understood (Fig. 3). Survivin has been found associating with heat shock proteins (Hsp), including Hsp70 and Hsp90, in the conditioned medium of serum-starved HeLa cells [22], indicating that it is released in response to a stressful environment. Exosomal survivin was demonstrated to be enriched in breast cancer cells treated with paclitaxel, and these exosomes strongly promote the survival and chemoresistance of cancer cells [83]. Sublethal proton irradiation (3 Gy) also resulted in a significant accumulation of survivin in the exosomal fraction from the conditioned medium of serum-starved HeLa cells [22]. Therefore, further studies are required to confirm the pathways by which EV survivin is released.
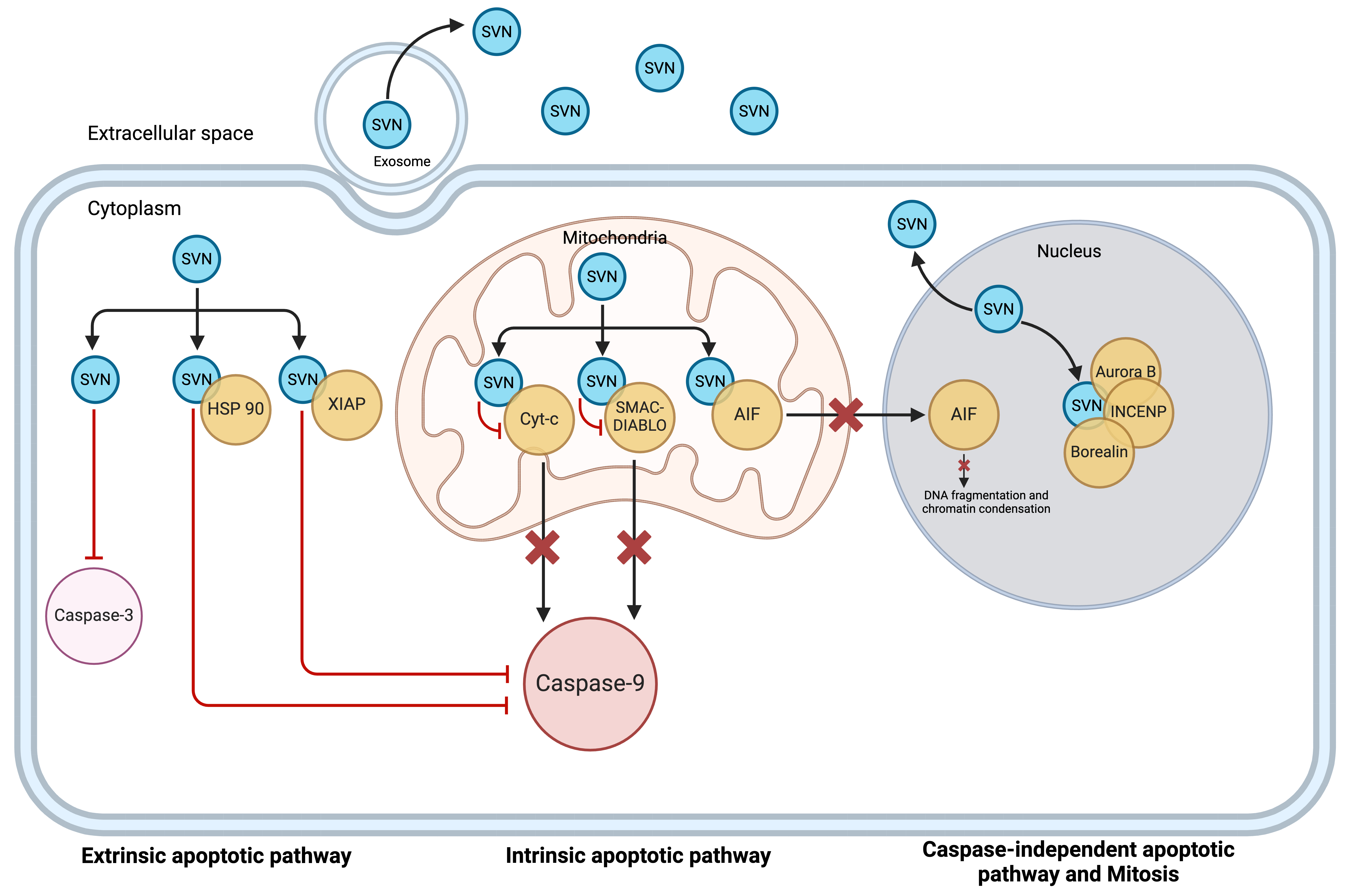
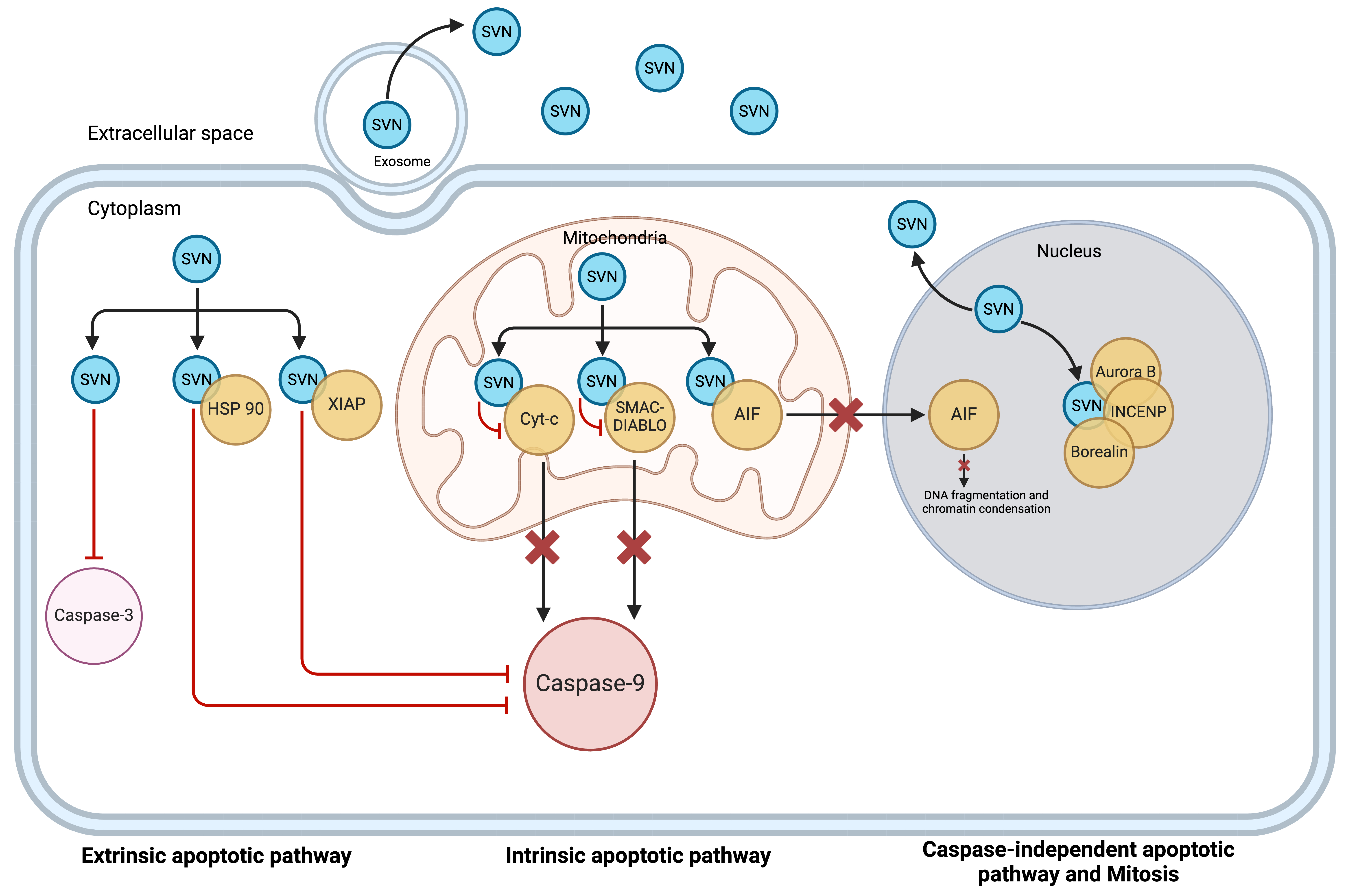 Fig. 2.
