

1 Nuclear Medicine Unit, Istituti di Ricovero e Cura a Carattere Scientifico (IRCCS) Ospedale Policlinico San Martino, 16132 Genoa, Italy
2 Department of Experimental Medicine, Human Anatomy, University of Genoa, 16132 Genoa, Italy
3 Molecular Pathology Unit, IRCCS Ospedale Policlinico San Martino, 16132 Genoa, Italy
4 Department of Health Sciences, University of Genoa, 16132 Genoa, Italy
5 Institute of BioImaging and Complex Biological Systems, National Research Council (CNR), 20054 Milan, Italy
Abstract
Background: Selective deprivation of glutamine has been shown to
accelerate the generation of reactive oxygen species (ROS) and to impair the
activity of a specific pentose phosphate pathway (PPP) located within the
endoplasmic reticulum (ER). The consequent oxidative damage suggests that glucose
flux through this reticular pathway might contribute to the redox stress of
breast cancer cells. We thus evaluated whether this response is reproduced when
the glutamine shortage is coupled with the glucose deprivation. Methods:
Cancer growth, metabolic plasticity and redox status were evaluated under
saturating conditions and after 48 h starvation (glucose 2.5 mM, glutamine 0.5
mM). The Seahorse technology was used to estimate adenosine triphosphate
(ATP)-linked and ATP-independent oxygen consumption rate (OCR) as well as proton
efflux rate (PER).
Keywords
- hormone-sensitive breast cancer
- glucose and glutamine metabolism
- cytosolic/reticular pentose phosphate pathway
- NADPH
- redox status
- hexose-6-phosphate-dehydrogenase
- glucose-6-phosphate dehydrogenase
After the pioneering discovery of Warburg [1, 2], cancer metabolism is currently thought to convert large amounts of glucose to lactate. Besides its energetic inefficiency, the high-rate hexose flux through glycolysis inevitably competes with the pentose phosphate pathway (PPP) activity, potentially hampering the capability to generate the nucleotides and the NADPH equivalents needed to preserve redox balance and the reductive biosyntheses associated with tumor growth [3, 4].
Among the variegate pathways able to overcome this limitation, extensive literature previously documented that cancer cells preserve the high proliferation rate by using large amounts of glutamine [5, 6]. On one side, the glutamine-derived glutamate can enter the mitochondrial matrix to feed the tricarboxylic acid (TCA) up the generation of NADPH through the degradation of the generated malate [7, 8]. On the other hand, a profound interreliance has been observed between the glutamine degradation and the catalytic function of hexose-6P-dehydrogenase (H6PD) that triggers a selective PPP within the reticular lumen [6].
In agreement with this “double addiction”, glucose deprivation simultaneously can impair cell proliferating activity and increases mitochondrial oxygen consumption rate (OCR) [9] while the selective deprivation of glutamine at least partially reproduces this pattern [6]. According to current models of energy metabolism, this response agrees with the expected enhancement of cell respiration induced by the limited adenosine triphosphate (ATP) generation through glycolysis. On the other hand, the direct link between glutamine deprivation and redox stress seems more complex and asks to hypothesize that the amino acid processing through the TCA cycle might be mostly dedicated to anaplerotic and reductive functions. These observations thus indicate a reciprocal interference between glucose and glutamine metabolism in the regulation of both cell redox state and energy asset. Accordingly, decreasing the availability of both substrates might elicit a largely different metabolic response with respect to the enhancement of redox stress observed under the selective deprivation of either one.
In the present study, we aimed to evaluate whether combined and prolonged glucose and glutamine depletion causes metabolic and redox changes in breast cancer cells. To this purpose, our analysis involved the evaluation of glucose metabolism within the endoplasmic reticulum (ER) as a primary regulatory of the complex interaction between the pathways processing these two substrates.
