

1 Department of Biological Sciences, University of Chicago, Chicago, IL 60637, USA
2 SynRx Therapeutics, Chicago, IL 60642, USA
3 School of Life Sciences, Westlake University, 310030 Hangzhou, Zhejiang, China
Abstract
Background: The switching/sucrose non-fermentable (SWI/SNF) Related, Matrix Associated, Actin Dependent
Regulator Of Chromatin, Subfamily A (SMARCA) member 2 and member 4 (SMARCA2/4)
are paralogs and act as the key enzymatic subunits in the SWI/SNF complex for chromatin remodeling. However, the role of
SMARCA2/4 in DNA damage response remains unclear. Methods: Laser
microirradiation assays were performed to examine the key domains of SMARCA2/4
for the relocation of the SWI/SNF complex to DNA lesions. To examine the key
factors that mediate the recruitment of SMARCA2/4, the relocation of SMARCA2/4 to
DNA lesions was examined in HeLa cells treated with inhibitors of
Ataxia-telangiectasia-mutated (ATM), Ataxia telangiectasia and Rad3-related
protein (ATR), CREB-binding protein (CBP) and its homologue p300 (p300/CBP), or
Poly (ADP-ribose) polymerase (PARP) 1/2 as well as in H2AX-deficient HeLa cells.
Moreover, by concomitantly suppressing SMARCA2/4 with the small molecule
inhibitor FHD286 or Compound 14, the function of SMARCA2/4 in Radiation sensitive
51 (RAD51) foci formation and homologous recombination repair was examined.
Finally, using a colony formation assay, the synergistic effect of PARP
inhibitors and SMARCA2/4 inhibitors on the suppression of tumor cell growth was
examined. Results: We show that SMARCA2/4 relocate to DNA lesions in
response to DNA damage, which requires their ATPase activities. Moreover, these
ATPase activities are also required for the relocation of other subunits in the
SWI/SNF complex to DNA lesions. Interestingly, the relocation of SMARCA2/4 is
independent of
Keywords
- DNA damage response
- DNA repair
- SWI/SNF complex
Genomic DNA constantly encounters genotoxic stresses that induce DNA damage. In response to DNA damage, the surveillance system of cells is quickly activated [1]. DNA lesions are detected by DNA damage sensors, which are associated with chromatin relaxation for DNA damage repair [2]. Several important chromatin remodeling complexes have been reported to participate in DNA damage response, one of which is the switching/sucrose non-fermentable (SWI/SNF) complex [3].
The SWI/SNF complex is evolutionarily conserved in eukaryotes and plays a key role in regulating the position of nucleosomes on genomic DNA [3, 4, 5, 6]. This complex is an ATP-dependent chromatin remodeling complex with multiple subunits. The key subunit in the SWI/SNF complex is subfamily A (SMARCA) member 2 (SMARCA2)/Brahma (BRM), an ATPase with the Asp-Glu-Gly-His (DEGH) helicase fold [7, 8]. It hydrolyzes ATP and provides the energy for the whole complex to disrupt the interaction between histones and DNA, thereby removing nucleosome barriers and exposing naked DNA for numerous biological activities, such as transcription and replication [9, 10]. SMARCA4/Brahma-related gene 1 (BRG1) is a paralog of SMARCA2, which contains a similar domain architecture and has a redundant function to SMARCA2 [2, 11, 12, 13, 14]. In addition to SMARCA2/4, the SWI/SNF complex also has up to 28 other subunits depending on different biological contexts [7]. However, nine of them are known as core subunits that are involved in all the known functions of the SWI/SNF complex [15].
It has been shown that the SWI/SNF complex participates in DNA damage repair. The complex is recruited to DNA double-strand breaks (DSBs) and participates in both non-homologous end joining and homologous recombination repair via sliding nucleosome barriers and exposing naked DNA ends to facilitate the loading of repair machinery [5, 6]. However, the recruitment mechanism of the SWI/SNF complex seems unclear. It has been reported that poly(ADP-ribosyl)ation (aka PARylation) signals or histone acetylation events mediate the recruitment of the SWI/SNF complex to DNA lesions [16, 17, 18, 19]. However, PARylation is mainly induced by DNA single-strand breaks [20], whereas the SWI/SNF complex is known to participate in DSB repair [5, 6]. It is also unclear if any specific histone acetylation events are significantly enriched at DNA lesions. Moreover, earlier studies have shown that loss of SMARCA2 is sufficient for impairing DSB repair [5, 6]. However, it was recently identified that SMARCA4, the paralog of SMARCA2, plays a redundant role in the SWI/SNF complex [21]. Since both SMARCA2 and SMARCA4 are ATPase in the SWI/SNF complex, only lacking both of them, but not either of them, can suppress the function of the SWI/SNF complex [21, 22]. The DSB repair relies on the chromatin remodeling activity of the SWI/SNF complex, during which the energy is derived from the ATPase of SMARCA2 or SMARCA4 in the SWI/SNF complex. With these unsolved questions, we have carefully examined the recruitment of SMARCA2 and SMARCA4 to DNA lesions as well as their functions in homologous recombination (HR) repair. We demonstrate that lacking both SMARCA2 and SMARCA4 inhibits HR repair. Moreover, the recruitment of SMARCA2 and SMARCA4 to DNA lesions is independent of PARylation and histone acetylation but requires the ATPase activities of SMARCA2 and SMARCA4.