Fig. 2.
Schematic presentation of survivin locations. Survivin is located intracellularly in the cytoplasm, mitochondria, and nucleus. In the cytoplasm and mitochondria, it functions as an anti-apoptotic protein. In the nucleus, it plays its role as a pro-mitotic protein. Survivin is also released into the extracellular space as a free soluble protein as well as via exosomes (reproduced with permission from Li et al., Expert Opinion on Biological Therapy; published by Taylor & Francis, 2021 [18]). SVN, Survivin; HSP, Heat shock protein; XIAP, X-linked inhibitor of apoptosis; AIF, apoptosis-inducing factor; INCENP, inner centromere protein.
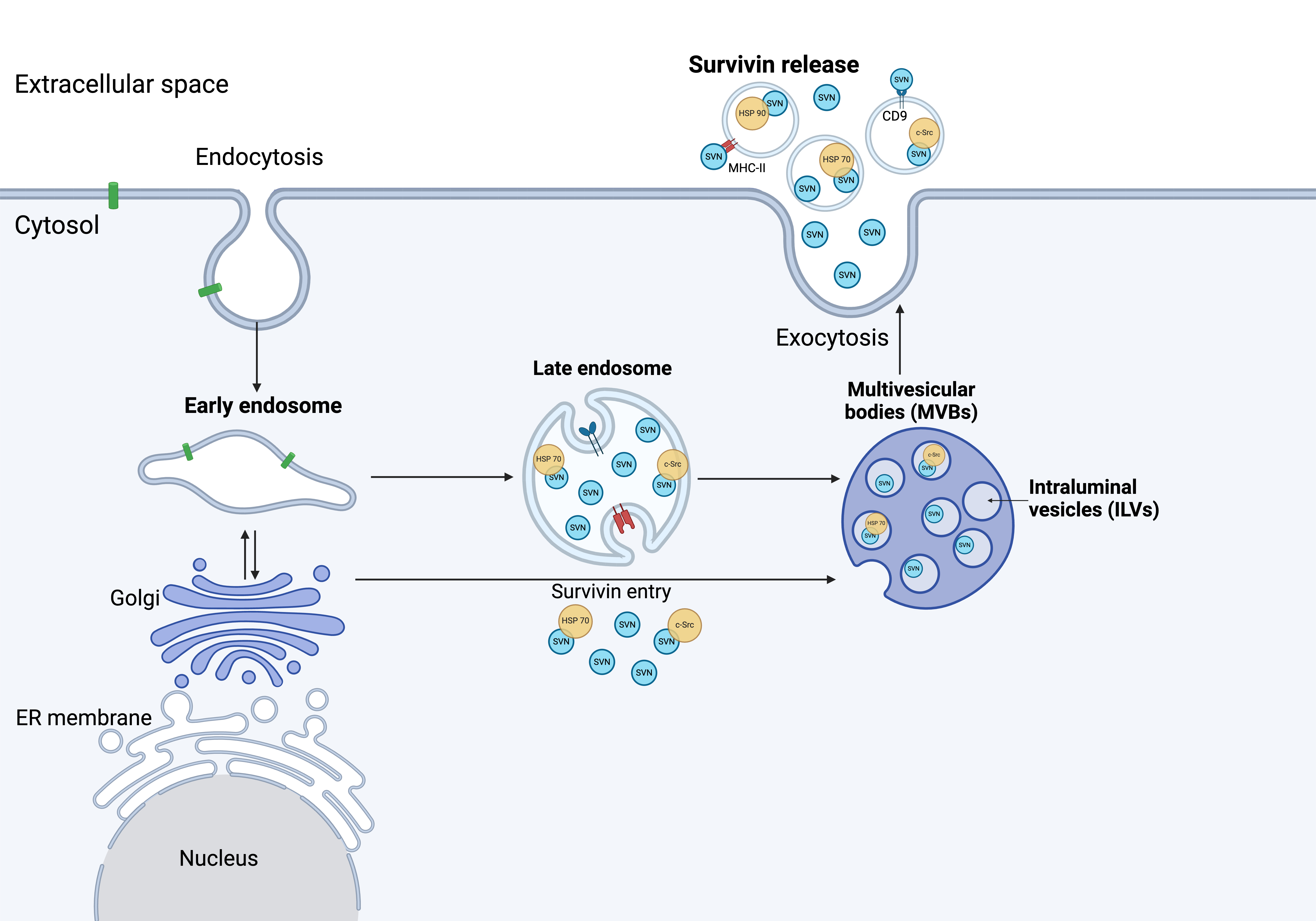
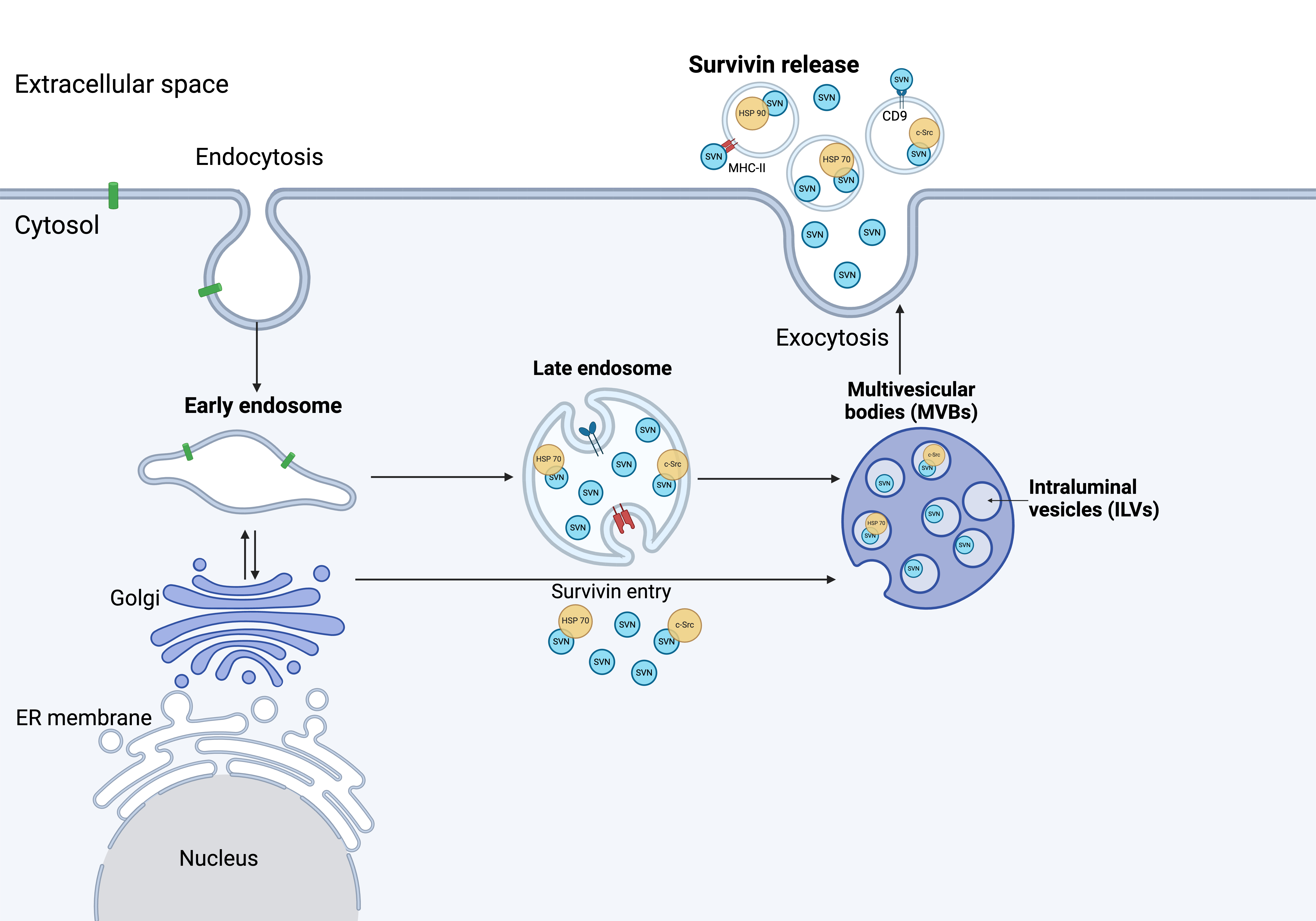 Fig. 3.
