

 , John M. Barbaro 2, Grace McDermott 3, Joan W. Berman 2,4
, John M. Barbaro 2, Grace McDermott 3, Joan W. Berman 2,41 Department of Medicine, Division of Infectious Diseases, Albert Einstein College of Medicine, Bronx, NY 10461, USA
2 Department of Pathology, Albert Einstein College of Medicine, Bronx, NY 10461, USA
3 Albert Einstein College of Medicine, Bronx, NY 10461, USA
4 Department of Microbiology and Immunology, Albert Einstein College of Medicine, Bronx, NY 10461, USA
Abstract
Autophagy is an evolutionarily conserved process in which intracellular macromolecules are degraded in a lysosomal-dependent manner. It is central to cellular energy homeostasis and to quality control of intracellular components. A decline in autophagic activity is associated with aging, and contributes to the development of various age-associated pathologies, including cancer. There is an ongoing need to develop chemotherapeutic agents to improve morbidity and mortality for those diagnosed with cancer, as well as to decrease the cost of cancer care. Autophagic programs are altered in cancer cells to support survival in genetically and metabolically unstable environments, making autophagy an attractive target for new chemotherapy. Antiretroviral drugs, which have dramatically increased the life- and health spans of people with human immunodeficiency virus (HIV) (PWH), have offered promise in the treatment of cancer. One mechanism underlying the antineoplastic effects of antiretroviral drugs is the alteration of cancer cell autophagy that can potentiate cell death. Antiretroviral drugs could be repurposed into the cancer chemotherapy arsenal. A more complete understanding of the impact of antiretroviral drugs on autophagy is essential for effective repurposing. This review summarizes our knowledge of the effects of antiretroviral drugs on autophagy as potential adjunctive chemotherapeutic agents, and highlights gaps to be addressed to reposition antiretroviral drugs into the antineoplastic arsenal successfully.
Keywords
- autophagy
- LC3
- p62
- age-related diseases
- cancer
- chemotherapy
- cell death
- antiretroviral drug
- HIV
- drug repositioning
Autophagy is a highly conserved proteolytic process that removes damaged and toxic macromolecules, protein aggregates, organelles, and infectious pathogens from the intracellular environment. There are three major types of autophagy: macroautophagy, microautophagy, and chaperone-mediated autophagy. Macroautophagy, henceforward termed autophagy, is the major type discussed in this review. In macroautophagy, intracellular substrates, or cargo, are incorporated inside a double membrane vesicle, called an autophagosome (APG), that fuses with a lysosome for cargo degradation, as detailed in Fig. 1. Briefly, an appropriate stimulus leads to phosphorylation of mTORC1, resulting in a series of phosphorylation events, protein complex recruitment, and phosphatidylinositol signaling that culminates in a new double membraned APG [1]. Cargo is incorporated inside the APG as its membrane is elongated. A fully formed APG fuses with a lysosome, and cargo inside is degraded [1]. The products of this degradation can be exported out of the lysosome to fulfill needs of the cell, such as production of energy substrates, macromolecules, and organelles. The protein microtubule-associated protein 1A/1B light chain 3B (LC3)-II, hereafter called LC3-II, associates with APG from nucleation to degradation, and is used commonly as a marker for assessing autophagy dynamics. APG can enclose around cytosolic cargo non-specifically, or cargo can be targeted to APG specifically, termed selective autophagy. Selective autophagy is mediated by autophagy receptors and adapters, such as p62 (Fig. 1). These contain protein binding domains that recognize degradation signals on cargo for cargo selection as well as LC3 interacting regions that facilitate cargo recruitment into forming APG. p62, which binds mostly ubiquitinated proteins, is degraded within the lysosome similarly to LC3-II, and as such, is another common marker used to assess autophagy.
