


1 Department of Medicine, Division of Hematology & Oncology, University of Pittsburgh Medical Center (UPMC), Pittsburgh, PA 15232, USA
2 Faculty of Medicine, Al-Azhar University, 11651 Cairo, Egypt
3 Department of Medicine, Ochsner Lafayette General Medical Center, Lafayette, LA 70503, USA
4 UPMC Hillman Cancer Center, Pittsburgh, PA 15232, USA
Abstract
Liver cancer, primarily hepatocellular carcinoma (HCC), is the second leading cause of cancer-related deaths globally. It is typically characterized by rapid progression, poor prognosis, and high mortality rates. Given these challenges, the search for molecular targets aiding early diagnosis and targeted therapy remains imperative. Glypican 3 (GPC3), a cell-surface glycoprotein, emerges as a promising candidate for addressing HCC Overexpressed in HCC tissues; GPC3 is a credible immunohistochemical marker for liver cancer diagnosis and a potential marker for liquid biopsy through soluble GPC3 in serum. Various immunotherapies targeting GPC3 have been developed, including vaccines, anti-GPC3 immunotoxins, and chimeric antigen receptor-modified cells. This review comprehensively covers the structure, physicochemical properties, biological functions, and clinical applications of GPC3. It explores diagnostic and treatment strategies centered around GPC3, offering hope for improved early detection and targeted therapies in the challenging landscape of HCC.
Keywords
- glypican 3
- GPC
- HCC
- liver cancer
- immunotherapies
Hepatocellular carcinoma (HCC) stands as the third leading cause of cancer-related mortality globally and is the most common primary liver cancer [1]. Its incidence is expected to rise in the coming years. HCC is often associated with chronic liver conditions such as hepatitis B and C, cirrhosis, and nonalcoholic steatohepatitis [2]. Despite significant efforts over the last decade, long-term survival rates for liver cancer patients have not seen substantial advancement despite the diversification of treatment protocols with various combinations. Surgical interventions, including liver transplantation and resection, are the mainstay of HCC treatment. However, these procedures are plagued by high postoperative recurrence rates, with about 40% of cases recurring within the first year and 5-year recurrence rates between 50% and 70% [3]. While liver transplantation patients have 5-year survival rates of 60% to 80%, recurrence remains a significant issue, affecting 10% to 20% of patients [4].
In contrast to normal hepatocytes, HCC cells exhibit elevated expression levels
of several surface molecules, including transferrin receptor (TfR) [5],
glypican-3 (GPC3) [6], asialoglycoprotein receptor (ASGP-R) [7], AF20 antigen
[8], somatostatin receptor (SSTR) [9], and lysosome-associated protein
transmembrane 4
In addition to exploring GPC3 as a molecular target, it is important to consider comprehensive gene sets associated with HCC to enhance the diagnostic and therapeutic perspectives. An advanced method was introduced for identifying reliable network-based biomarkers for HCC using phenotype-driven module detection and ranking, called mRank. This method identified 69 candidate biomarkers for HCC with high accuracy in distinguishing tumor samples from controls, including well-known HCC signatures such as AFP, SPP1, DCP, OPN, VEGF, ANG-2, and GP73. Incorporating this gene provides a more comprehensive understanding of HCC’s molecular landscape, supporting the diagnostic and therapeutic strategies centered around GPC3 [12].