MCF-7 (human hormone sensitive breast cancer cell line) was purchased from ATCC
(American Type Culture Collection). The cell line was maintained at 37
°C with 5% CO
MCF-7 cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) (#01-057-1A, Sartorius, AG, Göttingen, Germany) supplemented with 10% fetal bovine serum (#10270-106, Thermo Fisher Scientific , Waltham, MA, USA), 25 mM glucose, 2 mM glutamine, 1 mM pyruvate, 1% penicillin-streptomycin (control medium) (Thermo Fisher Scientific, Waltham, MA, USA).
Cells were seeded in DMEM medium and after five hours the cell cultures were exposed to either fresh control medium (CTR) or DMEM containing only 2.5 mM glucose and 0.5 mM glutamine (LB). After 48 h the Cells were harvested for measurements.
Cells were stained with 1 µg/mL propidium iodide (PI) (Enzo Life Sciences, Executive Blvd, Farmingdale, NY 11735, USA) and cell viability was evaluated by PI fluorescence measured using a FACScan Flow Cytometer (Becton Dickinson, Eysin, Swizzerland).
Cell proliferation rate was evaluated using carboxyfluorescein-diacetate-succinimidyl ester (CFSE) (Merck KGaA, Darmstadt, Germany). Briefly, cells were plated in 6-well plates and after 24 h later the cells were labeled with CFSE (0.3 µM in PBS). After 20 min at 37 °C the labeling solution was replaced with a fresh culture medium, cells were incubated for another 20 min at 37 °C and then cultured either with CTR or LB medium for 48 h. CFSE fluorescence was acquired on a FACScan flow cytometer and the data were analyzed by FlowJo software (Version 10.8.1, Becton–Dickinson, Eysin, Swizzerland).
OCR and ECAR were measured using Seahorse XFp Extracellular Flux Analyzer (Agilent Technologies, Santa Clara, CA, USA) following manufacturer’s istructions. 7000 cells per well were plated in XFp plates and incubated in control medium. The medium was replaced 5 h later and the cells were incubated with either CTR or LB medium for 48 h. Briefly, cells were incubated in Agilent Seahorse DMEM, pH 7.4 (#103575, Agilent Technologies, Santa Clara, CA, USA), enriched with nutrients mimicking the respective incubation medium conditions. OCR and ECAR were measured at baseline and after sequential injection of the selective glutaminase inhibitor Bis-2-(5-phenylacetamido-1,3,4-thiadiazol-2-yl)-ethyl sulfide (BPTES, 3 µM), the ATP-synthase inhibitor Oligomycin A (1.5 µM) and the ATP synthesis uncoupler carbonyl cyanide-4-trifluoromethoxyphenylhydrazone (FCCP, 1.25 µM).
OCR was expressed as picomol O
Lactate and pyruvate in cell lysates were measured in a spectrophotometric assay
following the reduction of NAD
Cells were lysed in RIPA buffer (#9806, Cell Signaling Technologies, Danvers, MA, USA) plus protease inhibitor cocktail (G135, Applide Biological Materials, Richmond, Canada). Western blot experiments were performed according to the standard procedure. Primary antibodies employed are the following: anti-hexose 6-phosphate dehydrogenase (H6PD, #ab170895, Abcam, Cambridge, UK), anti-glucose 6-phosphate dehydrogenase (G6PD, #ab124738, Abcam, Cambridge, UK) anti- glucose regulated protein 78 (GRP78, #ab21685, Abcam, Cambridge, UK) and anti-Actin (#MA1-140, ThermoFisher Scientific, Waltham, MA, USA). Relevant secondary antibodies (Sigma-Aldrich, St. Louis, MO, USA) were, diluted 1:10,000 in PBS-Tween, developed with chemiluminescence substrate (#170-5060, Bio-Rad, Hercules, CA, USA). Detection and densitometric analyses were performed using Alliance 6.7 WL 20M and UVITEC software (Alliance-1D software, UVITEC, Cambridge, UK), respectively. All the bands of interest were normalized with actin levels detected on the same membrane.
Cells were incubated for 20 min at 37 °C with 1 µM
ER-Tracker Red, 5 µM H
NADPH/NADP and NADH/NAD
In vitro
Lumped constant (LC) was calculated as described before [13].
