



 , Longyuan Li 2,†, Huan Yang 1, Chenxi Shi 1, Zhe Lei 1,*
, Longyuan Li 2,†, Huan Yang 1, Chenxi Shi 1, Zhe Lei 1,* , Lingchuan Guo 1,*, Yuhong Wang 1,*
, Lingchuan Guo 1,*, Yuhong Wang 1,*1 Department of Pathology, The First Affiliated Hospital of Soochow University, Soochow University, 215006 Suzhou, Jiangsu, China
2 Department of Neurosurgery & Brain and Nerve Research Laboratory, The First Affiliated Hospital of Soochow University, Soochow University, 215006 Suzhou, Jiangsu, China
†These authors contributed equally.
Abstract
The p53, a pivotal tumor suppressor, regulates various cellular responses, including DNA repair and apoptosis. Normally, p53 levels are low due to murine double minute clone 2 (MDM2) mediated polyubiquitination. However, stress signals disrupt p53-MDM2 interaction, stabilizing p53 and activating target genes. Dysfunctional p53 is common in cancers, especially colorectal cancer (CRC), with TP53 mutations in 43% of tumors. These mutations impair wild-type p53 function or confer novel activities, promoting cancer progression. Despite drugs targeting p53 entering trials, understanding wild-type and mutant p53 functions is crucial for novel CRC therapies. P53 mutations not only impact DNA repair and apoptosis but also play a crucial role in tumor immunotherapy. While rendering tumors resistant to chemotherapy, p53 mutations provide opportunities for immunotherapy due to neoantigen-rich tumors. Additionally, p53 mutations influence tumor microenvironment cells, such as fibroblasts and immunosuppressive cells, through p53-mediated signaling pathways. Investigating p53 mutations in tumor therapy is vital for personalized medicine and immunotherapy. In cancer treatment research, scientists explore drugs and strategies to restore or enhance p53 function. Targeting wild-type p53 aims to restore DNA repair and cell cycle control, while targeting mutant p53 seeks new drugs to inhibit its detrimental effects, advancing tumor treatment. Understanding p53 drugs and strategies is crucial for cancer therapy progress.
Keywords
- TP53
- p53
- mutation
- colorectal cancer
Colorectal cancer (CRC) is one of the common malignant tumors globally. According to statistics from the World Health Organization (WHO), it ranks third globally in cancer incidence, following only lung cancer and breast cancer [1], accounting for approximately 10% of all cancer cases [2]. Especially in developing countries, particularly in Asia, with the improvement of living standards and the adoption of Westernized diets, the incidence of CRC is gradually increasing [3]. Despite the improvement in living standards and advancements in medical technology, early screening and prevention awareness for CRC have been increasing. However, its incidence still shows an upward trend, posing challenges to public health and clinical medicine [4].
Various mutation patterns can be found in colorectal cancer (CRC), impacting the advancement of the illness and overall survival. Frequently, mutations occur in DNA mismatch repair system alongside alterations in oncogenes and/or tumor suppressor genes like NRAS, KRAS, APC, PIK3CA, and TP53 [5, 6]. Among these, TP53 plays a central role. The TP53 gene is crucial in human cells, encoding the protein p53, which is a highly unstable transcription factor [7]. Its primary function is to monitor DNA damage in cells and promote either repair or guide severely damaged cells into apoptosis (programmed cell death), thus preventing cancer development [8]. Many endogenous and exogenous stressors can activate p53, and activated p53 binds to p53 response elements located in its promoter region to activate downstream genes [9], triggering a series of cellular responses necessary for maintaining internal balance. The activation of p53 in various stress responses is crucial for the survival of normal cells and protecting them from tumorigenesis [10].
Under normal physiological conditions, the level of p53 is strictly regulated by MDM2 [11]. Murine double minute clone 2 (MDM2) is an E3 ubiquitin ligase, and its C-terminal triggers the degradation of p53 through the ubiquitin-proteasome-dependent pathway, maintaining low cellular p53 levels [11] (Fig. 1). Additionally, MDM2 itself is a target of the transcription factor p53, forming a negative feedback regulatory pathway [12]. Additionally, the transcription factor activity of p53 is hindered by the MDM4 protein (also referred to as MDMX), serving as an extra natural inhibitor of p53’s functioning [13]. As a key transcription factor, p53 is activated under various stimuli and serves as a major regulatory factor in various cellular activities, including DNA damage repair, proliferation, senescence, differentiation, and apoptosis [14].