Over the past decade, GPC3 has emerged as a prominent molecule closely correlated with the onset and progression of HCC [11]. During embryonic development, GPCs are remarkably expressed at high levels [13, 14, 15]. Different expression patterns distinguish the GPC family across different developmental stages and tissues. GPC1 is present in various tissues, including muscle, epithelium, kidney, bone, and bone marrow. However, GPC2 is expressed predominantly within the nervous system. On the other hand, GPC3 and GPC6 show widespread expression on various cell surfaces during embryonic development. GPC4 finds expression in the lung, kidney, and brain, while GPC5 is exhibited in the kidney, liver, brain, lung, and limbs. Notably, in adults, the expression profiles of GPCs undergo substantial alterations [13, 14, 15]. While GPC1, GPC4, and GPC6 are expressed across various tissues, GPC2 is restricted to the nervous system, GPC3 to the ovary, and GPC5 specifically to the brain. Recent investigations have revealed a significant correlation between GPCs and cancer development. Specifically, GPC3 is expressed in liver cancer tissues and peripheral blood samples of HCC patients while absent in healthy liver tissues and pathological samples of cirrhosis, fatty liver, hepatitis, or injury. Due to this expression profile, GPC3 surpasses alpha-fetoprotein (AFP) as a reliable tumor marker. Furthermore, GPC3 exhibits limited expression in normal adult tissues other than liver cells, rendering it suitable for targeted anticancer therapy as a tumor antigen [14].
Given these attributes, this review examines the diagnostic and treatment strategies centered around GPC3, recognizing its pivotal role in advancing diagnostic and therapeutic approaches for HCC.
The GPC3 gene, located on the X chromosome at 26q, consists of 11 exons
and produces a 2130 bp transcript encoding a 580-amino acid protein with a
molecular weight of approximately 70 kDa. Proteolytic cleavage of the
GPC3 protein results in N-terminal (40 kD) and C-terminal (30 kD)
fragments connected by disulfide bonds. The N-terminal fragment can further
cleave to form soluble GPC3 (sGPC) in peripheral blood. Heparan sulfate
modification occurs at Cys495 and Cys508, while Ser560 anchors the protein to the
cell membrane [15]. GPC3 is highly conserved across human, mouse, alpaca, and
camel species. The signal peptide sequence predicted using SignalP 4.1 spans the
initial 24 amino acids. Physicochemical analysis characterizes GPC3 as an acidic
protein with a molecular weight of 65,967.09 Da and a theoretical isoelectric
point of 6.01, with leucine being the most abundant amino acid (10.3%).
Secondary structure prediction by DNAstar indicates a prevalence of
Under normal physiological conditions, GPC3 is abundantly expressed on the membranes of embryonic cells but is noticeably absent in the adult liver. Mutations in the GPC3 gene lead to Simpson-Golabi Behmel Syndrome (SGBS) in humans, characterized by macrosomia and dysplasia of multiple organs and skeletons. Knockout experiments in mice demonstrate that the absence of GPC3 mirrors the symptoms of SGBS, highlighting its pivotal role in regulating body growth and development [16]. Despite lacking an intracellular signaling segment, GPC3, a membrane protein, constitutes a significant component of the extracellular matrix (ECM). It interacts with various growth factors, chemokines, and cytokines, forming concentration gradients on the cell membrane [11]. This ability to recruit extracellular signals across multiple pathways, such as WNT and Hedgehog, underscores GPC3’s crucial role in maintaining ligand concentrations and facilitating ligand-receptor interactions during development [17].
In 1997, Hsu HC et al. [18] published a seminal report revealing a very high expression of MXR7 (GPC3) cDNA among patients with liver cancer. MXR7 mRNA was seen in 74.8% of the samples from 154 patients with primary or recurrent HCC, significantly higher than serum AFP elevations [18]. They identified MXR7 mRNA as a potential early marker for HCC and later renamed GPC3 [18]. GPC3 demonstrates high expression levels in over 70% of HCC samples, as indicated by tissue microarray data and immunohistochemical analyses. Notably, this expression is absent in normal liver tissues, liver cirrhosis, benign liver lesions, or hepatitis tissues [19, 20, 21].
Moreover, the expression level of GPC3 has been significantly correlated with prognosis across various studies [20, 21, 22]. Beyond HCC, GPC3 exhibits high expression in other cancers, including lung squamous cell carcinoma [23], hepatoblastoma [19], melanoma [20], ovarian yolk sac tumor [21], and urothelial carcinoma [22]. This multifaceted expression profile underscores GPC3 as a specific biomarker and prognostic factor for HCC, as well as a promising target for a wide range of tumor treatments [11].