Briefly, LC is obtained as the ratio between the metabolic rate of glucose
(MRGlu*) and glycolytic-related glucose disposal (MRGlu). Were MRGlu* is defined
as the product of retained FDG fraction and the total available glucose (5
Data are presented as mean
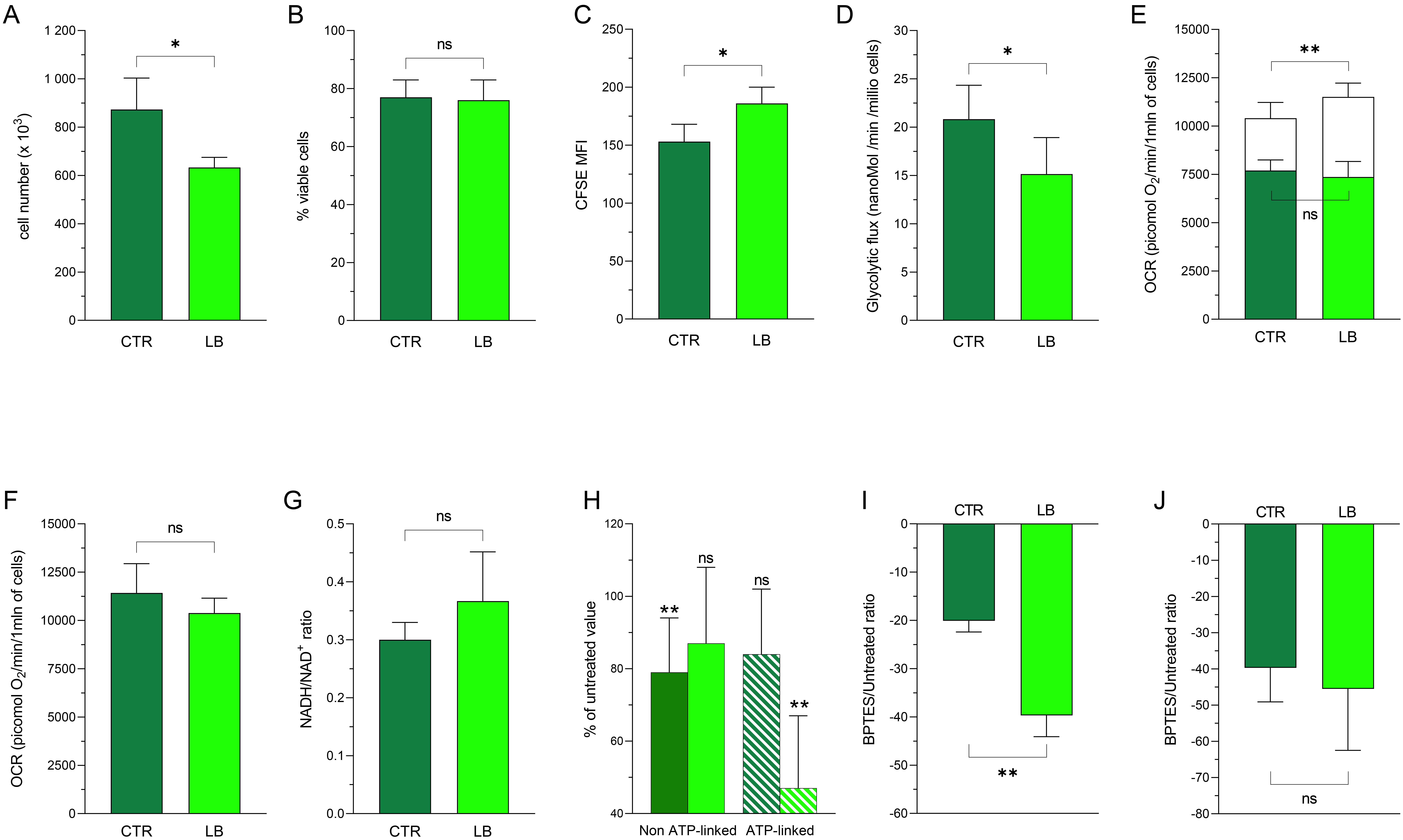
Prolonged (48 hours) glucose-glutamine shortage slightly, yet significantly, decreased the growth curve of MCF-7 cells (Fig. 1A). This effect was not related to any increase in mortality rate, since propidium iodide negative cells remained invariant during nutrient stress (Fig. 1B, Supplementary Fig. 1A). By contrast, it reflected a reduced MCF-7 cells proliferation rate, as documented by the CSFE dilution (Fig. 1C, Supplementary Fig. 1B).