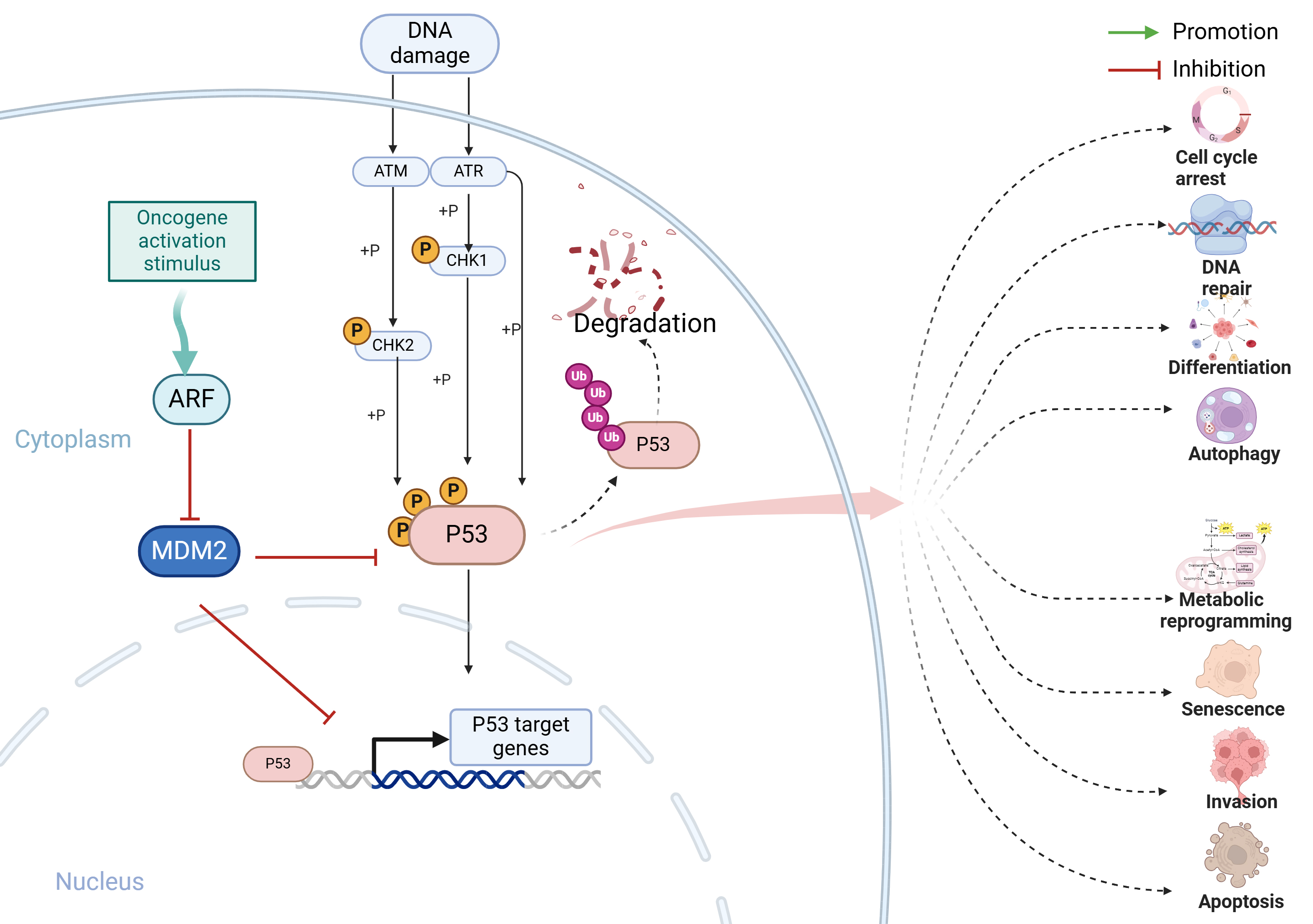 Fig. 1.
Fig. 1.TP53 signaling pathway. p53, an essential factor for suppressing tumors, is tightly controlled by MDM2 in normal physiological situations. MDM2 serves as an E3 ligase that degrades p53 through the ubiquitin-proteasome pathway. Furthermore, p53 has the ability to activate MDM2 expression by interacting with its promoter, creating a feedback mechanism that leads to p53 degradation and keeps cellular levels of p53 low. Under cellular stress, DNA damage activates DNA damage response kinases ATM and ATR, along with their substrates CHK2 and CHK1. These kinases phosphorylate p53, stabilizing it by antagonizing the p53-MDM2 interaction. The primary function of activated p53 is to regulate various cellular responses by activating transcription of target genes. These responses typically include halting the cell cycle, inducing senescence, repairing DNA, and triggering apoptosis. Additionally, p53 can influence other processes such as promoting autophagy, facilitating cell differentiation, and promoting ferroptosis. It also inhibits invasion, metastasis, and metabolic reprogramming. Together, these p53-mediated responses play a crucial role in suppressing the growth of tumors. MDM2, Murine Double Minute Clone 2; ATM, Ataxia Telangiectasia Mutated; ATR, Ataxia Telangiectasia and Rad3-Related Protein; ARF, Alternative Reading Frame; CHK1, Checkpoint Kinase 1; CHK2, Checkpoint Kinase 2. Created with BioRender.com.
However, in tumors, TP53 commonly undergoes mutations in the majority of cases, losing its role in suppressing tumors and even conferring functions required for tumor growth [15]. The loss of the p53 tumor suppressor factor is one of the key processes in the transition from benign adenomas to invasive carcinomas, driving tumors towards a more aggressive direction [16]. In addition to the loss of p53’s tumor-suppressive role, it has been demonstrated that some missense mutations in the DNA-binding domain of the TP53 gene induce gain-of-function (GOF) effects [17]. In such cases, TP53 mutations may confer resistance to systemic therapy, profoundly impacting patient treatment response and outcomes [15]. In this review, we summarized the research progress of mutated TP53 in CRC, elucidating the role of different types of TP53 mutations in CRC, and helping researchers and clinical practitioners systematically understand the role of p53 in CRC.