Numerous studies have highlighted GPC3’s superior sensitivity over AFP in liver cancer diagnosis. It is expressed on tumor cell surfaces and released into the peripheral circulation in a soluble form (sGPC3). Combining GPC3 with AFP enhances non-invasive HCC diagnosis [24]. It was demonstrated by Capurro M et al. [25] that GPC3 was expressed in 72% of HCC samples but not in normal or benign liver tissue cells and exhibited higher sensitivity than AFP in both surgically resected and biopsy samples. While GPC3 is a specific marker for HCC, its sensitivity alone may not meet clinical requirements [26]. Combining multiple markers, such as GPC3+Heat Shock Protein 70(HSP70)+Glutamine Synthetase(GS), has been suggested to distinguish between hepatocellular nodules, early liver cancer, and AFP-negative small liver cancer with high specificity. Other potential marker combinations, like GP (golgi protein)7373+GPC3+CD34 and GPC3+CK19, have been proposed for improved diagnostic accuracy [11].
The expression of GPC3 in the serum of liver cancer patients is crucial for non-invasive diagnosis. The soluble form of GPC3 (sGPC3) in serum serves as an important marker [27]. Studies by Capurro M et al. [25] and Abdelgawad IA et al. [28] reported increased sGPC3 levels in HCC patients, with a sensitivity and specificity reaching 95%. However, another group of researchers [29] reported lower positivity (28%) in HCC patients, possibly due to the use of different antibodies. A meta-analysis by Yang SL et al. [30] indicated that serum GPC3 is a specific biomarker for HCC when combined with other markers, yielding a sensitivity of 69% and specificity of 93%. Despite its importance, sGPC3 should be used in conjunction with other markers for comprehensive diagnosis. Additionally, comparing the effectiveness and specificity of different commercial antibodies is crucial to determining the most suitable antibody for clinical diagnosis, requiring large sample sizes for validation [31].
GC33 is a monoclonal antibody designed to target the terminal carboxyl subunit of the GPC3 molecule, exhibiting notable efficacy in inhibiting tumor growth (Fig. 1a) [32]. Its primary mechanism of action involves antibody-dependent cell-mediated cytotoxicity (ADCC), which underlies its tumor inhibition effect [33]. Notably, GC33 is the sole candidate progressing to the clinical trial stage [27, 34, 35]. In a phase I clinical trial, GC33 demonstrated favorable safety, with the high-dose group showing no significant adverse reactions. Moreover, patients with high GPC3 expression had a longer median time to progression compared to those with low GPC3 expression (twenty-six weeks vs. seven weeks) [36]. While nearly half of the patients exhibited disease stability (7 out of 13), the phase II clinical trial results fell short of the assumed goals. In patients with GPC3-positive HCC, treatment with GC33 showed no significant benefits, potentially due to the specific immune microenvironment of liver cancer, suggesting that antibody therapy targeting GPC3 alone is of limited efficacy [11].