To express green fluorescent protein (GFP)-tagged SMARCA2, SMARCA4, SMARACC1 and SMARCD1, the cDNAs of SMARCA2, SMARCA4, SMARACC1 and SMARCD1 were synthesized by Tsingke Biotechnology (Beijing, China) based on the sequences from NCBI (NM_003070 for SMARCA2, NM_001128844 for SMARCA4, NM_003074 for SMARCC1, and NM_003076 for SMARCD1). The complementary DNAs (cDNAs) were then cloned into pEGFP-C1 (Clontech, Beijing, China) to generate GFP-SMARCA2, GFP-SMARCA4, GFP-SMARCC1, and GFP-SMARCD1. For the expression of truncation mutants of SMARCA2, N-terminal (N-Ter, amino acids 1-653), helicase domain (Hel, amino acids 654-1383), and bromo domain (BRD, amino acids 1384-1590) were cloned into pEGFP-C1 to generate GFP-N-Ter, GFP-Hel and GFP-BRD. The K755A mutant of SMARCA2 and the K785A mutant of SMARCA4 were generated by site-directed mutagenesis from the wild type SMARCA2 and SMARCA4, respectively [21, 22].
HeLa, HCC1187, MDA-MB-468 and MDA-MB-231 cells were purchased from American Type
Culture Collection. HeLa cells were cultured in Dulbecco’s Modified Eagle Medium
(DMEM) (Catalog number: 11965092; Thermo Fisher Scientific, Waltham, MA, USA),
HCC1187 cells were cultured in Roswell Park Memorial Institute (RPMI)-1640 medium
(Catalog number: 11875168; Thermo Fisher Scientific, Waltham, MA, USA), and
MDA-MB-468 and MDA-MB-231 cells were cultured in Leibovitz’s L-15 Medium (Catalog
number: 21083027; Thermo Fisher Scientific, Waltham, MA, USA), containing 10%
fetal bovine serum and 1% penicillin-streptomycin (Catalog number: 15140122;
Thermo Fisher Scientific, Waltham, MA, USA) at 37 °C with 5% CO
To ectopically express GFP-tagged proteins, transfection of the cells with all pEGFP constructs was carried out using Lipofectamine 2000 (Catalog number: 11668500; Thermo Fisher Scientific, Waltham, MA, USA) according to the manufacturer’s protocol.
To generate HeLa SMARCA2 or HeLa SMARCA4 knockout cells, CRISPR/Cas9-mediated
gene editing was performed. Cells were transfected with single guide RNA (sgRNA)
against SMARCA2, constructed in pSpCas9(BB)-2A-Puro (PX459 V2.0) vector (Addgene
plasmid ID: 62988). The sgRNA sequence targeting SMARCA2 was
5
The following antibodies were purchased from their respective companies:
anti-
Laser microirradiation assays were performed according to a previous study [23].
GFP-tagged constructs were transfected into the specified cells and plated on
35-mm glass-bottom dishes. Cellular DNA damage in the nuclei of cultured cells
was induced using microirradiation with a pulsed nitrogen laser (Spectra-Physics;
365 nm, 10 Hz pulse). This laser system was directly integrated into the