TP53 is the gene that undergoes the highest frequency of mutations in
human cancers [18, 19]. Emerging evidence suggests that TP53 mutations
can acquire oncogenic properties through a mechanism known as ‘gain of function’,
thereby contributing to carcinogenesis [8, 20]. Those missense mutated proteins
generally exhibit diminished or absent DNA binding ability. Studies using mouse
models containing such mutated TP53 and cell experiments conducted
in vitro have demonstrated that this incomplete p53 may facilitate the
transformation of premalignant cells and promote tumor growth [21]. In non-highly
mutated human colon cancers, it is worth mentioning that around 80% of tumors
with missense mutations showed loss of heterozygosity (LOH), leading to the
elimination of the TP53 gene’s wild-type form [22]. A recent study has
revealed that the combined impact of Trp53R270H gene mutation and loss
of heterozygosity (LOH) can enhance activin A-induced partial epithelial
mesenchymal transition (EMT), leading to the development of multiple projections
and an increased occurrence of metastasis [23]. The result suggests that
TP53 GOF/LOH represents a crucial genetic state in triggering partial
EMT and malignant progression induced by the TGF-
Furthermore, numerous studies have demonstrated a strong correlation between inactivated TP53 mutations and the aggressive nature as well as the responsiveness to treatment of cancer [24]. The latest research suggests that p53 inactivation is a primary contributor to genomic instability, with TP53 mutations typically occurring prior to other genomic reorganization events. Researchers have discovered that the absence of p53 in cells triggers a predictable, organized, and deterministic evolution of the cancer genome while simultaneously inducing genetic instability. The development of cancer does not occur solely due to the absence of p53; rather, it is the combination of p53 deficiency and a gradual accumulation of genetic changes in a methodical fashion that leads to uncontrollable genomic instability, ultimately giving rise to cancer [25]. Interestingly, in another recent study, researchers discovered that p53 mutants exhibit the anticipated carcinogenic effect in the distal gut; however, they exert a significant tumor suppressor effect in the proximal gut and tumor organoids. Nevertheless, this tumor suppressor effect is completely abrogated by the presence of the gut microbiome [26].
In various cellular activities within cancer, p53 has been reported to play a crucial role. p53 can induce autophagy through multiple mechanisms, directly enhancing autophagy by binding to various autophagy-related factors (such as Atg2, Atg4, Atg7, and Atg10) [27]. In addition to its direct effects, p53 can also indirectly regulate autophagy through the AMPK/mTOR pathway or the BNIP3 pathway, thereby inhibiting cancer cell proliferation [28, 29]. Moreover, p53 also plays a significant role in maintaining metabolic homeostasis. Tumor cells typically require a large amount of energy to meet their growth demands, and most tumor cells rely on aerobic glycolysis for energy supply, a phenomenon known as the Warburg effect [30]. p53 can transcribe genes necessary for oxidative phosphorylation, such as Synthesis Of Cytochrome C Oxidase 2 (SCO2), to maintain metabolic stability, and it inhibits glycolysis by interacting with the key rate-limiting enzyme glucose-6-phosphate dehydrogenase (G6PDH) in the pentose phosphate pathway, thereby shifting tumor cell metabolism towards oxidative phosphorylation to suppress tumor growth [31, 32]. Conversely, mutant p53 can bind to key factors in other metabolic pathways, leading to metabolic plasticity in tumor cells, enhancing mitochondrial function, and promoting tumor development and metastasis [33]. Additionally, TP53 can regulate ferroptosis through direct or indirect pathways. For instance, p53 can inhibit the transcription of Solute Carrier Family 7 Member 11 (SLC7A11), leading to elevated glutathione/glutathione disulfide (GSH/GSSG) levels, thereby promoting the accumulation of lipid peroxides to facilitate ferroptosis [34]. Mutant forms of p53, however, lose their ability to regulate SLC7A11 transcription, thereby promoting ferroptosis [35].
Colorectal cancer stands out as the tumor type with the highest incidence of TP53 mutations, with approximately 74% of tumor samples carrying TP53 mutations (Fig. 2, Ref. [36], cBioPortal, https://www.cbioportal.org/; CRC (MSK, JNCI 2021)). Most of these mutations occur in the central DNA-binding domain. Unlike other tumor suppressor genes, the predominant type of TP53 mutation in CRC is missense mutations rather than truncating mutations, indicating alterations in the amino acid sequence of the encoded p53 protein [37]. The mutation sites of TP53 are predominantly located in the central DNA-binding domain, with most identified recurrent hotspot amino acids also located in this region, especially R175, G245, R248, R273, and R282. Truncating mutations occurring at the R196 and R213 sites are also relatively common (R196, 32 of 1516 samples; R213, 50 of 1516 samples) [38]. The high frequency of TP53 mutations in CRC is attributed to various complex factors, with several environmental mutagens identified to induce mutations in specific DNA sequences within the TP53 gene [37]. For instance, aflatoxin acting on the R249S codon (a region rich in G-C) has been established as a definitive carcinogen in liver cancer [39], while mutations in variants R248W and R282W are associated with solar ultraviolet induction in non-melanoma skin cancer [40]. However, in CRC, no definitive tissue-specific environmental mutagens contributing to TP53 mutations have been identified.