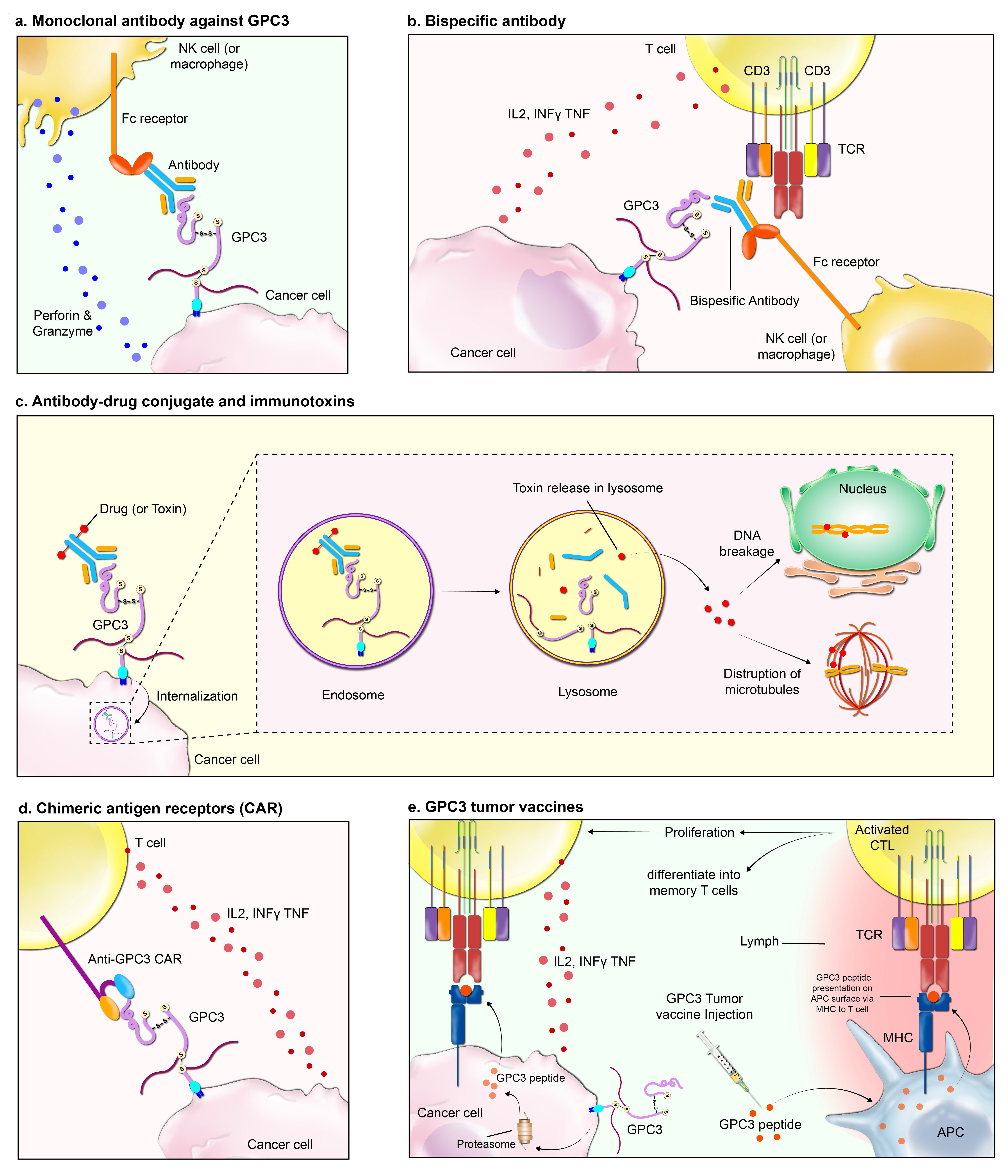 Fig. 1.