epifluorescence microscope’s imaging pathway, focusing through a Plan-Apochromat
63
Cells growing on glass coverslips were irradiated with 5 Gy and allowed to
recover for 12 h before immunofluorescence. Then, cells were treated with
fixation buffer (4% PFA, 0.1% Triton X-100). The slides were incubated with
different primary antibodies in 1% BSA at room temperature for 2 h or 4
°C overnight. Subsequently, the slides were washed with bovine serum albumin (PBS) and
incubated with a secondary antibody. DNA was stained, and coverslips were mounted
with 4
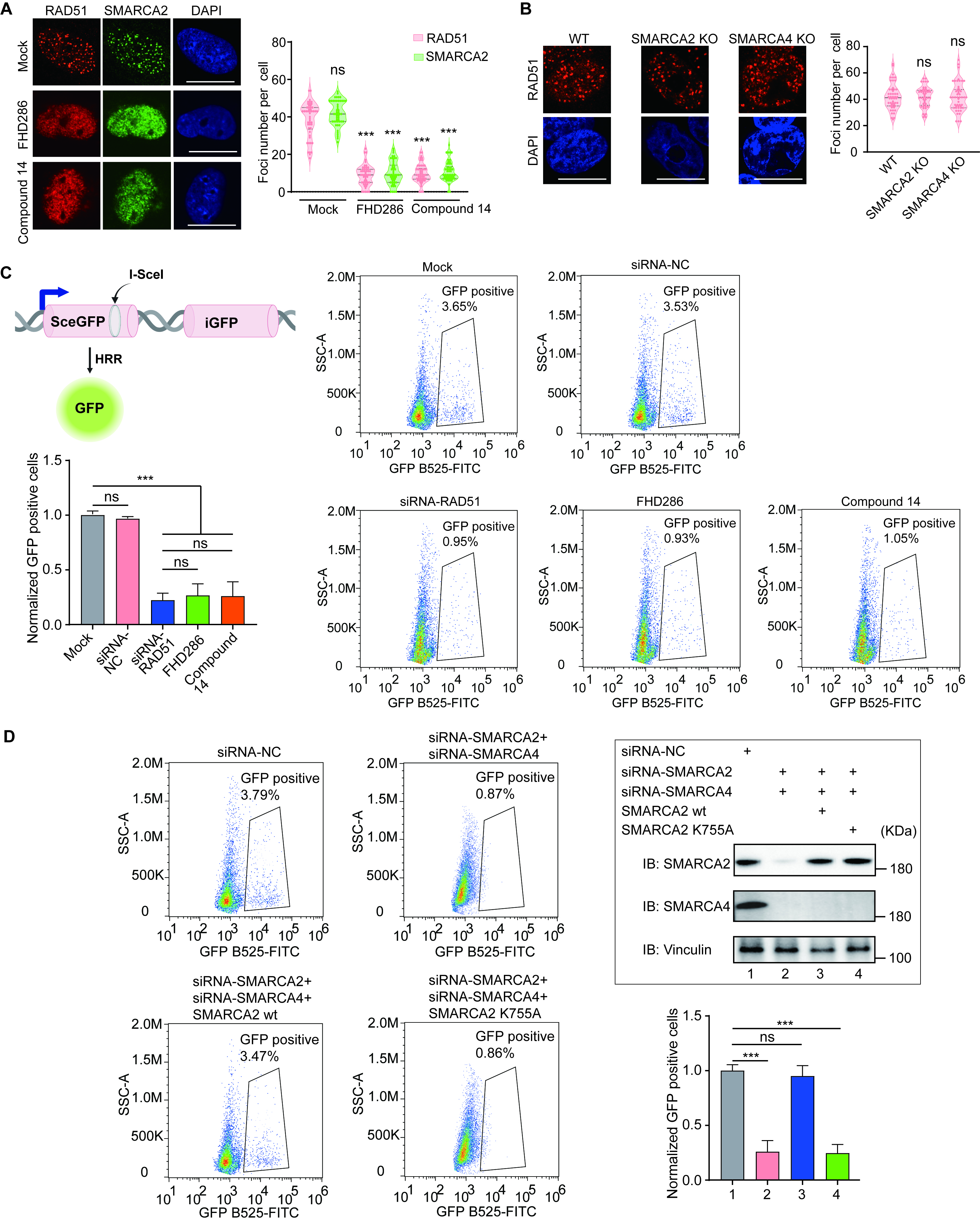
The DR-GFP reporter assay system was provided by Dr. Xiaochen Gai [25] at Westlake University. The assay was performed as described in a previous study [25]. The DR-GFP reporter system was specifically designed to quantify homologous recombination repair, where the iGFP gene fragment functions as a template for the HR process following an I-SceI-induced DSB in the upstream SceGFP cassette. To initiate this process, the HA-I-SceI-GR pCW tet-on plasmid was transfected into the cells. Doxycycline is then administered to trigger the expression of I-SceI for 18 hours. Acetonide treatment facilitates the translocation of I-SceI from the cytoplasm to the nucleus. Subsequently, I-SceI cuts the targeting site, generating a DSB, which upon repair, leads to the expression of GFP. Flow cytometry was performed to examine the efficiency of homologous recombination repair by calculating the GFP positive populations. For RAD51 knockdown, cells with DR-GFP insertion were transfected with negative control siRNA (siRNA-NC) or siRNA-RAD51 for 24 hours following HA-I-SceI-GR pCW tet-on plasmid transfection. For inhibitor treatment, cells with DR-GFP insertion were pretreated with or without FHD286 (1 µM) or Compound 14 (1 µM) for 2 hours before acetonide treatment.
SMARCA4 and SMARCA2 double knockdown was carried out by siRNA transfection
directed against SMARCA2 (siRNA-SMARCA2): 5
For the clonogenic cell proliferation assay, cells were seeded in 6-well plate (1000 cells/well). The cells were then treated with either a vehicle control or drugs with the indicated concentration on the next day, and culture medium was refreshed every 3 days for a total of 14 days in total. At the endpoints of the colony formation assays, the colonies were washed with PBS and fixed with 70% methanol for 10 minutes. This was followed by staining with 0.1% crystal violet. The number of colonies was counted by ImageJ software, and survival graphs were generated from three replicate wells, with colony numbers normalized to untreated controls.