Fig. 3.
Survivin release from cancer cells. The mechanism of survivin release has yet to be fully understood. Several studies have shown survivin associated with Hsp70, Hsp90, and c-Src upon release. Immuno-magnetic extraction of HeLa cell-secreted exosomes coupled to human MHC Class II (HLA-DR) or CD9 aldehyde-sulfate latex beads, exhibited a phenotype with high expression of LAMP1 and Hsp70, as well as expression of CD54, CD9 and survivin (Created with BioRender.com). ER, endoplasmic reticulum.
| Cancer site | Cancer type | Cell line | Vesicle type | Studies |
| Brain | Glioblastoma | SNB-19 | EV | Hurwitz et al., 2016 [78] |
| Glioblastoma | U251 | EV | Hurwitz et al., 2016 [78] | |
| Glioblastoma | Obtained from surgical resections | Small EV | Skog et al., 2008 [81] | |
| Glioblastoma | U87 | Small EV | Kreger et al., 2016 [83] | |
| Glioblastoma | A-1207 | Small EV | Figel et al., 2021 [84] | |
| Breast | Breast adenocarcinoma | MDA-MB-468 | EV | Hurwitz et al., 2016 [78] |
| Breast adenocarcinoma | MDA-MB-231 | Small EV | Li et al., 2020 [79]; Kreger et al., 2016 [83]; Chang et al., 2021 [80]; | |
| Ductal carcinoma | BT-549 | Small EV | Li et al., 2020 [79] | |
| Breast adenocarcinoma | SKBR3 | Small EV | Kreger et al., 2016 [83] | |
| Colon | Colorectal adenocarcinoma | HCT-116 | EV | Hurwitz et al., 2016 [78] |
| Colorectal adenocarcinoma | HCT-15 | EV | Hurwitz et al., 2016 [78] | |
| Colorectal adenocarcinoma | KM12 | EV | Hurwitz et al., 2016 [78] | |
| Colorectal adenocarcinoma | SW620 | EV | Hurwitz et al., 2016 [78] | |
| Colorectal adenocarcinoma | SW840 | Small EV | Hong et al., 2009 [82] | |
| Leukaemia | Acute lymphoblastic leukaemia (ALL) | CCRF-CEM | EV | Hurwitz et al., 2016 [78] |
| Lung | Squamous cell carcinoma | NCI-H226 | EV | Hurwitz et al., 2016 [78] |
| Large cell carcinoma | NCI-H460 | EV | Hurwitz et al., 2016 [78] | |
| Lung adenocarcinoma | NCI-H23 | Small EV | Chang et al., 2021 [80] | |
| Alveolar cell carcinoma | A549 | Small EV | Chang et al., 2021 [80] | |
| Melanoma | Melanoma | UACC-62 | EV | Hurwitz et al., 2016 [78] |
| Melanoma | YUSAC 2 | Small EV | Aspe, 2014 [85] | |
| Ovary | High-grade ovarian serous adenocarcinoma | OVCAR-5 | EV | Hurwitz et al., 2016 [78] |
| High-grade ovarian serous adenocarcinoma | OVCAR-8 | EV | Hurwitz et al., 2016 [78] | |
| High-grade ovarian serous adenocarcinoma | NCI-ADR-RES | EV | Hurwitz et al., 2016 [78] | |
| Cervix | Cervical adenocarcinoma | HeLa | Small EV | Khan et al., 2011 [22]; Gonda et al., 2018 [23] |
| Prostate | Prostate carcinoma | DU145 | EV | Hurwitz et al., 2016 [78] |
| Prostate carcinoma | PC3 | Small EV | Khan et al., 2011 [22] | |
| Pancreas | Epithelioid adenocarcinoma | PANC-1 | Small EV | Khan et al., 2011 [22]; Chang et al., 2021 [80] |
| Pancreatic adenocarcinoma | MIA PaCa-2 | Small EV | Aspe, 2014 [85]; Chang et al., 2021 [80] |
EV, extracellular vesicle.