Fig. 1.The figure depicts a suite of immunotherapeutic interventions targeting glypican-3 (GPC3), a cell surface proteoglycan commonly upregulated in various malignancies. The methodologies are as follows: (a) Monoclonal Antibody Therapy: An antibody specific to GPC3 identifies and binds to the antigen on tumor cells, enabling immune effector cells, such as Natural Killer (NK) cells and macrophages, to recognize and eliminate the tagged cells via Fc receptor-mediated mechanisms. (b) Bispecific Antibody Approach: Utilizing a dual-affinity construct, this antibody simultaneously engages GPC3 on the tumor cells and CD3 on T cells. This dual binding facilitates the T cells’ cytotoxic attack on the tumor cells. (c) Antibody-Drug Conjugates and Immunotoxins: Antibodies conjugated to cytotoxic agents target GPC3, facilitating internalization into the tumor cell. Subsequent intracellular release of the toxin leads to cell death through mechanisms such as DNA damage or microtubule disruption. (d) Chimeric Antigen Receptor (CAR) T-Cell Therapy: T cells are genetically engineered to express CARs that specifically recognize GPC3, empowering them to directly target and lyse GPC3-positive tumor cells. (e) Tumor Vaccination Strategy: Vaccination with GPC3-derived peptides aims to prime the immune system, sparking a cascade that culminates in the activation and proliferation of cytotoxic T lymphocytes that can seek out and destroy GPC3-presenting tumor cells. Abbreviations: TCR, T cell receptor; MHC, major histocompatibility complex; CTL, cytotoxic T cell; APC, antigen-presenting cell; IL2, interleukin 2; INF, interferon; TNF, tumor necrosis factor.
Beyond GC33, scientists have explored a wide range of therapeutic antibodies
targeting GPC3. Building upon the foundation of GC33, Ishiguro T et al.
[37] devised a bispecific antibody named ERY974, targeting both GPC3 and CD3
(Fig. 1b). ERY974 demonstrated a potent killing effect on a wide range of
GPC3-expressing tumors, capable of transforming “cold tumors” into “hot tumors”.
This promising development has advanced to the phase I clinical trial stage
(NCT02748837i) [37]. Additionally, Zhang YF and Ho M [38] identified the
YP7 antibody, targeting the carboxyl-terminal epitope of GPC3. The scFv-Fc
antibody derived from YP7 demonstrated a superior anti-tumor effect in a nude
mouse liver cancer xenograft model [38]. Feng M et al. [39] also
identified a high-affinity antibody known as HN3, specifically targeting
full-length GPC3. This could impede the proliferation of GPC3-positive cells by
inducing cell cycle arrest in the G1 phase. Furthermore, it potently inhibits the
growth of HCC xenografts in mice nuclei [39]. Gao W et al. [36]
discovered the antibody HS20, explicitly targeting the GPC3 heparan sulfate
chain. This antibody could block Wnt/
An antibody-drug conjugate (ADC) represents a therapeutic drug formed by integrating monoclonal antibodies, cytotoxic molecules, and adaptors, facilitating their connection (Fig. 1c) [11]. This unique design allows ADCs to concentrate highly toxic drugs at tumor sites, enhancing the killing of tumor cells while reducing adverse effects. By precisely targeting tumor cells, ADCs broaden the therapeutic window for small molecular toxins, significantly reducing adverse effects [40]. Building upon the YP7 antibody, Fu Y et al. [41] developed antibody-drug conjugates (ADCs) by coupling it with Duocarmycin SA or pyrrolobenzodiazepine dimer - hYP7-DC and hYP7-PC. Notably, these two drugs effectively eliminate tumor cells at concentrations as low as pMol [42].
Immunotoxins are potent biological preparations formed by combining antibodies with toxins, targeting specific cells for destruction. Pseudomonas exotoxin A (PE-A) is frequently utilized due to its ability to inhibit protein synthesis and cause cell death [1]. Gao W et al. [43] developed immunotoxins YP7-PE38 and HN3-PE38, displaying effective anti-tumor activity and regression of GPC3-positive xenografts. Truncated prime editor A (PE-A) yielded immunotoxin HN3-mPE24, significantly extending the survival of tumor-bearing mice. YP7 scFv coupled with PE38KDEL demonstrated potent anti-tumor effects in mice. Photoimmunotherapy, pioneered by the Kobayashi team in 2011 [44], involves near-infrared dye IR700 coupled with antibodies, inducing cell death upon irradiation. IR700-YP7 and IR700-HN3 photoimmunotherapy agents effectively inhibit tumor growth [11].