Statistical analyses were performed using GraphPad Prism7 (GraphPad Software,
Inc., San Diego, CA, USA). The statistical significance was assessed using a
two-tailed unpaired Student’s t-test. All of the data were presented as
mean
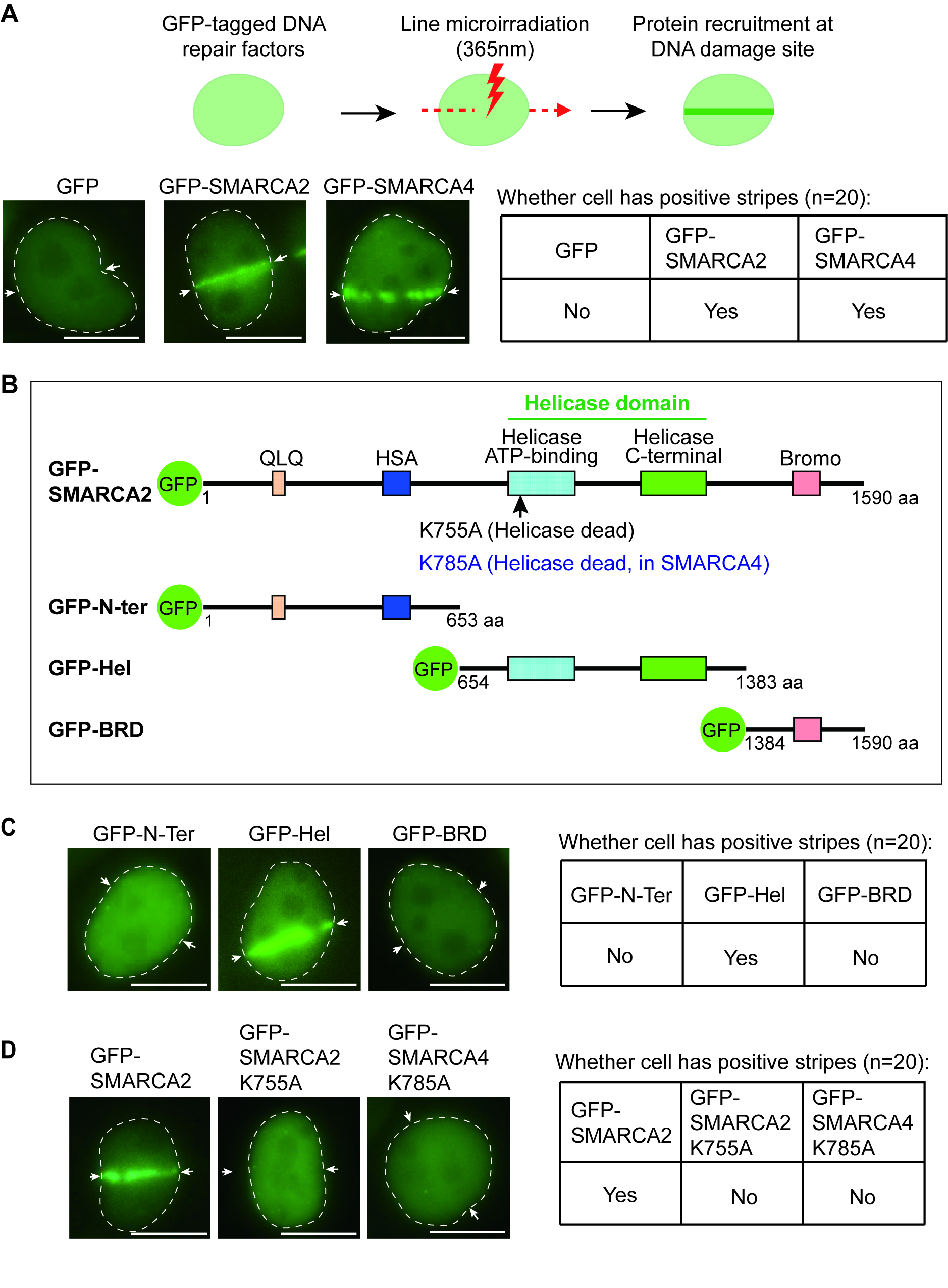
To study the molecular mechanism by which the SWI/SNF complex is recruited to DNA lesions, we examined SMARCA2/4, the key enzymatic subunits in the SWI/SNF complex. Using laser microirradiation assay and observation by fluorescence microscopy, we found that both GFP-tagged SMARCA2/4 relocated to DNA lesions (Fig. 1A). We also measured the recruitment kinetics and found that within five seconds, SMARCA2/4 was recruited to DNA lesions (Supplementary Fig. 1).
 Fig. 1.
Fig. 1.The relocation of the switching/sucrose non-fermentable (SWI/SNF) Related, Matrix Associated, Actin Dependent Regulator Of Chromatin, Subfamily A (SMARCA) member 2 and member 4 (SMARCA2/4) to DNA lesions. (A) pEGFP-C1-SMARCA2, pEGFP-C1-SMARCA4, or pEGFP-C1 was transfected into HeLa cells for 24 hours. Following laser microirradiation, the laser stripes of green fluorescent protein (GFP)-SMARCA2/4 were observed using fluorescence microscopy. The laser stripe is indicated with white arrows. The table on the right presents the cellular status, specifically indicating the presence or absence of stripes. Scale bar: 10 μm. (B) Schematic of the deletion mutants of SMARCA2. (C) GFP-tagged truncation mutants of SMARCA2 were transiently expressed in HeLa cells. Following laser microirradiation, the laser stripes of GFP-N-Ter, GFP-Hel and GFP-bromo domain (BRD) were examined using fluorescence microscopy. The laser stripe is indicated with white arrows. The table on the right presents the cellular status, specifically indicating the presence or absence of stripes. Scale bar: 10 μm. (D) GFP-SMARCA2-K755A or GFP-SMARCA4-K785A were transiently expressed in HeLa cells. Following laser microirradiation, the laser stripes of these enzymatically dead mutants were examined. Scale bar: 10 μm.