Survivin expression was shown to be specific for exosomes as MVs isolated from the same cells lacked detectable levels of this protein [83]. Although survivin is present at low levels in exosomes from dimethylsulfoxide (DMSO)-treated control cells, its expression increased sharply in exosomes from MDA-MB-231 cells treated with paclitaxel. However, the presence of survivin in the largest extracellular vesicle, apoptotic bodies, has not been shown. The location of survivin was also demonstrated with immunoelectron microscopy to be on the surface of exosomes, being associated with Hsp70, in the conditioned medium taken from survivin-releasing HeLaS/POZnSurvivin cervical cancer cell line engineered to overexpress a FLAG/hemagglutinin (HA)-tagged survivin [22]. Moreover, immuno-magnetic extraction of HeLa cell-secreted exosomes coupled to human MHC Class II (HLA-DR) or CD9 aldehyde-sulfate latex beads, exhibited a phenotype with high expression of LAMP1 and Hsp70, as well as expression of CD54, CD9 and survivin. In addition, inhibitors of apoptotic proteins (IAPs), such as survivin, cIAP1, cIAP2 and XIAP, have been identified to be differently expressed in a panel of tumour cell lines: DLCL2, HeLa, MCF-7, Panc-1, and PC3 [86]. Therefore, it could be suggested that survivin is localised in or on the surface of exosomes in cancer cells.
EVs containing survivin can be taken up by surrounding cells and induce a pro-survival field effect that promotes proliferation and survival in recipient cells [87]. This re-entry process begins when recipient cells take up EVs via endocytosis. Exosomes can fuse with the endosomal membranes of recipient cells, releasing their cargo, including survivin, directly into the cytoplasm [88]. Survivin-containing exosomes released by breast cancer cells were shown to be internalised by fibroblasts. Survivin subsequently up-regulates superoxide dismutase 1 (SOD1) expression in fibroblasts and converts them into myofibroblasts. In turn, myofibroblasts promote proliferation, epithelial-to-mesenchymal transition (EMT), and the stemness of breast cancer cells [79].
Cancer cells’ release of EVs containing survivin and other anti-apoptotic proteins might be a last attempt to protect themselves from stresses within the TME [24]. The amounts and contents of EVs released by cancer cells vary depending on their cell of origin, stage of development, and response to therapy [83, 89, 90, 91, 92, 93]. The presence of survivin inside EVs will influence the approaches towards utilising it as a diagnostic marker or therapeutic target and have implications in the strategies of targeting the TME for the treatment of solid tumours.
EVs, loaded with proteins, genetic materials, and lipids, are present in human serum and represent their cell of origin. Serum/plasma survivin has been clinically detected using commercially available enzyme-linked immunosorbent assay (ELISA) in patients with various malignancies. Higher survivin levels have been correlated with unfavourable clinicopathological features, including lower response to therapy and metastasis [94, 95, 96, 97, 98, 99, 100, 101]. Survivin has also been shown to be a potential biomarker for early diagnosis [102, 103]. However, the presence of survivin inside EVs implies that measurement of free serum survivin alone does not represent the true amount of survivin in patients’ sera and might not be sufficient. EV survivin measurements may provide a more accurate picture of survivin amounts in cancer patients’ sera.