The Chimeric Antigen Receptor (CAR) is a genetically engineered cell surface fusion protein comprising an antigen recognition fragment, a co-stimulatory signal molecule, and an immune cell receptor activating molecule. CARs enable the precise identification and targeted direction of immune cells like T cells or natural killer (NK) cells to eliminate tumor cells following their transfection to express these chimeric antigen receptors on their surface (Fig. 1d) [45].
Several research groups, including Chen C et al. [46], Pan Z et al. [47], Li K et al. [23], and Jiang Z et al. [45], have successfully engineered Chimeric Antigen Receptor T cells (CAR-T) based on the GC33 antibody. These CAR-T constructs have significantly killed patient-derived xenografts (PDX) and GPC3-positive cell lines. Efforts to enhance CAR-T specificity and mitigate off-target effects risk include the development of a GPC3/ASGR1 bispecific CAR-T by Chen C et al. [46], enabling selective killing of GPC3+ASGR1+ HCC cells by this innovative design. Additionally, Pan Z et al. [47] implemented co-expression of a soluble programmed death protein 1 (sPD1) fragment alongside CAR to augment the efficacy of GPC3 CAR-T, guarding against T cell depletion induced by the PD1/PDL1 signaling pathway during co-incubation with target cells. Guo X et al. [48] found that using CAS9 to knockout PD1 in CAR-T cells can enhance Akt phosphorylation and the anti-apoptotic protein Bcl-xL expression, ultimately improving the anti-tumor effectiveness of CAR-T. Zhao R et al. [49] enhanced CAR-T cells killing activity by introducing the intracellular domain of DAP10 into CAR.
Despite the promising results observed in cancer therapy with CAR-T, this approach is not without side effects, including cytokine storms, off-target effects, and a significantly high risk of graft-versus-host disease (GVHD). Researchers have turned their attention to Natural Killer (NK) cells [50], which have a short physiological cycle (1~2 weeks), possess potent tumor-killing abilities, and can eliminate tumor cells independent of the major histocompatibility complex (MHC), inducing target cell apoptosis through various mechanisms including releasing granzyme and perforin [27], Antibody-Dependent Cell-Mediated Cytotoxicity (ADCC), and TNF expression. However, despite all these capabilities, primary NK cells show several drawbacks, specifically limited in vitro amplification and significantly low transfection efficiency [51]. Due to the limitations of primary NK cells, numerous research studies have focused on NK-92 cell lines. NK-92 is a highly cytotoxic NK cell line that undergoes regular and uniform amplification and can effectively establish stable Chimeric Antigen Receptor Natural Killer (CAR-NK) cell lines [52]. Notably, NK-92 has exhibited excellent safety profiles in various trials, making it suitable for therapeutic applications [11]. Scientists have developed several CAR-NK products targeting GPC3. Yu M et al. [53] created a CAR-NK cell line targeting GPC3 based on the hu9f2 antibody, demonstrating robust killing activity in vitro and in vivo studies.
GPC3 is a pivotal serological marker for diagnosis and prognosis in patients with HCC, offering molecular targets for imaging and therapeutic interventions. A spectrum of immunotherapies targeting GPC3 has emerged, including genetic therapies, humanized anti-GPC3 cytotoxic antibodies, peptide/DNA vaccines, and immuno-toxin therapies. Various GPC3-targeted imaging techniques, such as magnetic resonance imaging, near-infrared imaging, and positron emission tomography, have been explored for early-stage HCC detection [54]. In combination with regimens for advanced HCC, GPC3 exhibits promising results, especially when paired with anti-angiogenic drugs and anti-PD-1/anti-PD-L1 agents. Clinical trials involving the adoptive transfer of GPC3-specific CAR-T cells have shown promising outcomes [55]. To overcome the limitations of conventional CAR-T cells, split anti-human Glypican-3 (anti-hGPC-3) CAR-T cells have been developed, demonstrating efficient recognition and lysis of HCC cells with reduced cytokine release. Ongoing clinical trials, potential combination strategies, and barriers in CAR-T therapy are discussed, emphasizing the need for validated extracellular HCC-specific antigens and exploring GPC3 as a versatile and safer alternative for HCC treatment [56]. These trials aim to assess the safety, tolerability, and preliminary efficacy of these therapies for GPC3-positive advanced HCC and pediatric solid tumors (Table 1).