 Fig. 1.
Fig. 1.Cell proliferation rate and metabolic pattern of MCF-7 under
prolonged starvation. (A–C) Cell numbers (A), percent of viable cells (B) and
CFSE mean fluorescence intensity (MFI, C) of MCF-7 cultures under CTR (dark
green) or LB (light green) medium. (D–G) Glycolytic flux (D), baseline OCR
(Non-ATP-linked (solid columns) and ATP-linked (white columns), E), OCR in
response to FCCP (F) and NADH/NAD
The metabolic reaction to energy substrate depletion implied a different
response by glycolysis and cell respiration. As expected, the ten-fold shortening
of glucose availability decreased lactate release. Nevertheless, PER slowed down
by only 30% (Fig. 1D), apparently suggesting a metabolic shift increasing the
glucose fraction channeled to the glycolysis. Baseline OCR was apparently not
affected by the prolonged nutrient deprivation, although its ATP-synthase
dependent fraction was higher in starved than in control cultures (Fig. 1E). This
respiratory reaction agreed with the relatively preserved response to
FCCP-induced uncoupling (Fig. 1F) and matched a slight and not significant
increase in NADH/NAD
We thus hypothesized a possible enhancement of glutamine channeling to the Krebs cycle. To confirm this hypothesis, we evaluated the response of OCR and PER to the inhibition of glutamine conversion into glutamate. BPTES did not influence lactate release (data not shown) regardless of the nutrient availability. By contrast, the decrease in overall OCR induced by the inhibition of glutamine conversion to glutamate reflected two different mechanisms in the two experimental models: a selective lessening of oligomycin-insensitive OCR in control cultures as opposed to a selective slowing down of oxygen usage dedicated to ATP regeneration in starved ones (Fig. 1H).
This finding thus suggested that glutamine-derived carbons contribute to energy assets during prolonged nutrient deprivation, while their contribution to ATP synthesis is less represented under control conditions. This concept was at least partially confirmed by the estimation of intracellular levels of pyruvate and lactate. Indeed, cell content of both metabolites comparably decreased under starvation (Fig. 1I,J), suggesting that the incomplete glucose degradation was not caused by any increase in the catalytic function of LDH. On the other hand, BPTES administration decreased pyruvate levels to a greater extent with respect to lactate, suggesting a possible differential fate of glutamine-derived pyruvate equivalents compared to glycolysis-generated ones.