Recent evidence, summarised in Table 2 (Ref. [24, 25, 26, 80, 104, 105]), has
demonstrated the usefulness of EV survivin for early detection, diagnosis,
prognosis, and monitoring of cancer progression [24, 25, 26, 106]. In the studies
reviewed and described here, exosomes might more accurately be understood as
small EVs (EVs
| Studies | Cancer Type | No. of patients | Samples | Methods of exosome isolation | Methods of exosomal survivin measurement | Findings |
| Khan et al., 2012 [24] | Prostate cancer (PCa) | 39 | Western blot with proportion analysis of survivin density to LAMP1 density | |||
| Khan et al., 2014 [25] | Breast cancer | 40 | ||||
| Khan et al., 2017 [104] | Prostate cancer | 41 African-American (AA) men and 31 European-American (EA) men | Plasma and serum | ExoQuick (SBI, USA) | Western blot with densitometric analysis of survivin to LAMP1 | |
| Chang et al., 2021 [80] | Pancreatic ductal adenocarcinoma (PDAC) | 13 | Serum | Differential centrifugations, ultracentrifugation (120,000 g, 4 h) | Western blot with densitometric analysis of survivin to Flotillin-2 | Survivin expression was increased in the exosomes from 8 out of 13 PDAC patients and was significantly higher compared to non-PDAC patients |
| Yildirim et al., 2022 [26] | Invasive ductal breast cancer | 55 | Serum | ExoQuick (SBI, USA) | ||
| Galbo et al., 2017 [105] | Recurrent high-grade glioblastoma (GBM) (WHO grade III and IV), following survivin vaccination (SurVaxM) | 8 | Serum | Differential centrifugations and ultracentrifugation (100,000 g, 80 min, 4 °C) | Imaging flow cytometry as a percentage of all CD9+ events | |
ELISA, enzyme-linked immunosorbent assay; GBM, glioblastoma; HER2, human epidermal growth factor receptor 2; PCa, prostate cancer; PFS, progression-free survival; EV, extracellular vesicle.
Exosomal survivin and its alternative splice variants, i.e.,
survivin-
The role of exosomal survivin in breast cancer diagnosis was also examined by
disrupting the membrane integrity of serum-derived exosomes and measuring
survivin concentration using an ELISA [26]. Survivin concentration in lysed sera,
presumably containing free and exosomal survivin, was significantly higher among
invasive ductal breast cancer patients (2.48
Exosomal survivin has also been detected in the sera of patients with pancreatic
ductal adenocarcinoma (PDAC). Chang et al. (2021) [80] isolated exosomes
from the serum samples of 13 PDAC patients and 5 non-PDAC patients and using a
western blot compared the amounts of exosomal survivin followed by densitometric
analysis of survivin relative to Flotillin-2 band density. A parallel experiment
using pancreatic cancer cells that express oncogenic Kirsten rat sarcoma (KRAS) mutants confirms the
KRAS-dependent mechanisms of exosomal survivin production. Survivin expression
increased in the exosomes from 8 out of 13 patients and was significantly higher
than controls (p
In terms of disease monitoring, survivin-positive exosomes have demonstrated potential use among glioblastoma patients receiving the immunotherapeutic survivin peptide vaccine (SurVaxM) [105]. Samples were derived from a phase I clinical trial involving survivin-positive malignant glioma patients whose tumours had recurred or progressed following standard therapy [118]. In this study, patients with malignant gliomas exhibit survivin-positive (CD9+/GFAP+/SVN+ and CD9+/SVN+) exosomes being released into the circulation. Early depletion of survivin-positive exosome levels was associated with longer progression-free survival among those patients [105]. Recently, the United States Food and Drug Administration (FDA) granted the fast-track designation (FTD) to an immunotherapeutic survivin peptide vaccine (SurVaxM) for the treatment of patients with newly diagnosed glioblastoma [119]. As survivin is present in a high proportion of cancers, this vaccine could potentially be used in other cancers. There is a high likelihood that other survivin-based vaccines might soon appear and advance through clinical studies. Considering this, exosomal survivin might be beneficial for monitoring therapy response among patients receiving these survivin-based vaccines.