| Clinical Trial | Trial ID | Phase | Agent | Status |
| Anti-GPC3 CAR-T for Treating GPC3-positive Advanced Hepatocellular Carcinoma (HCC) | NCT03084380 | I | CAR-T cell | Not yet recruiting |
| Glypican 3-specific Chimeric Antigen Receptor Expressing T Cells for Hepatocellular Carcinoma (GLYCAR) | NCT02905188 | I | CAR-T cell product (Glycar) | Completed accrual |
| Glypican 3-specific Chimeric Antigen Receptor Expressed in Autologous T Cells as Immunotherapy for Patients with Pediatric Solid Tumors [GAP] | NCT02932956 | I | CAR-T cell product (GAP T cells) | Ongoing, Currently enrolling |
| To Evaluate the Safety, Tolerability and Preliminary Efficacy of EU307 | NCT05783570 | I | Autologous Glypican 3 (GPC3) Targeted CAR-T cell [EU307] | Ongoing, Currently enrolling |
| GPC3-targeted CAR-T Cell for Treating GPC3 Positive Advanced HCC | NCT04121273 | I | CAR-T cell product | Ongoing, not currently enrolling. |
| Interleukin-15 Armored Glypican 3-specific Chimeric Antigen Receptor Expressed in Autologous T Cells for Solid Tumors | NCT05103631 | I | CAR-T cell product [GPC3-CAR] with IL-15 [CATCH T cells] | Ongoing, Currently enrolling |
| Interleukin-15 and -21 Armored Glypican-3-specific Chimeric Antigen Receptor Expressed in T Cells for Pediatric Solid Tumors | NCT04715191 | I | CAR-T cell product [GPC3-CAR] with IL-15 and IL-21 | Not yet enrolling |
| A Study of GPC3 Redirected Autologous T Cells for Advanced HCC (GPC3-CAR-T) | NCT02715362 | I/II | CAR-T cell product [TAI-GPC3-CAR-T cell] | Ongoing, Currently enrolling |
Abbreviations: CAR-T, Chimeric Antigen Receptor T cells.
A tumor vaccine, harnessing tumor antigens such as tumor-specific antigen (TSA) and tumor-associated antigen (TAA), shows promise in activating the immune system against cancer cells. The focus on GPC3, a TSA specifically expressed in hepatocellular carcinoma (HCC) cells, has yielded significant findings (Fig. 1e). Researchers have identified specific GPC3 epitopes, including GPC3 298-306 and GPC3 144-152, which induce immune responses in both mice and HCC patients, demonstrating effectiveness without triggering autoimmune reactions [11].
In a phase I clinical trial involving 33 advanced HCC patients, the GPC3 vaccine proved well-tolerated and successfully induced GPC3-specific immune responses in the majority of participants. Notably, 19 patients had stable disease, with some experiencing tumor necrosis or regression [38]. In a notable case, a 62-year-old sorafenib-resistant liver cancer patient exhibited significant intrahepatic tumor necrosis post-GPC3 vaccination, highlighting the vaccine’s targeted impact on cancerous tissues while sparing normal liver cells. Furthermore, the vaccine showed positive responses in clinical trials involving GPC3-positive ovarian cancer patients. Two chemotherapy-resistant patients exhibited encouraging results, including partial response and stability for over a year, indicating potential efficacy beyond HCC [11]. Phase II trials for HCC further support the vaccine’s anti-tumor effectiveness, as evidenced by lower recurrence rates post-surgery compared to surgery alone. These findings collectively suggest the GPC3 vaccine holds promise for enhancing cancer immunotherapy and warrants further exploration in larger clinical trials [42].