SMARCA2/4 has several distinct domains, including the N-terminal QLQ motif and HAS domain, which mediate the interaction with other subunits of the SWI/SNF complex; the ATPase/helicase domain in the middle, which is the solo enzymatic domain in the whole complex; the C-terminal bromodomain that is known to recognize acetylated histones [10, 22] (Fig. 1B). Since SMARCA2/4 shares an identical domain architecture, we generated deletion mutants of SMARCA2 and found that only the ATPase/helicase domain (indicated as GFP-Hel), but not other domains (indicated as GFP-N-Ter or GFP-BRD), was able to relocate to DNA lesions (Fig. 1C). Moreover, we established the ATPase-dead constructs which mutated the key lysine residue into alanine in ATPase/helicase domains of SMARCA2 (K755A) and SMARCA4 (K785A) to abolish their ATP-binding activities [22, 26], and found that these ATPase activities were required to target SMARCA2/4 to DNA lesions (Fig. 1D).
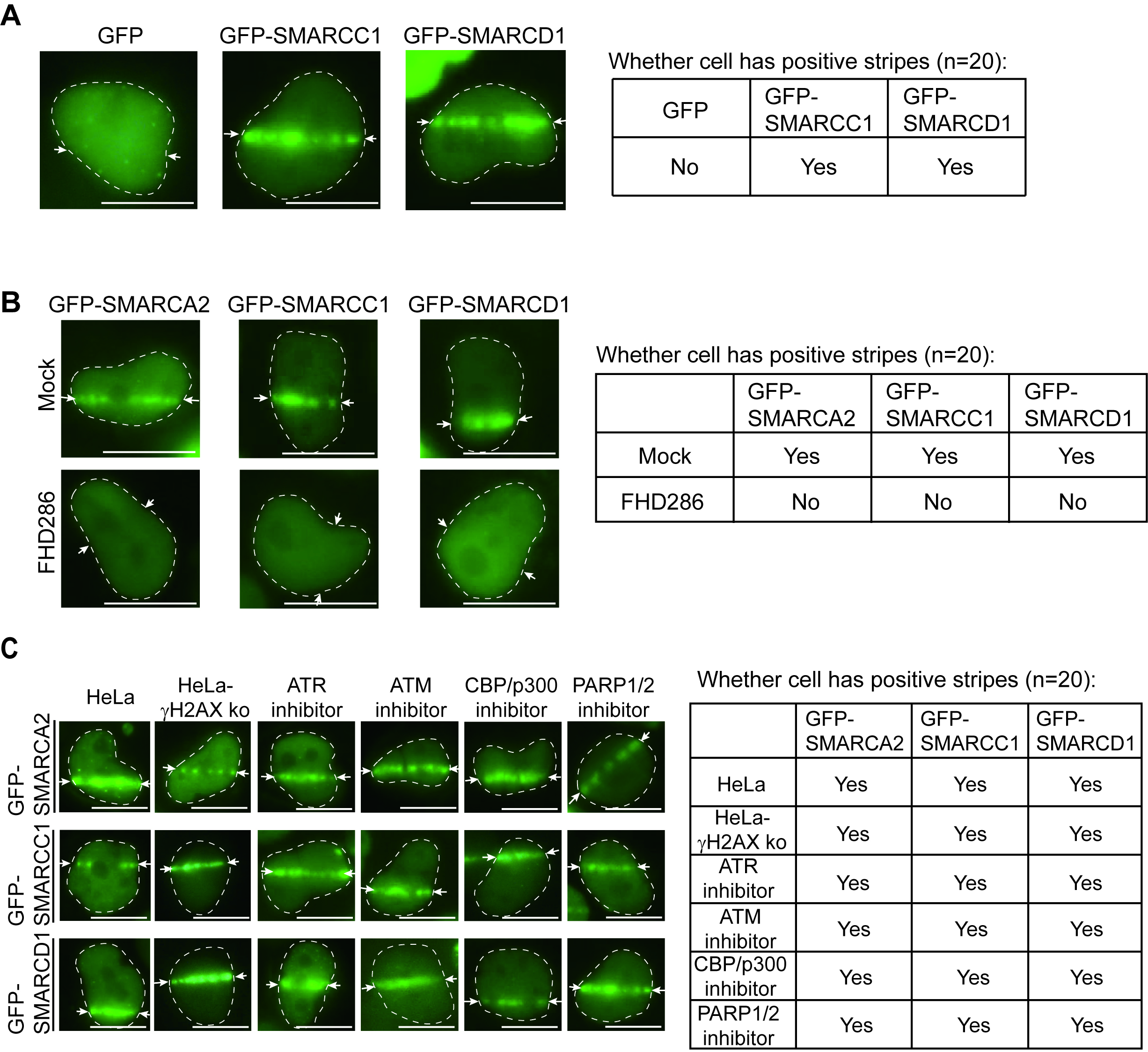
Next, we examined other core subunits of the SWI/SNF complex, including SMARCC1 and SMARCD1. We show the first evidence that, similar to SMARCA2/4, both SMARCC1 and SMARCD1 relocated to DNA lesions when cells were treated with laser microirradiation (Fig. 2A). Interestingly, when we pre-treated cells with FHD286, a specific dual inhibitor to kill both ATPase activities of SMARCA2/4 [27], SMARCC1 and SMARCD1 were no longer recruited to DNA lesions in the laser microirradiation assays (Fig. 2B). This suggests that the ATPase activity of SMARCA2/4 is also required for targeting other subunits of the SWI/SNF complex to the sites of DNA damage.