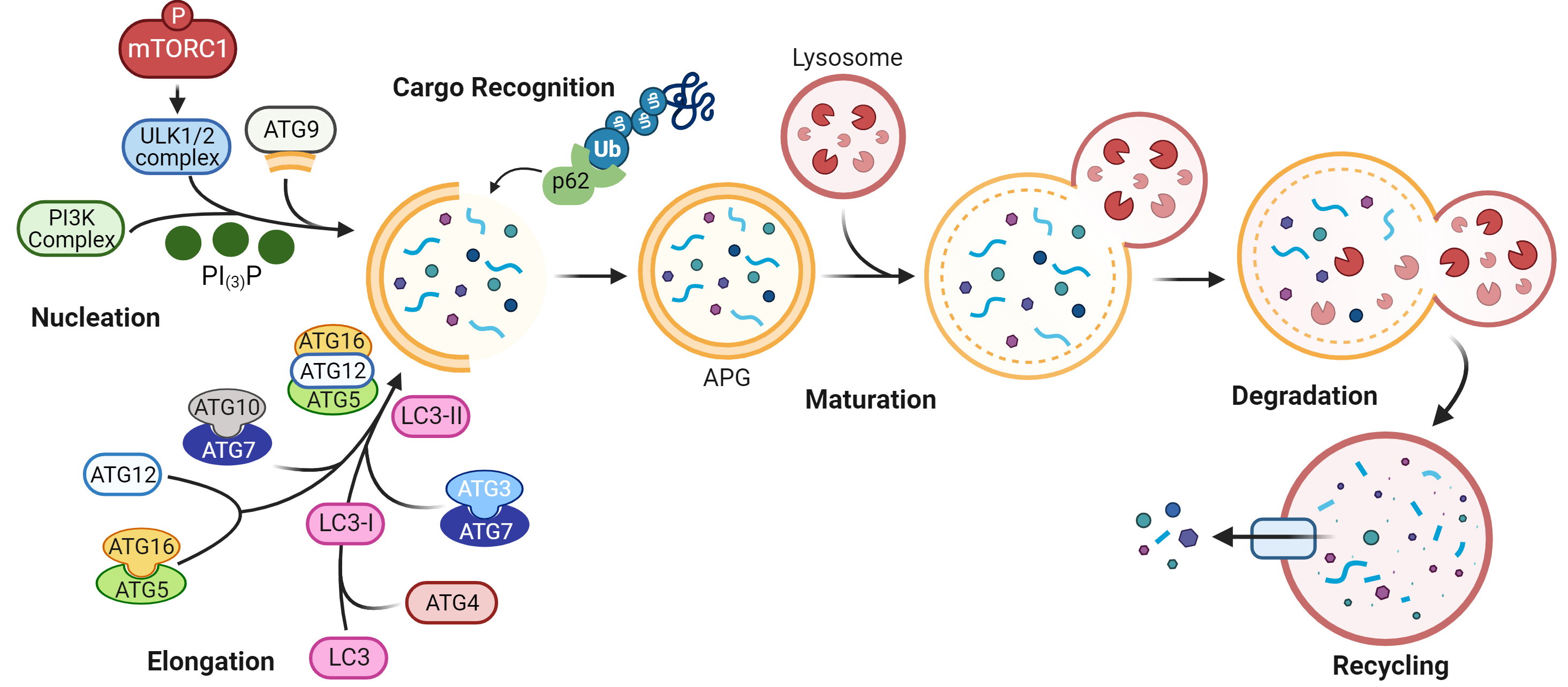 Fig. 1.
Fig. 1.Schematic of macroautophagy. More than 30 proteins are involved
in this dynamic process. Autophagy is initiated upon phosphorylation of mTORC1,
inactivating the complex with which it associates. This enables a pre-initiation
complex that includes UNC-51 like kinase family proteins (ULK1/2), to recruit a
class III phosphatidylinositol 3-kinase (PI3K) complex. Phosphatidylinositol‐3‐phosphate (PI
Autophagy is constitutive, and is also dynamic, increasing in response to homeostatic imbalance. While the first recognized purpose of autophagy was protection from nutrient-deprivation, it is now well-recognized to participate in many cell processes, including various modes of cell death. Extensive evidence from genetic and pharmacologic models across species demonstrates that alterations in autophagic activity can drive cell, tissue, and organism dysfunction, thus negatively impacting health- and lifespan, including contributing to aging and the pathogenesis of age-related diseases such as cancer. The relationship between autophagy and tumorigenesis is extraordinarily complex. Broadly, autophagy can both suppress tumorigenesis as well as promote tumor progression. Once a tumorigenic clone has emerged, autophagic processes support cancer cell metabolism, increase cancer cell resistance to inhospitable conditions such as hypoxia, reactive oxygen species (ROS), limited nutrients and trophic factors, and anticancer therapeutics, and can undermine the immune response to cancer cells [2, 3]. Altering autophagy in cancer cells can result in cancer cell death [4, 5, 6]. This makes modulation of autophagy an appealing target for chemotherapy.
Cancer is a leading cause of death in adults worldwide, second only to heart diseases [7]. Approximately 40% of U.S. men and women will be diagnosed with any type of cancer at some point in their lifetime [8], although advancing age is the single greatest risk factor for cancer development, with approximately 80% of all cancers being diagnosed in people aged 55 years or greater [8].