The future of GPC3 as a molecular target in HCC is promising and multifaceted. Enhancing diagnostic accuracy through multi-marker approaches and advanced imaging techniques will be critical. Combining GPC3 with other biomarkers like AFP, HSP70, and CK19 could improve diagnostic specificity and sensitivity. Integrating GPC3-targeted imaging modalities with AI can further enhance tumor localization and staging.
Optimizing therapeutic strategies involves exploring combination therapies, such as pairing GPC3-targeted treatments with immune checkpoint inhibitors and anti-angiogenic agents to achieve synergistic effects. The development of bispecific and trispecific antibodies targeting multiple pathways offers another promising direction.
Advancing CAR-T and CAR-NK cell therapies is crucial. Enhancing CAR-T cell efficacy through immune checkpoint integration, costimulatory domain modification, and genome editing to knock out inhibitory receptors will improve outcomes. CAR-NK cells, especially NK-92, present a safer alternative with fewer risks, and future research should focus on optimizing their engineering and scalability.
The development of next-generation immunotoxins and ADCs targeting GPC3 should continue, with an emphasis on enhancing specificity and reducing immunogenicity. Personalized GPC3 vaccines tailored to individual tumor profiles, potentially combined with other immunotherapies, offer another promising avenue.
Collaborative efforts in clinical trials and translational research will be essential to translate these promising strategies into clinical practice, ultimately improving outcomes for patients with HCC.
GPC3, initially identified as a liver cancer marker in 1997, has evolved into a crucial clinical indicator, surpassing traditional markers like AFP. Its potential in in-vivo imaging aids pre-treatment localization, staging, and surgical planning with the assistance of AI technology. Secretory GPC3 offers a non-invasive method for early tumor screening, while GPC3 antibodies and immunotherapy offer promising liver cancer treatment. However, technical challenges persist as GPC3’s role in liver cancer remains incompletely understood. It may act as a “chameleon molecule”, its function dependent on the membrane-matrix interface. Single anti-GPC3 treatments have limitations, and therapeutic strategies like CAR-modified immune cells face obstacles in the immunosuppressive liver cancer microenvironment. Strategies to enhance anti-GPC3 CAR efficacy include overcoming immunosuppression, adapting to tumor metabolism, and combining therapies. Addressing GPC3-negative tumor growth under treatment pressure is crucial, highlighting the need to explore new therapeutic targets and understand GPC3 expression regulation to prevent drug resistance.
Conceptualization: AnwS; resources: AnwS; writing original draft: AHH; writing: AT, AHH, AliS, MS, AnwS; literature and data acquisitin: AT, MS, AHH; analysis: AT, AliS, MS; review and editing: AT, AliS, MS, AnwS; supervision: AnwS; visualization: AT. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Graphical illustrations displayed in Fig. 1 are illustrated by Arezoo Sadeghipour, Medical Illustrator (http://www.sciencewish.com/ (accessed on 4 April 2024)).
This research received no external funding.
Anwaar Saeed reports a leadership role with Autem therapeutics, Exelixis, KAHR medical and Bristol-Myers Squibb; consulting or advisory board role with AstraZeneca, Bristol-Myers Squibb, Merck, Exelixis, Pfizer, Xilio therapeutics, Taiho, Amgen, Autem therapeutics, KAHR medical, and Daiichi Sankyo; institutional research funding from AstraZeneca, Bristol-Myers Squibb, Merck, Clovis, Exelixis, Actuate therapeutics, Incyte Corporation, Daiichi Sankyo, Five prime therapeutics, Amgen, Innovent biologics, Dragonfly therapeutics, Oxford Biotherapeutics, Arcus therapeutics, and KAHR medical; and participation as a data safety monitoring board chair for Arcus therapeutics. The remaining authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.