Survivin has recently become an attractive target in the treatment of cancer for
the following reasons: (1) higher expression in tumour cells relative to normal
cells, (2) significant role for cancer cells survival, and (3) universal
expression in
Since survivin is associated with resistance to therapy, later stages of cancer, and higher grades of tumour, its release in EVs might be one of the last resort mechanisms employed by cancer cells to evade the immune system. As survivin is hidden inside EVs, it is not protected from the cellular and humoral immune responses induced by survivin-based vaccines. Moreover, exosomes can also promote cancer cells’ immune escape by modulating the activity of immune cells, resulting in an environment supportive of tumour development [137]. Hence, targeting free survivin and survivin-containing exosomes could be a crucial approach in anti-survivin cancer therapeutics. One option would be to combine anti-survivin therapy and exosome-targeting agents, i.e., exosome inhibitors. Exosomes are targeted through inhibition of their uptake by cancer cells or inhibition of their biogenesis and release, with the latter being more promising. Numerous compounds, drugs, and antibodies that inhibit exosomes have been described [138, 139].
Recently, the potential efficacy of adding exosome inhibitors to existing therapeutic regimens has been reported. Numerous studies revealed the potential of exosomal PD-L1 inhibition in increasing responsiveness to immune checkpoint inhibitors (ICI). Genetic inhibition of genes involved in exosome biogenesis and release, including Rab27a [74], nSMase [140], endothelin A receptor antagonists (ETA) [141], and lysine-specific demethylase 1 (LSD1) [142], increased antitumor immunity. Treatment with an exosome secretion inhibitor (nSMase inhibitor), GW4869, demonstrated a synergistic effect when combined with anti-PD-L1 therapy [140]. Endothelin A receptor (ETA) antagonists, sulfisoxazole (SFX) and macitentan (MAC) were revealed to improve the efficacy of anti-PD-L1 therapy in breast, lung, and colon cancer models [141, 143]. These findings indicate the potential of combining cancer therapeutics with exosome inhibitors.
Cancer cell release of survivin as a free protein and packaged within EVs (especially small EVs or exosomes), will impact the approaches required for its use as a biomarker or therapeutic target. Several recent studies have demonstrated that EV survivin levels are significantly higher among cancer patients and its expression is associated with unfavourable clinicopathologic parameters. Therefore, EV survivin may be useful as a diagnostic, prognostic, and monitoring marker for various types of cancer, including breast cancer, prostate cancer, pancreatic cancer, and glioblastoma. One plausible method to target EV survivin is the combination of anti-survivin therapy, including survivin-inhibiting molecules and survivin-based immunotherapy, with exosome-targeting therapy. However, this concept may take many years before its realisation as exosome-targeting therapeutics are still in early pre-clinical phases.
Future studies should attempt to further explore the role of EV survivin as a diagnostic, prognostic, and monitoring marker in other cancer types. The levels of EV survivin should also be compared with those of free extracellular serum survivin to provide a detailed picture of how EV survivin can enhance the diagnostic utility of existing plasma/serum survivin assays. Furthermore, in the future, survivin-based therapies should also be tested in combination with exosome inhibitors to impede their roles in the tumour microenvironment completely.
WW: Conceptualization, Data curation, Writing – original draft, Writing – review & editing; SMP: Conceptualization, Resources, Supervision, Writing – review & editing; SJ: Conceptualization, Resources, Supervision, Writing – review & editing. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
The authors express gratitude to the Jardine Foundation for its support by providing scholarship for the first author.
This research received no external funding.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.