Cancer death rates in the U.S. decreased on average by 2.3% per year in men, and 1.9% per year in women from 2015 to 2019 [9]. There were almost 17 million cancer survivors in 2019, and this is projected to increase to 22.2 million by 2030 [8]. These rates are afforded by ever-improving cancer detection methods and treatment modalities. Despite these positive trends, there are 2 million new cancer diagnoses, and over 600,000 deaths projected in the U.S. alone for 2023 [8]. The national patient economic burden associated with cancer care in 2019 was over $21 billion [8], and as the population both ages and grows, and as new chemotherapeutic agents are developed, the cost of cancer care is expected to increase.
There is an ongoing need to develop therapies to improve morbidity and mortality, as well as cost of care, for people diagnosed with cancer and undergoing treatment. An understanding of the cellular processes that lead to cancer as well as the development of drug resistance underpins the advancement of novel and effective chemotherapeutic agents. Dysregulation of many cell processes leads to cancerous transformation. Autophagy is one such process, and altering autophagy in cancer cells can lead to cancer cell death [2, 3, 4, 5, 6]. Autophagy is therefore an attractive target for chemotherapy drug development. However, discovery and testing of novel agents, regardless of whether they target autophagy, requires significant time (years) and cost (billions).
Drug repurposing, or drug repositioning, is a strategy by which new uses for approved drugs are identified; these uses are distinct from that of the original purpose of the drug. There are many advantages of drug repurposing, including reduced time frames to authenticate a new indication, because the safety and formulations of a repurposed drug are already established. Reduced time frames translate to faster access to efficacious treatments, as well as to decreased research and development costs. A detailed review of these concepts and more can be found in [10, 11, 12].
Antiretroviral drugs, developed for treatment of infection with Human Immunodeficiency Virus (HIV), have shown promise in the treatment of a variety of cancers. Their antineoplastic effect is distinct from antiretroviral activity. There are several mechanisms that underlie their antitumor actions, including modulation of autophagy. Because of the importance of autophagy to cancer cell survival, the impacts of antiretroviral drugs on autophagy may be exploited to reposition them as adjunctive chemotherapeutic agents.
Amazing strides have been made in the management of HIV infection since the beginning of the HIV epidemic 40 years ago. Prior to antiretroviral therapy (ART), infection with HIV conferred high morbidity and swift mortality. As a result of the greater than 35 different antiretroviral drugs that have been developed to treat HIV (Table 1), people with HIV (PWH) now have significantly improved quality and quantity of life.
| Entry Inhibitors |
Fusion Inhibitor |
Capsid Inhibitor |
Rev. Transc. Inhibitors |
Integrase Inhibitors |
Protease Inhibitors | |
| Maraviroc | Enfuvirtide | Lenacapavir | NRTI |
Raltegravir | Saquinavir | |
| Ibalizumab | Zidovudine | Elvitegravir | Indinavir | |||
| Fostemsavir | Didanosine | Dolutegravir | Nelfinavir | |||
| Cenicriviroc |
Zalcitabine | Bictegravir | Ritonavir | |||
| Stavudine | Cabotegravir | Lopinavir | ||||
| Lamivudine | Amprenavir | |||||
| Abacavir | Atazanavir | |||||
| Tenofovir | Fosamprenavir | |||||
| Emtricitabine | Tipranavir | |||||
| Dapivirine | Darunavir | |||||
| NNRTI |
||||||
| Nevirapine | ||||||
| Delavirdine | ||||||
| Efavirenz | ||||||
| Etravirine | ||||||
| Rilpivirine | ||||||
| Doravirine | ||||||
| NRTTI |
||||||
| Islatravir |
||||||
PWH who begin and maintain an ART regimen have a rapid reduction in viral load, steady T cell recovery both in naïve and memory cell compartments, and eventually, sustained immune reconstitution. Associated with immune restoration is a decreased incidence of Acquired Immunodeficiency Syndrome (AIDS)-associated malignancies, particularly Kaposi’s Sarcoma, Non-Hodgkin’s lymphomas, and anogenital tumors, tumor regression, prolonged time to treatment failure, and longer survival. The major attribution to these findings is a return of immunologic control of the viruses, Human Herpesvirus-8, Epstein-Barr Virus, and Human Papilloma Virus, that are associated with these malignancies, respectively. However, several studies have shown that the anti-tumor effects of antiretroviral drugs are not well correlated with markers of immune reconstitution, suggesting there are additional anti-tumor mechanisms. Many antiretroviral drugs have pleiotropic anti-tumor effects. They block telomerase activity, inhibit the proteosome, Akt signaling, matrix metalloproteases, and angiogenesis, and may alter epitope processing to modulate antitumor immune responses, and impact autophagy, among other mechanisms. These impacts are important for optimizing antiretroviral repositioning, and have been reviewed elsewhere. To facilitate research of anti-cancer mechanisms, examples of these effects are in Supplementary Table 1, and we refer the reader to the following reviews: [13, 14, 15, 16, 17].