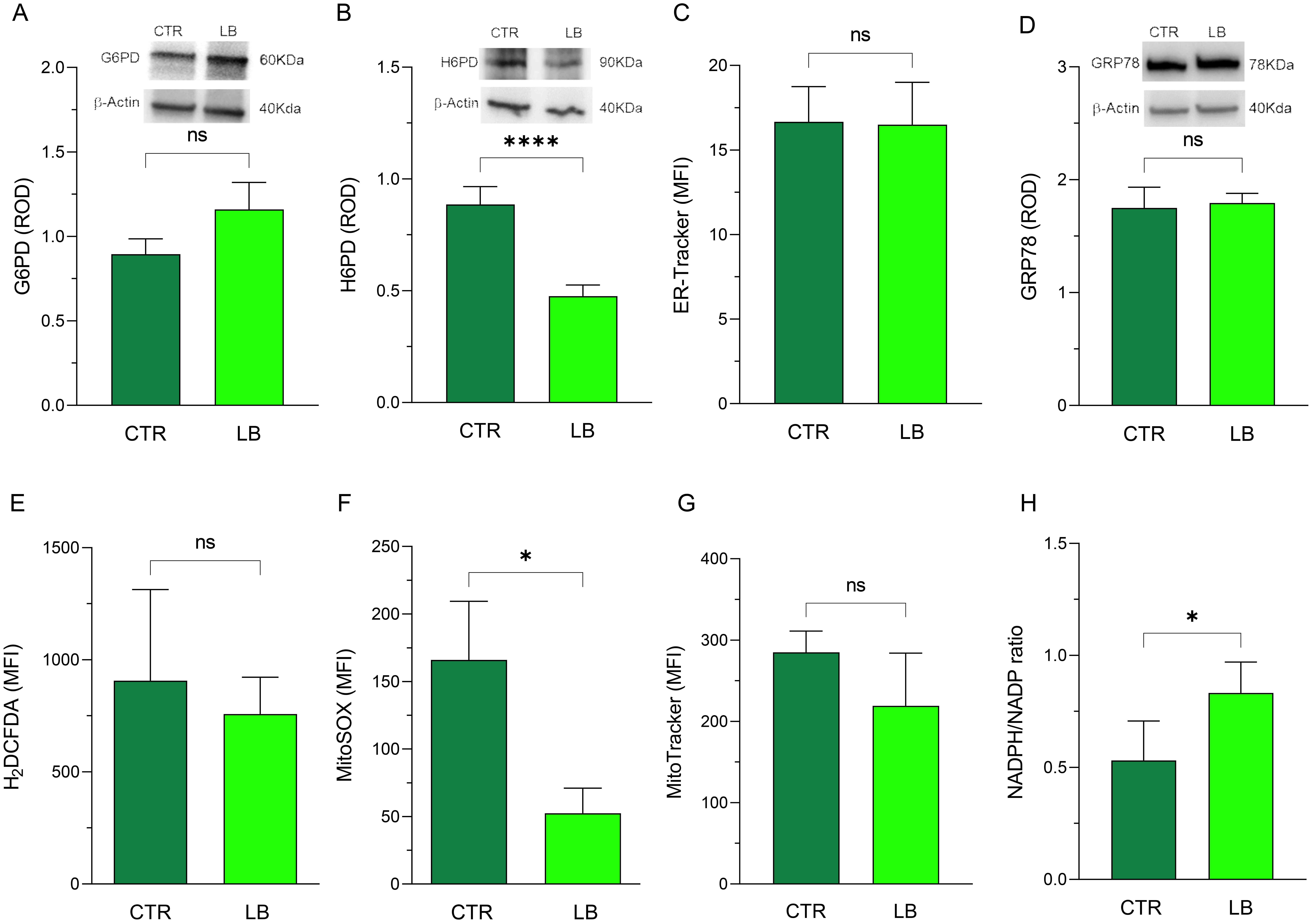
Altogether, these data suggested that the combined shortage of glucose and glutamine enhanced the incomplete degradation of glucose to lactate, apparently indicating a profound decrease in glucose utilization through the PPP. We thus aimed to verify whether this response was caused by an impairment of the catalytic function of either G6PD or H6PD. The abundance of the house-keeping protein ß-Actin was not affected by starvation (Fig. 2A,B,D and Supplementary Fig. 2). G6PD expression was slightly—though not significantly—increased after starvation (Fig. 2A). By contrast, its reticular counterpart H6PD was markedly decreased (Fig. 2B). This response was not the consequence of an ER shrinkage since fluorescence intensity after incubation with ER-Tracker was superimposable in both experimental models (Fig. 2C, Supplementary Fig. 3A). Similarly, it was not associated with a significant stress, since GRP78 abundance was similar in both control and starved cultures (Fig. 2D).
 Fig. 2.
Fig. 2.Effect of glucose-glutamine depletion on redox homeostasis.