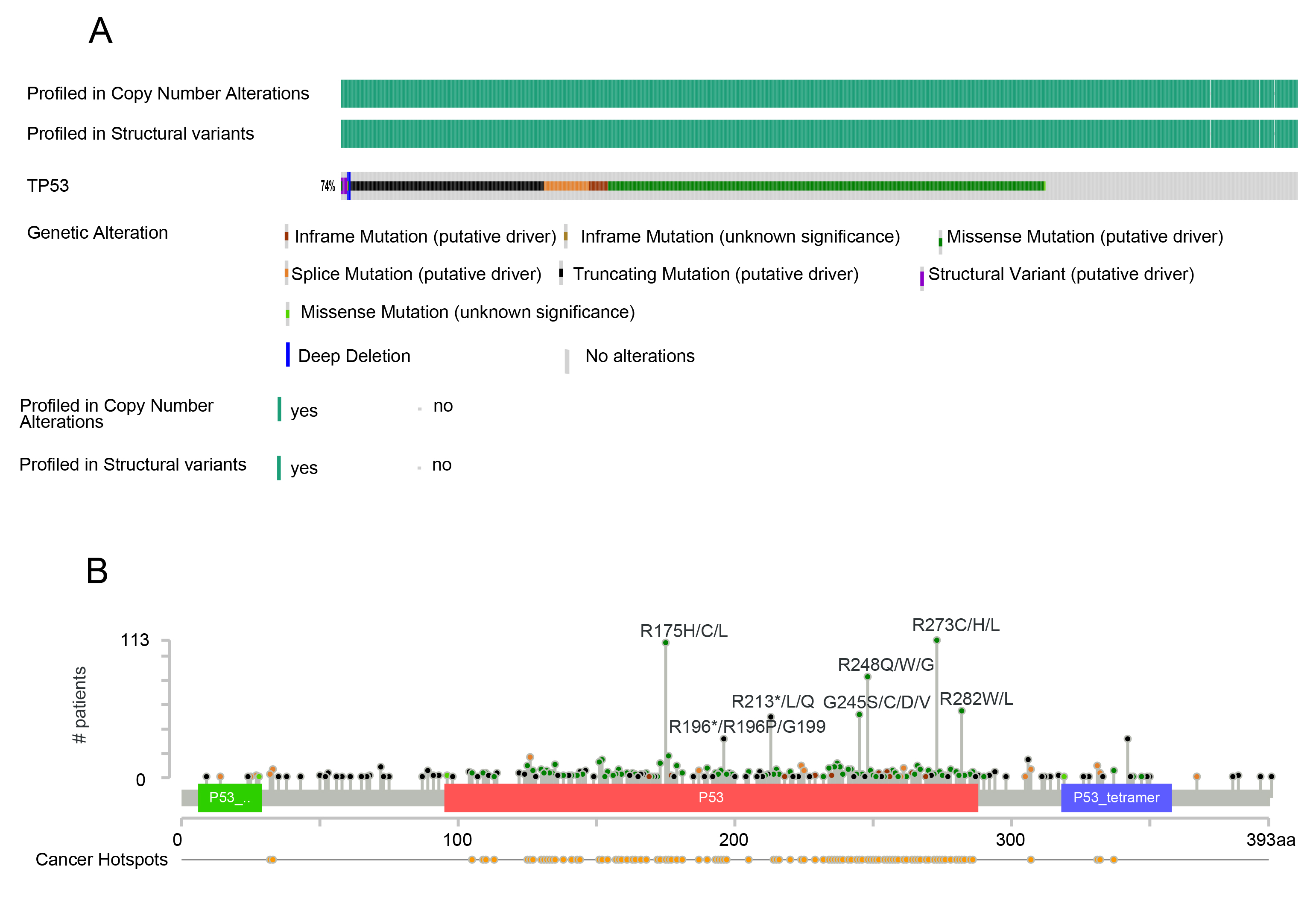 Fig. 2.
Fig. 2.Mutational status of TP53 in colorectal cancer. In cBioPortal (https://www.cbioportal.org/), using colorectal cancer sample data (MSK, JNCI 2021) [36], there are a total of 1516 samples, with the majority of mutations occurring in the central DNA binding region. (A) Overall mutation status of TP53 in 1516 colorectal cancer samples. (B) Sites of mutations in TP53 in the 1516 colorectal cancer samples.
It’s worth noting that there are abundant genes exhibiting both high and low methylation levels in CRC, with these epigenetic changes occurring even more frequently than genetic mutations [41]. Almost all hotspot codons in the TP53 gene contain Unmethylated cytosine-phosphate-guanine (CpG) dinucleotides, suggesting that the presence of these methylated sites may be one of the reasons for TP53 mutations in CRC [42].
Different types of TP53 mutations in CRC exhibit distinct characteristics. CRC with mutations at the R273 site is more prone to developing metastatic cancer, with significantly reduced survival rates [43]. Knock-in mice carrying the p53 R273H mutation have a higher incidence of cancer and lymphoma [44]. Similarly, the R248Q and R248 hotspot mutation variants also exhibit similar promotion of colon cancer cell dissemination, invasion, and metastasis [43].