 Fig. 2.
Fig. 2.Relocation of the switching/sucrose non-fermentable (SWI/SNF) to DNA lesions. (A) pEGFP-C1-SMARCC1, pEGFP-C1-SMARCD1, or pEGFP-C1 was transfected into HeLa cells for 24 hours. Following laser microirradiation, the laser stripes of GFP-SMARCC1 or GFP-SMARCD1 were observed using fluorescence microscopy. The laser stripe is indicated with white arrows. The right table presents the cellular status, specifically indicating the presence or absence of stripes. Scale bar: 10 μm. (B) GFP-SMARCA2, GFP-SMARCC1 or GFP-SMARCD1 was transiently expressed in HeLa cells. The cells were pre-treated with or without 1 µM FHD286 for two hours followed by laser microirradiation. The localization of SMARCA2, SMARCC1, and SMARCD1 was examined. The laser stripe is indicated with white arrows. The right table presents the cellular status, specifically indicating the presence or absence of stripes. Scale bar: 10 μm. (C) GFP-SMARCA2, GFP-SMARCC1 or GFP-SMARCD1 was transfected into HeLa cells for 24 hours. Cells were treated with 1 µM Ataxia-telangiectasia-mutated (ATM) inhibitor (ku55933), 1 µM Ataxia telangiectasia and Rad3-related protein (ATR) inhibitor (VE-821), 1 µM CREB-binding protein (CBP) and its homologue p300 (CBP/p300) inhibitor (CBP/p300-IN-21) or 1 µM Poly (ADP-ribose) polymerase (PARP) inhibitor (olaparib) for two hours followed by laser microirradiation. The localization of SMARCA2, SMARCC1 and SMARCD1 was examined. The localization of the subunits of the SWI/SNF complex was also examined in the H2AX KO HeLa cells. The laser stripe is indicated with white arrows. The right table presents the cellular status, specifically indicating the presence or absence of stripes. Scale bar: 10 μm.
It has been reported that several DNA repair factors may mediate the recruitment
of the SWI/SNF complex to DNA lesions, such as
Earlier studies have shown that the enrichment of DNA damage response factors,
such as
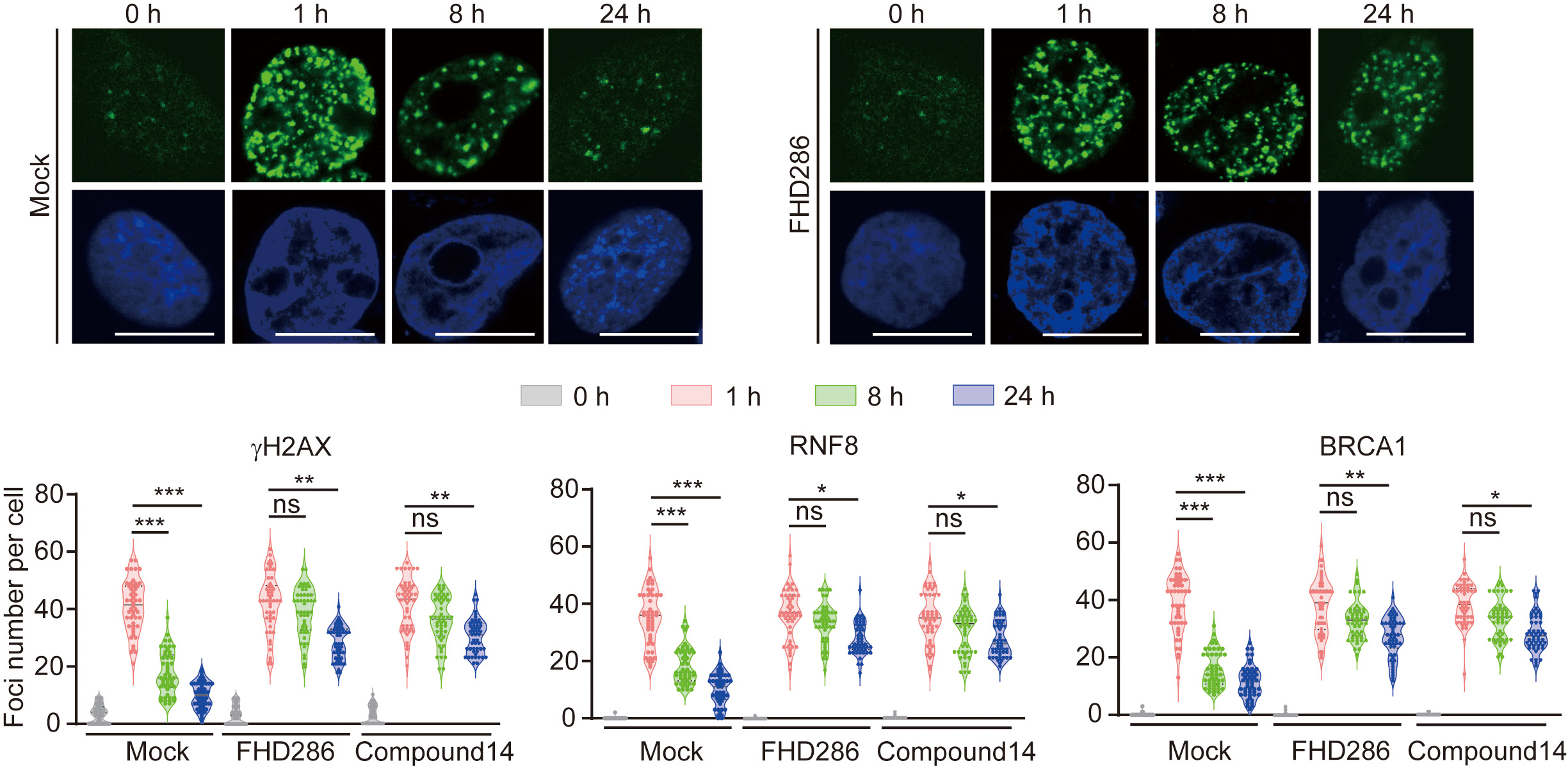
 Fig. 3.