(A,B) Relative optical density (ROD) (left) and a representative Western blot
(right) of G6PD (A) and H6PD (B) normalized versus
We thus hypothesized that the decreased fuel to PPP, combined with the decreased expression of H6PD might have enhanced the oxidative stress of starved cells. Nevertheless, cellular and mitochondrial oxidative status remained unchanged or even decreased, as described by the DCF (Fig. 2E, Supplementary Fig. 3B) and MitoSOX staining (Fig. 2F, Supplementary Fig. 3C). On the other hand, MitoTracker staining at least partially reproduced the analysis of OCR, and documented a relative integrity of mitochondrial membrane polarization (Fig. 2G, Supplementary Fig. 3D). This behavior was coherent with the response of the NADPH/NADP ratio that even slightly increased after prolonged nutrient depletion (Fig. 2H). Accordingly, these data suggested that the combined shortage of glucose and glutamine eventually resulted in a reduced ROS generation.
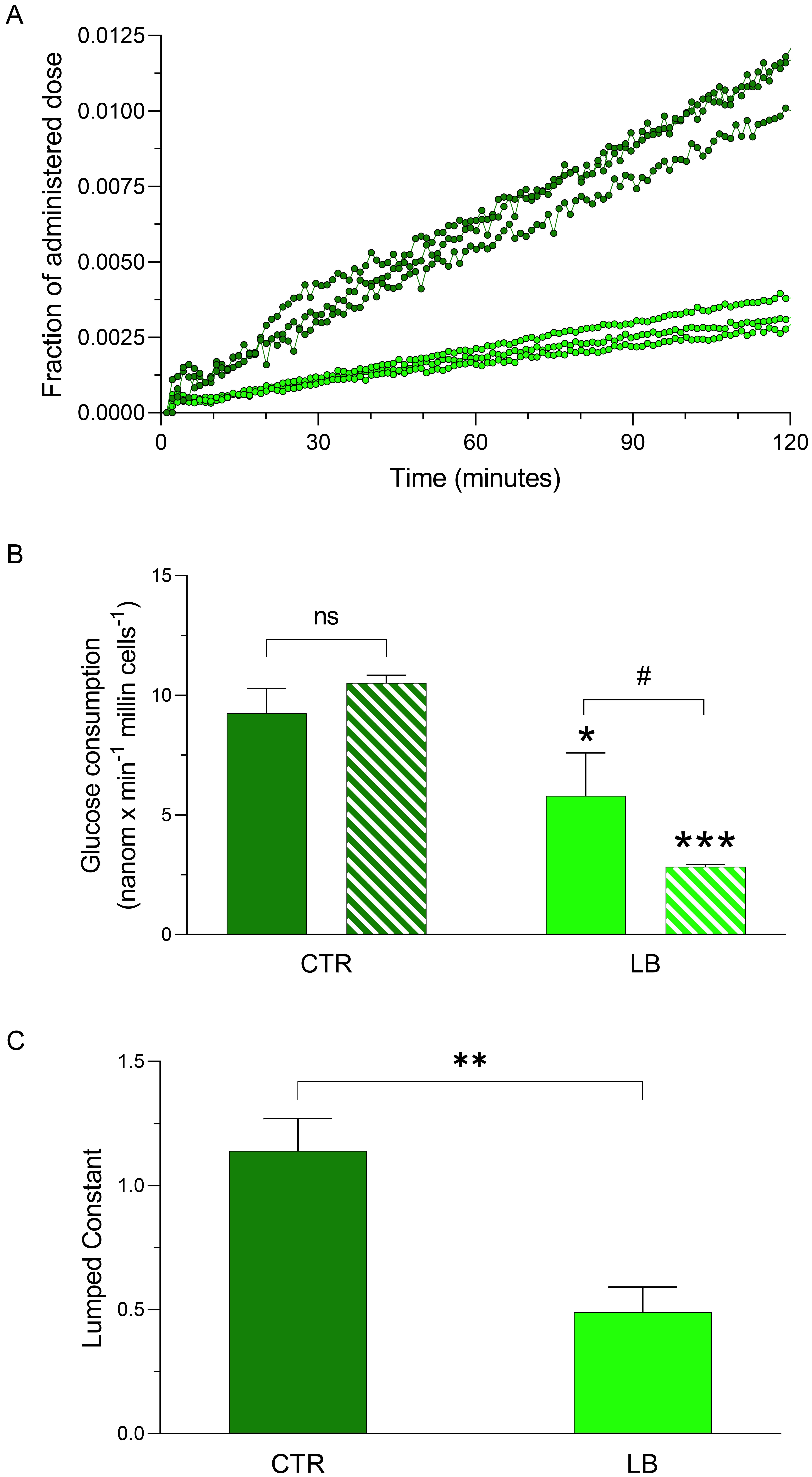
According to the previous analyses, the combined glucose and glutamine shortage also decreased the ROS generation in the whole cell and in the mitochondria, respectively. We thus aimed to verify whether this response also characterized the reticular lumen, modifying the activity of the local PPP. To this purpose, we exploited the strict link of FDG uptake with the expression and the catalytic function of H6PD. As expected, FDG uptake showed a linear increase under both experimental conditions (Fig. 3A). Nevertheless, despite the marked decrease in the concentration of the competing unlabeled glucose, the rate of tracer retention was significantly lower in starved cells than in control ones (Fig. 3A). Tracer-based assessment of glucose consumption thus provided a divergent picture with respect to the direct measurement of hexose disappearance from the incubation medium. Indeed, the estimated MRGlu* was superimposable to the directly estimated MRGlu under control condition (Fig. 3B). By contrast, this proportionality was virtually lost in starved cultures (Fig. 3B). As a consequence, the selective impairment of FDG uptake induced by nutrient deprivation markedly decreased the lumped constant (the ratio MRGlu*/MRGlu) (Fig. 3C).