Due to the very high rate of TP53 mutations in CRC, numerous studies have attempted to tackle the challenges of CRC treatment from the perspective of p53 mutants. The variability in the forms of mutations of the tumor suppressor p53 not only leads to the loss of tumor-suppressive properties but also often confers characteristics that promote tumor development. Moreover, the structural differences in mutant p53 proteins resulting from mutations make them difficult to target. Currently, major research directions targeting p53 can be broadly categorized into two main classes: restoring wild-type p53 function and eliminating the effects caused by mutant p53.
Under the pressure of cancer, activated p53 can regulate various cellular processes such as DNA repair, cell cycle arrest, aging, apoptosis, etc., through multiple pathways such as phosphorylation, acetylation, ubiquitination, and methylation [45, 46, 47, 48]. In cases of cancer occurrence, intact TP53 is often present in the genome [49]. However, missense mutations in p53 may lead to loss of function (LOFs) required for tumor suppression mediated by wild-type p53, and through the formation of tetramers, activate MDM2, thus inhibiting the function of wild-type p53, demonstrating dominant negative effects (DNE) [50]. Earlier studies indicated that amplification of MDM2 seemed to lead to similar clinical outcomes in tumor patients with mutant p53 [51]. Consequently, restoring the function of wild-type p53 has become a focus of research for translating the p53 signaling pathway into clinical applications. Several clinical studies have demonstrated the significant potential of such drugs in cancer treatment, and perhaps in the future, they may be used for treating CRC (Table 1).
| Drug name | NCT | Inclusion of population | Mechanism of action | Study start | Study completion | Study status |
| ALRN-6924 | NCT03654716 | Pediatric Cancer | Stapled peptide dual MDM2–MDMX inhibitor | 2018/11/1 | 2023/7/17 | Completed but no publications available |
| DS-3032b | NCT02579824 | Relapsed and/or Refractory Multiple Myeloma | MDM2 inhibitor | 2016/8/30 | 2019/11/7 | Termination but no publications available |
| APG-115 | NCT03781986 | Salivary Gland Cancer | MDM2/P53 inhibitor | 2019/10/28 | 2025/1 | Suspend |
| RO6839921 | NCT02098967 | Acute Myeloid Leukemia | MDM2 inhibitor | 2014/4/21 | 2018/5/7 | Completed |
| RO5045337 | NCT01677780 | Previous Roche-sponsored Cancer | MDM2/P53 inhibitor | 2012/11/28 | 2017/6/26 | Completed but no publications available |
| Milademetan | NCT03671564 | Relapsed or Refractory Acute Myeloid Leukemia | MDM2 inhibitor | 2018/8/23 | 2019/9/11 | Completed with results |
| RO5045337 | NCT01605526 | Soft Tissue Sarcoma | MDM2/P53 inhibitor | 2012/3 | 2013/6 | Completed but no publications available |
| Milademetan Tosylate | NCT03634228 | Recurrent or Refractory Acute Myeloid Leukemia | MDM2 inhibitor | 2018/12/17 | 2022/4/3 | Completed |
| Idasanutlin | NCT03566485 | Stage IV or Unresectable Recurrent Estrogen Receptor Positive Breast Cancer | P53–MDM2 Interaction inhibitor | 2018/7/10 | 2020/12/10 | Termination with results |
| Idasanutlin | NCT02545283 | Acute Myeloid Leukemia | P53–MDM2 Interaction inhibitor | 2015/12/30 | 2020/4/24 | Termination with results |
| RO5503781 | NCT01462175 | Advanced Malignancies Except Leukemia | MDM2 antagonist | 2011/11 | 2014/7 | Completed but no publications available |
| APG-115 | NCT03611868 | Metastatic Melanomas or Advanced Solid Tumors | MDM2/P53 inhibitor | 2018/8/29 | 2025/3/30 | Recruiting |
| ALRN-6924 | NCT05622058 | TP53-Mutant Breast Cancer | P53–MDM2 Interaction inhibitor | 2023/1/9 | 2023/2/22 | Termination but no publications available |