Fig. 3.Lacking the enzymatic activities of SMARCA2/4 extends the
accumulation of
Upon SMARCA2/4 inhibition, the prolonged accumulation of DNA damage response
factors, such as
 Fig. 4.
Fig. 4.Suppression of the enzymatic activities of SMARCA2/4 impairs
RAD51-dependent homologous recombination (HR) repair. (A) HeLa cells were pre-treated with or without
FHD286 (1 µM) or Compound 14 (1 µM) for two hours, followed by 5 Gy
of IR. Foci formation of RAD51 and SMARCA2 was examined by immunofluorescence
staining with anti-RAD51 or anti-SMARCA2 antibodies. The cells were also
counterstained by DAPI. The number of foci per cell was analyzed. p
values were calculated using Student’s t-test. “ns” indicates
nonsignificant, ***p
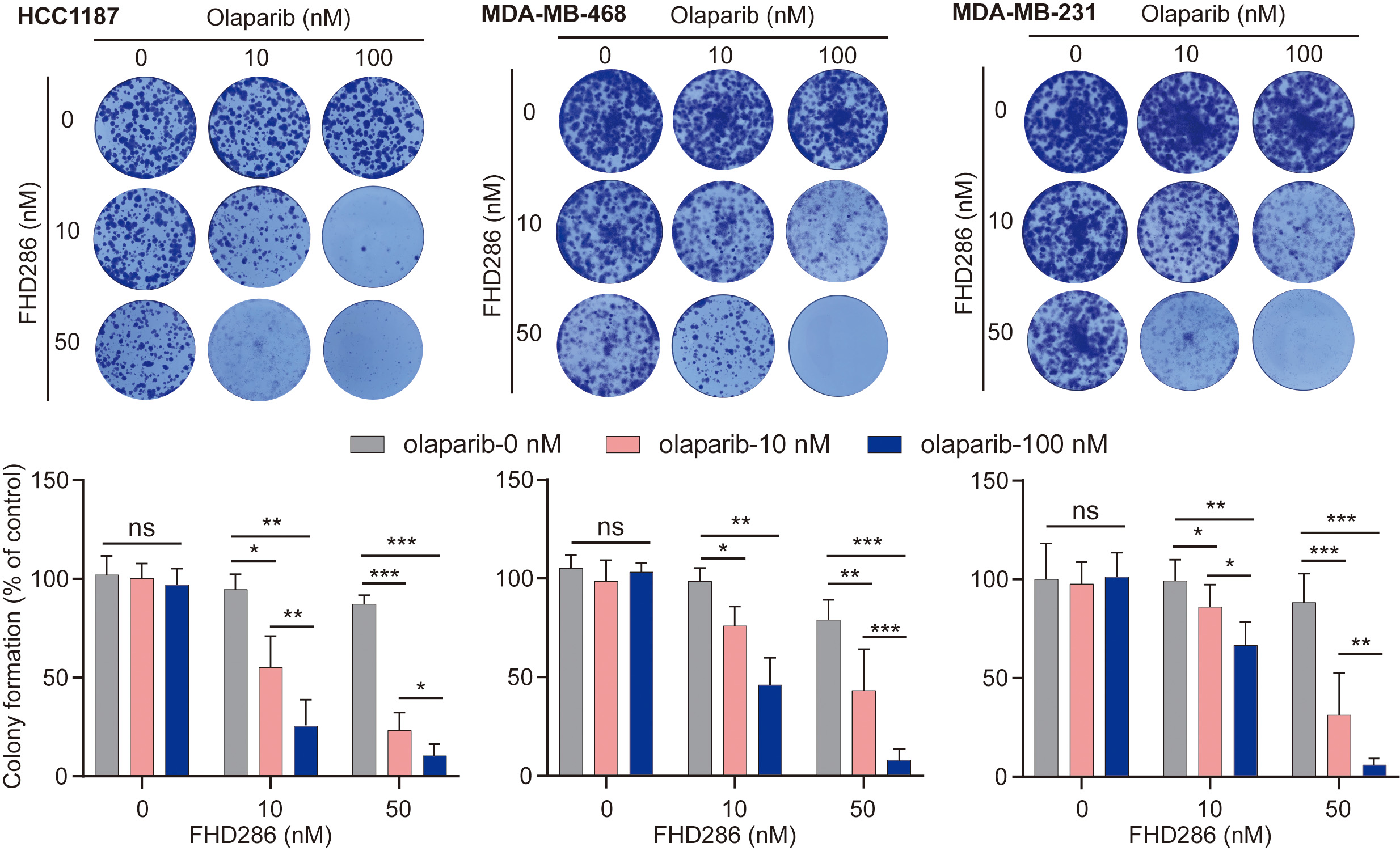
HR-deficient cells are often hypersensitive to PARP inhibitor treatment. To date, PARP inhibitors have been used for the treatment of breast, ovarian, pancreatic, and prostate cancers with HR deficiency. Interestingly, FHD286 is currently in clinical trials for cancer treatment. Since FHD286 treatment impairs HR repair, we ask if FHD286 treatment can sensitize tumor cells to PARP inhibitor treatment. We selected three breast cancer cell lines (HCC1187, MDA-MB-468 and MDA-MB-231) that are not sensitive to PARP inhibitor olaparib. Interestingly, with 50 nM of FHD286, all these cells line were sensitive to 100 nM olaparib treatment (Fig. 5), indicating that FHD286 may have an additive effect with PARP inhibitors for cancer treatment in the future.
 Fig. 5.
Fig. 5.FHD286 sensitizes tumor cells to the Poly (ADP-ribose) polymerase (PARP) inhibitor treatment.
A total of 1000 cells were seeded in each well and treated with indicated doses
of FHD286 and/or PARP inhibitor olaparib. Colony formation was examined at Day 14
with 0.1% crystal violet staining. Statistical analyses are shown in the lower
panel. p values were calculated using Student’s t-test. “ns”
indicates nonsignificant, *p
In this study, we have demonstrated that the enzymatic activity of SMARCA2/4 plays a key role in the recruitment of the SWI/SNF complex to DNA lesions, facilitating DSB repair. Earlier studies have shown that other factors, such as PARylation, acetylation, or phosphorylation events, mediate the recruitment of the SWI/SNF complex [16, 17, 18, 19]. Here, we have shown that only the ATPase activities of SMARCA2/4 are essential for the recruitment of this chromatin remodeling complex. Notably, chromatin remodeling should be one of the early DNA damage response events. Only after the removal of the nucleosome barrier can the repair machinery access DNA lesions for repair [8, 30]. Since the ATPase domains of SMARCA2/4 directly