 Fig. 3.
Fig. 3.Endoplasmic reticulum (ER) metabolism under a nutrient
shortage. (A)
In the present study, the balanced shortage of glucose and glutamine decreased the proliferation rate of MCF-7 breast cancer cells and simultaneously increased the glucose channeling toward its incomplete degradation to lactate despite the relative energetic inefficiency of glycolysis. This response modified the biochemical role of glutamine oxidation/decarboxylation that was shifted from a preferential use through energy-independent processes to pathways dedicated to ATP-regeneration by OXPHOS.
The balanced nutrient shortage also decreased the redox stress, not only in the cytosol and mitochondria, rather also in the ER. On one side, the chaperone of the heat-shock protein family GRP78, directly involved in the unfolded protein response and ER-stress [13], was not altered by nutrient shortage. Indeed, the decreased ratio between FDG and glucose uptake displayed by starved cells coupled with the downregulation of H6PD expression indicates a significant slowdown of glucose metabolism within the reticular lumen.
Since the discovery of the Warburg effect [1, 14], cancer metabolism has been mostly studied in cultured cells exposed to extremely high concentrations of nutrients. On the other hand, the adaptation to the harsh conditions of the tumor microenvironment has been most often evaluated by studying the metabolic response to the depletion of single substrates, most often glucose or glutamine [15]. Nevertheless, recent evidence suggests that cancer metabolic plasticity might also derive from the dynamic changes in metabolite quantity, reaction rates and directions of the active pathways. Since this network of interactions is strictly dependent on the ratio between the abundance of the several substrates, the metabolic responses to balanced or selective nutrient deprivations might be largely different [16].