| Brigimadlin (BI 907828) | NCT05512377 | People with Cancer in the Biliary Tract, Pancreas, Lung or Bladder | MDM2/P53 antagonist | 2022/11/25 | 2027/3/25 | Recruiting |
| Milademetan | NCT02319369 | Acute Myelogenous Leukemia (AML) or High-Risk Myelodysplastic Syndrome (MDS) | MDM2 inhibitor | 2014/11/25 | 2020/8/21 | Termination with results |
| Lamivudine | NCT03144804 | Colorectal Cancer | transcriptase inhibitors | 2017/10/31 | 2022/9/27 | Completed |
| Ad-P53 | NCT02842125 | Colorectal Cancer hepatocellular carcinoma | P53 inhibitor | 2018/5/1 | 2020/4/1 | Termination with results |
| mRNA vaccine | NCT02316457 | Triple-negative breast cancer | mRNA target to 20 antigens including P53 | 2016/10 | 2023/5/17 | Completed |
| Erlotinib | NCT00642746 | Colorectal Cancer | P53 inhibitor | 2008/5 | 2011/12 | Termination with results |
| Atorvastatin Calcium | NCT04767984 | Colorectal Carcinoma | P53 inhibitor | 2021/9/24 | 2024/12/11 | Recruiting |
| Ulcerative Colitis | ||||||
| ALT-801 | NCT00496860 | Progressive Metastatic Malignancies | P53 inhibitor | 2007/5 | 2009/10 | Completed |
| Idasanutlin + ICI atezolizumab | NCT03555149 | Metastatic Colorectal Cancer | P53–MDM2 Interaction inhibitor | 2018/9/27 | 2022/9/26 | Terminated |
| KT-253 | NCT0577540 | Advanced solid cancers | MDM2 Degrader | 2023/5/15 | 2024/11 | Recruiting |
| Idasanutlin | NCT03362723 | Solid Tumors | P53–MDM2 Interaction inhibitor | 2017/11/27 | 2019/6/11 | Completed but no publications available |
| KRT-232 | NCT04640532 | JAK Inhibitor Intolerant Myelofibrosis. | MDM2 inhibitor | 2020/11/17 | 2025/7/24 | Recruiting |
| Siremadlin | NCT05447663 | Acute Myeloid Leukemia | MDM2 inhibitor | 2023/2/23 | 2023/10/26 | Termination but no publications available |
| Siremadlin | NCT05155709 | Acute Myeloid Leukemia | MDM2 inhibitor | 2022/5/17 | 2024/5/28 | Active, not recruiting |
RG7112, the initial MDM2 blocker tested in medical experiments, belonged to the nutlin family and showed enhanced strength and pharmaceutical qualities. In its interaction with p53, RG7112 tightly attaches to MDM2. By turning on the p53 pathway, RG7112 induces cell cycle halt and cancer cell death in cases where p53 is expressed as wild-type [52] (Fig. 3). APG-115 is the first MDM2 inhibitor to enter clinical trials in China. It can block the p53-MDM2 interaction, increasing the abundance of MDM2 in T cells. Concurrently, it plays a significant biological role in maintaining T cell stability, survival, and anti-tumor immunity [53]. APG-115 stands as the pioneering MDM2 inhibitor to undergo clinical trials in China. It effectively disrupts the p53-MDM2 interaction, leading to an increase in MDM2 levels within T cells. This mechanism plays a pivotal biological role in sustaining T cell stability, enhancing survival rates, and bolstering anti-tumor immune responses [54]. On the other hand, NVP-CGM097 represents a novel dihydroisoquinolinone MDM2 inhibitor meticulously crafted through screening and refinement processes. Apart from mimicking the three crucial amino acids responsible for mediating the p53-MDM2 interaction, its molecular structure also forms hydrogen bonds with Tyr100, Gln24, and Phe55. Consequently, NVP-CGM097 induces a conformational change in Phe55, facilitating its interaction with the dihydroisoquinolinone core. Notably, this drug exhibits exceptional selectivity towards wild-type p53 and exerts potent anti-proliferative effects against colorectal and osteosarcoma cells carrying the wild-type p53 mutation [55].
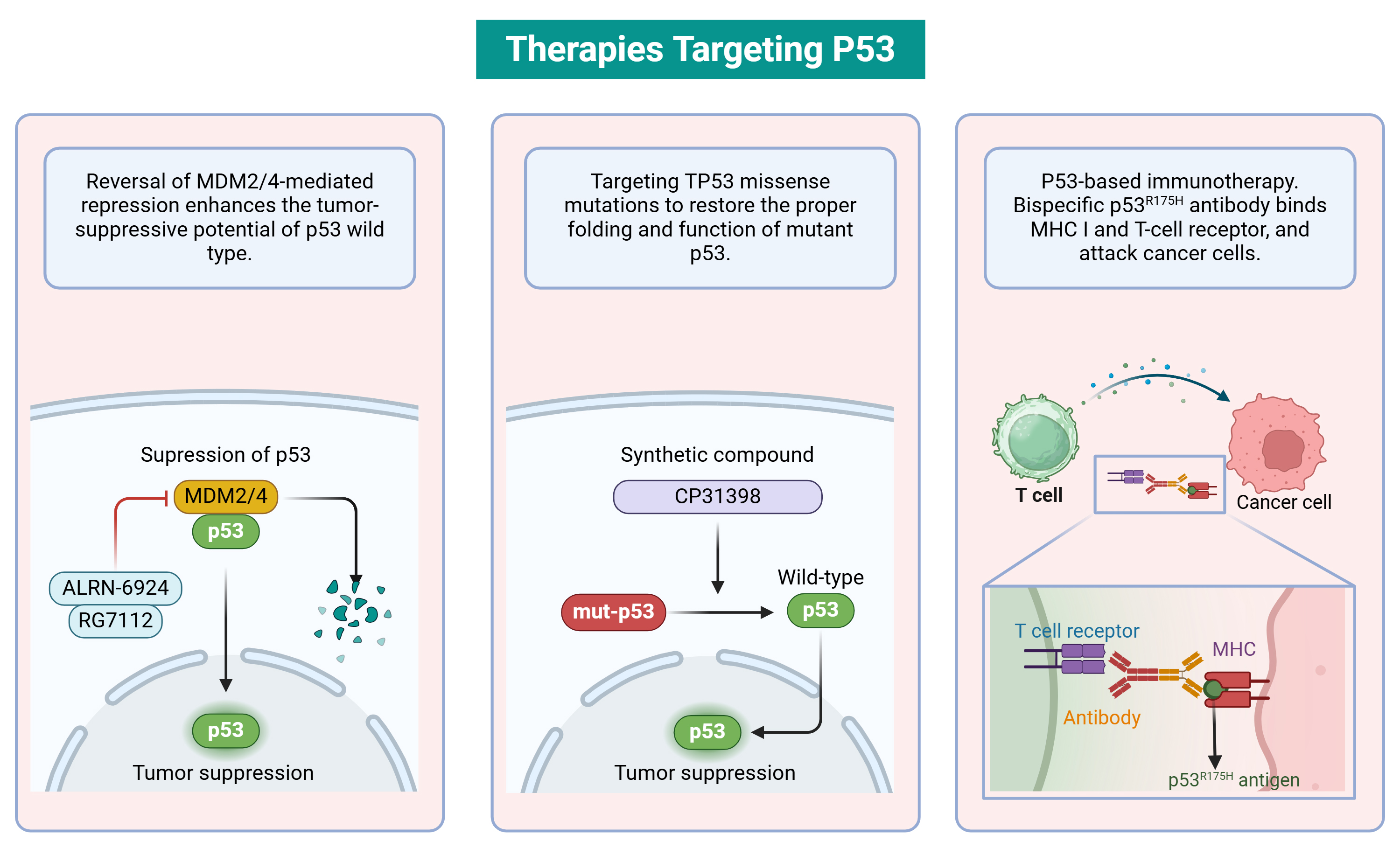 Fig. 3.