interact with genomic DNA [10, 13], it is possible that the SWI/SNF complex constantly scans genomic DNA and is able to detect DSBs. It also allows for quick chromatin remodeling for damage repair in response to DSBs. Laser microirradiation induces the rapid relocation of SMARCA2/4 within five seconds, confirming their quick response to DNA damage. Thus, we favor the possibility that SMARCA2/4 are able to quickly and directly recognize DNA lesions as DNA damage sensors.
Earlier studies have shown that the lack of SMARCA2 is sufficient to impair HR repair [5, 6]. However, numerous analyses have suggested that SMARCA2 and SMARCA4 are paralogs and have redundant functions in the SMARCA2/4 [2, 3, 11, 22]. In our study, we have demonstrated that abolishing both enzymatic activities of SMARCA2/4 is necessary to suppress HR repair. Notably, the SMARCA4 gene is frequently deleted during tumorigenesis [21, 31]. Therefore, if the cells only contain SMARCA2, ablating SMARCA2’s enzymatic activity is sufficient to disrupt the function of the SWI/SNF complex and impair HR repair.
In our study, we have demonstrated that suppressing SMARCA2/4 did not hinder the
foci formation of several upstream DNA damage response factors, such as
Collectively, our studies provide an in-depth understanding of the biological functions of SMARCA2/4 in the context of DNA damage repair.
The SMARCA2 and SMARCA4 proteins play a crucial role as DNA damage sensors. The ATPase activities are essential for recruiting the SWI/SNF complex to DNA lesions. Consequently, the loss of SMARCA2 and SMARCA4 impairs HR repair, a vital process for maintaining genome integrity. This finding suggests that treatment with SMARCA2 and SMARCA4 inhibitors may sensitize tumor cells to PARP inhibitors, a class of drugs used in cancer therapy.
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
LY and DW designed the research project, performed the experiments, analyzed the data, and wrote the manuscript. Both authors have thoroughly reviewed and approved the content of the final manuscript. Both authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
We apologize to all authors whose work could not be mentioned due to space limitations. Figures were created with BioRender.com.
This work was supported in part by grants from Hangzhou City Leading Innovation and Entrepreneurship Team (TD2020004), “Pioneer” and “Leading Goose” R&D Program of Zhejiang (2024SSYS0033), Westlake University Education Foundation, and Westlake Laboratory of Life Sciences and Biomedicine.
The authors declare no conflict of interest. Duo Wu is from the SynRx Therapeutics.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.