Besides the expected slowing in anabolic functions, the combined deprivation of
glucose and glutamine eventually resulted in a selective decrease of pyruvate
cell content coupled with relatively preserved levels of lactate. This finding
agrees with a recent study by Gkiouli and coworkers who, in MCF-7 cells exposed
to a comparable nutrient stress, documented a 3-fold decrease of pyruvate content
facing a relatively less evident decrement of lactate [17]. In our experiments,
this finding was not attributable to a modified LDH activity since starvation
left invariant both the substrate/product ratio of its catalyzed reaction and the
NADH/NAD balance. By contrast, it agrees with the notion of a variegate source of
intracellular pyruvate. In experiments using
In agreement with this large literature body, the present data indicate that the response to the nutrient shortage is not limited to the activation of mitochondrial glutaminolysis but may also modulate its role. Indeed, the inhibition of glutamine conversion to glutamate by BPTES slowed down the oligomycin-insensitive OCR in control cultures as opposed to a selective impairment of its ATP-related fraction in starved ones. Accordingly, feeding condition seems to be a factor able to modulate the anaplerotic or energy-related fate of glutamine carbon through the Krebs cycle.
Besides the modulation of cytosolic and mitochondrial use of glutamine and glucose, the present data shed new light on a possible involvement of the ER. Indeed, the preferential channeling to glycolysis inevitably decreased the PPP rate during starvation. This response eventually resulted in a downregulation of H6PD expression facing a slight and not significant increase of its cytosolic counterpart G6PD. Although the mechanisms underlying this response cannot be fully elucidated, it seems conceivable that this reaction was at least partially related to the decreased redox status of starved cultures. Indeed, prolonged starvation of both glucose and glutamine decreased both DCF and MitoSOX MFI as also confirmed by the significant increase in NADPH/NADP ratio. This finding mismatches the enhanced oxidative stress, described in cells exposed to a selective depletion of either substrate [20, 21, 22, 23, 24] and suggests a profound interference between glutamine metabolism and PPP activity. Further studies are needed to understand how and through which mechanisms this cross-talk is modulated by the availability of both nutrients and the ratio of their extracellular concentrations.
The decreased glucose flux in starved cultures also involved the reticular counterpart of PPP. Indeed, FDG uptake rate was markedly lower in cultures exposed to the nutrient stress than in control ones, despite the ten-fold lower concentration of unlabeled glucose competing for transmembrane transport and hexokinase-catalyzed trapping mechanism [25, 26, 27]. This response was not caused by a severe slowdown of glucose consumption since glycolytic flux, measured with the Seahorse approach, was scarcely affected by starvation indicating only a slight decrease in glucose consumption. Accordingly, the profound decrease in the lumped constant confirms previous studies by us and other groups, documenting that FDG uptake is loosely linked to glycolysis and rather reflects the activity of the reticular PPP [10, 28, 29]. Although the activity of this pathway was not directly evaluated, its link with the decreased tracer uptake is strongly corroborated by the evidence that starvation and the decreased proliferation rate were indeed associated with a significantly downregulated expression of its triggering enzyme H6PD. This finding thus extends the link between H6PD activity and FDG uptake as an index of cancer growth rate reproducing the observation also observed in the triple negative breast cancer cell line MDA-MB-231 [9].
In conclusion, the simultaneous reduction of glucose and glutamine does not seem cytotoxic, at least in this hormone-sensitive breast cancer cell line. Its expected consequence, the decrease in proliferating activity, is associated with a decreased redox stress that besides cytosol and mitochondria extends to involve the ER and its local PPP. This link confirms a relevant role of glucose metabolism within the reticular lumen as a pivotal determinant of cancer growth, suggesting the need for studies aiming to characterize the role of ER-PPP in the response to the oxidative damage as well as in the maintenance of the redox signaling needed to activate cell proliferation and cancer growth.
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
SCar, CM, GS, MG and FV conceived and designed the study. SCar, MG, VC, FV, SL, NB, SChi and AMO performed the experiments and verified data quality. SCar, SR, SL, ED and CM analyzed the data. GS, SCar, and CM wrote the paper. MR, MB and JM performed and analyzed all experiments with the radioactive glucose analog. MB reviewed and edited the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have significantly contributed to this work and agreed to be accountable for all its aspects.
Not applicable.
We thank all members of the laboratory for useful discussions.
This research was funded by the grant AIRC (IG 23201) for the project
“Chemotherapy effect on cell energy metabolism and endoplasmic reticulum redox
control”, by the Italian Ministry of Health 5
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/j.fbl2907251.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.