Fig. 3.Therapies targeting p53. The E3 ubiquitin ligase MDM2 mediates the ubiquitination of p53, leading to its degradation via the proteasome pathway. Ubiquitination by MDM2 depends on its direct interaction with p53. Small molecule compounds that inhibit the MDM2-p53 interaction can stabilize p53 and restore its function, serving as potential drugs to enhance p53 activity. Structural or conformational variants resulting from missense mutations in TP53 are considered potential targets for small molecules, aiming to restore proper folding and function of mutant p53. For instance, CP31398 stabilizes the wild-type conformation of p53 and prevents its degradation by inhibiting its ubiquitination. Bispecific antibodies exhibit high affinity binding to both p53 (R175H) peptide-HLA complexes on cancer cells and TCR-CD3 complexes on T cells. These bispecific antibodies can overcome the lack of neoantigen presentation and selectively redirect T cells to recognize cancer cells presenting mutated peptides. CP31398, a styrylquinazoline compound; HLA, Human Leukocyte Antigen; TCR-CD3, T cell receptor-CD3 complex; MHC, Major Histocompatibility Complex. Created with BioRender.com.
However, another perspective raises concerns about the application of MDM2 inhibitors in tumors. In patients with p53 mutations, inhibiting the degradation of p53 could potentially increase the levels of mutant p53 protein, exacerbating its gain-of-function (GOF) effects, thus promoting the development of tumors towards worse characteristics [42]. Research in myeloid malignancies has shown that in mice with missense mutation TP53 generated through CRISPR-Cas9, the dominant negative effects (DNE) of mutant p53 confer selective advantages on hematopoietic cells’ DNA damage [50], providing robust theoretical evidence for MDM2 inhibitors and similar drugs.
PRIMA-1 (a compound Mutant acts as p53 reactivator) was initially discovered through screening a library of low molecular weight compounds because they were able to restore the wild-type characteristics of mutant p53. Although there are many missense mutations in p53, research results show that PRIMA-1 can restore specific DNA binding functions in 13 different p53 mutant proteins. The only exception is the protein carrying the phe176 mutation. Therefore, regardless of the mutation location or whether it is a structural or contact mutation, PRIMA-1 seems to have the ability to restore some wild-type p53 characteristics [56, 57].
R175H is a common mutation in the p53 protein, which affects the Zn
The p53 Y220C mutation exhibits a certain level of specificity. Due to the significant spatial separation between Y220 and the DNA binding site of p53, substitution of tyrosine with cysteine in the S7/S8 region induces the creation of a hydrophobic cavity on the surface of p53 [62, 63]. Consequently, this resulting hydrophobic pocket presents an optimal target for drug intervention aimed at restoring proper folding while preserving its DNA binding capability [64].
C14586 is the first orally available selective reactivator of mutant p53 protein, selectively binding to the crack produced by the p53 Y220C mutation, thereby restoring the structure and anti-tumor function of wild-type p53 protein [19]. Currently, phase 1/2 clinical trials are underway to evaluate the safety, tolerability, and anti-tumor activity of C14586 in adult patients with advanced or metastatic solid tumors (including those with the Y220C mutation) [19, 65].
PK7088 interacts with the Y220C mutant protein, raising its melting temperature and restoring some of the wild-type transcriptional activity. When treating Y220C mutant cells, PK7088 induces an increase in the expression of p53 target genes p21 and NOXA, leading to cell cycle arrest and apoptosis [66]. The potential of PK7088 to exhibit anti-cancer effects in animal models has not been investigated thus far.
Histone deacetylase (HDAC) and Heat Shock Protein 90 chaperone axis (HDAC/Hsp90) inhibitors: Mutant p53 aggregates not only induce aggregation of wild-type p53 but also co-aggregate with p63 and p73 [67, 68]. This p53 aggregation leads to upregulation of Hsp70 and Hsp90 expression [69]. By inhibiting the ubiquitination degradation mediated by MDM2, Hsp70 can temporarily expose the adhesion sequence of mutant p53, thereby promoting the formation of p53 aggregates [70]. In addition, the interplay between Hsp90 and p53 with genetic alterations can prevents its protein from being ubiquitinated and degraded [71]. Therefore, HDAC inhibitors or Hsp90 inhibitors disrupt the HDAC6/Hsp90 complex, inducing degradation of mutant p53.
HSP90 inhibitor 17AAG releases mutant p53 degradation by disrupting the complex between mutant p53 and HSP90, thereby reducing the levels of mutant p53 [72]. Histone deacetylase HDAC6 positively regulates HSP90 chaperone activity by modulating HSP90 deacetylation. Selective degradation of mutant p53 in human cancer cells is induced by SAHA, an inhibitor of histone deacetylase (HDAC), through inhibition of the chaperone axis HDAC6-HSP90 [73].
ADH-6: ADH-6 is a positively charged tripyridinamide obtained through screening of oligomeric pyridinamide libraries [74]. Through nuclear magnetic resonance spectroscopy analysis, ADH-6 not only binds to the susceptible aggregation sites of p53 but also interacts with multiple regions of p53-DBD. These regions include loops 1, 3, 4, 6, 7, and helix 2. Furthermore, subsequent investigations revealed that ADH-6 effectively disrupts aggregates formed by mutant p53 and selectively triggers apoptosis in various cancer cells prone to mutant p53 aggregation [74].
Due to the increased reliance of mutp53 tumors on intra-S and G2 arrest, researchers have identified certain molecules that regulate these checkpoints (such as ATR, CHK1, MK2 (Mitogen-Activated Protein Kinase-Activated Protein Kinase 2), and Wee1) [75, 76, 77, 78]. ATR functions as a sensor for specific sites of single-strand DNA damage by phosphorylating CHK1 to control cell cycle progression and response to DNA damage [79]. M6620 is an experimental inhibitor of ATR that has been tested in human trials. Recent phase 2 clinical studies involving M6620 have demonstrated improved efficacy when combined with gemcitabine compared to gemcitabine using gemcitabine alone, suggesting the potential of ATR inhibitors to augment the efficacy of existing chemotherapy interventions [79, 80].
CHK1 inhibitors (NCT01870596, NCT02797964, and NCT02797977) have completed three clinical trials of ATR inhibitors. This could be attributed to the fact that inhibiting ATR results in considerable impairments in the separation of chromosomes within healthy cells, while the substantial molecular weight of ATR hampers the feasibility of conducting compound screening [75, 81]. The activation of the p38MAPK/MK2 pathway, which controls the G2/M checkpoint, occurs in response to agents that cause DNA damage [82]. A study conducted recently showed that using a novel arabinose analogue F-Se-Ara-C induces synthetic lethality, targeting MK2 in prostate cancer with p53 mutations [78].
The kinase family known as WEE1 comprises three types of kinases, namely WEE1, WEE1B, and a kinase known as PKMYT1 that inhibits the activity of cdc2 by targeting tyrosine and serine residues on the cell membrane [82]. WEE1 and PKMYT1 play crucial roles in regulating the transition from G2 to M phase. The former hinders the initiation of mitosis by adding a phosphate group to CDK1 at Tyr15, whereas the latter facilitates the onset of mitosis through its ability to target both Thr14 and Tyr15 [83]. MK-1775 is a WEE1 inhibitor known for its potent radiosensitizing effects in human cancer cells, occurring only in tumors lacking p53 [84].
As the gene exhibiting the most pronounced mutation frequency in CRC, the significant contribution of TP53 in the development and advancement of cancer has gained extensive acknowledgement. However, despite the clear therapeutic potential demonstrated by targeting the p53 signaling pathway or addressing p53 mutations, there are currently no drugs that have been clinically validated for both safety and efficacy in CRC. Numerous clinical trials are underway, but in the future, there is a need for more evidence-based medicine to substantiate the specific therapeutic effects targeting p53 in CRC patients.
Conceptualized and designed the study: LG, ZL and YW; WL, LL and YW significantly contributed to the study’s design and conceptualization; sorted out the figures and tables: HY, CS; supervised the study: YW. All authors have participated sufficiently in the work to take public responsibility for appropriate portions of the content and agreed to be accountable for all aspects of the work in ensuring that questions related to its accuracy or integrity. All authors read and approved the final manuscript. All authors contributed to editorial changes in the manuscript.
Not applicable.
Not applicable.
This work was supported by Suzhou Basic Research Key Project (No. SKY2023009), Suzhou health youth backbone talent “national tutorial system” training project (Qngg2023005) and Beijing Xisike Clinical Oncology Research Foundation (Y-tongshu2021/qn-0366).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.