It is known that antiretroviral drugs impact autophagy [18]. However, knowledge of these impacts is limited in the context of cancer. A comprehensive understanding of the effects of antiretrovirals specifically on autophagy in the context of cancer will help guide their repositioning as antineoplastics. In this review, we focus on changes in autophagy mediated by antiretroviral drugs of different classes in various cell types to contribute to the arrest of cancer cell proliferation or to cause cell death (Table 2, Ref. [19, 20, 21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35, 36, 37, 38, 39, 40, 41, 42, 43, 44, 45, 46, 47, 48, 49, 50, 51, 52]). The ultimate goal is to improve not just the clinical outcomes for people with cancer by overcoming chemotherapy resistance or chemotherapy failure through autophagy modulation, but also decrease the financial burden of cancer care by repurposing antiretroviral drugs.
| Antiretroviral drug | Malignancy | Cell type/Model | Effect on autophagy |
Ref. | |
| Protease Inhibitors | |||||
| Nelfinavir | Non-small cell lung | H157, AnNCr-nu/nu mice | Increased autophagy | [19] | |
| Medullary Thyroid | TT, MZ-CRC-1 | Increased autophagy | [20] | ||
| Prostate | DU145, PC-3 | Increased autophagy | [21] | ||
| CLL |
Primary human CLL | Increased autophagy | [22] | ||
| Bladder, pancreas | Mouse embryonic fibroblasts | Increased autophagy | [23] | ||
| Multiple Myeloma | NCI-H929 | Decreased LC3/LAMP2 colocalization | [24] | ||
| Cervical | ME-180 | Increased LC3-II, p62 protein, puncta | [25] | ||
| Breast | MDA-MB-453 | Increased LC3-II | [26] | ||
| Breast | MDA-MB-453 | Increased LC3-II | [27] | ||
| Breast | MCF-7 | Increased LC3-I | [28] | ||
| +2,5-dimethyl-celecoxib | Breast | MDA-MB-231 | Increased LC3-II | [29] | |
| Non-small cell lung | A549 | Increased LC3B | [30] | ||
| +metformin | Cervical | SiHa, HeLa | Increased APG, decreased LC3-I/LC3-II | [31] | |
| Pediatric ALL |
SEM, Molm13 | No change in LC3-II | [32] | ||
| Non-small cell lung | H157, A549 | No change in LC3-II | [33] | ||
| Ovarian | CAOV3 | No change in LC3-II | [34] | ||
| +lenalid., dexameth. |
Multiple Myeloma | Primary Human PBMC |
Increased LC3-II | [35] | |
| Saquinavir | Ovarian | A2780, SKOV3, CAOV3 | APG on EM, increased LC3 puncta | [36] | |
| Ovarian | SKOV3 | Increased mTOR, BECN1 RNA, protein | [37] | ||
| Cervical | NIKS16 | Decreased LC3, p62 puncta | [38] | ||
| Lopinavir | Gastric | AGS | Decreased autophagy | [39] | |
| Lopinavir analog | Glioblastoma | LN-229, U-251 | Increased late-stage autolysosomes | [40] | |
| Melanoma | B16, B16F10, A375 | Increased late-stage autolysosomes | [41] | ||
| Indinavir analog | Lung | A549 | Increased LC3-II | [42] | |
| Ritonavir | Glioblastoma | pGBC |
Increased LC3-II, vacuoles | [43] | |
| Two Darunavir analogs | Hepatocellular | HepG2 | Increased LC3-II | [44] | |
| Rev. Transc. Inhibitors | |||||
| Efavirenz | Prostate | PC3, LNCaP, PNT2 | Increased autophagy | [45] | |
| +metformin, fluoxetine | Colon | HCT116 | Increased p62 | [46] | |
| Dapivirine | Glioblastoma | U87 | Increased LC3-II/I ratio | [47] | |
| Zalcitabine | Pancreas | PANC1, Capan2 | Increased autophagy | [48] | |
| Abacavir | Medulloblastoma | DAOY, MEB-Med8a, D283-Med | Increased “autophagy activity factor” | [49] | |
| Tenofovir | Breast | DMBA |
Increased LC3, Beclin-1 | [50] | |
| Etravirine | Ovarian | A2780, A2780-ADR | Increased autophagy | [51] | |
| Entry Inhibitors | |||||
| Maraviroc | Prostate | C4-2 injected BALB/c-nude mice | Decreased autophagy | [52] | |
Protease inhibitors (PI) (Table 1) were introduced in 1995, and remain an important component of modern ART regimens. PI inhibit the viral aspartyl protease, preventing the processing of viral polyproteins into functional forms that comprise mature virions. Concomitant with widespread use of PI, the incidence of AIDS-associated malignancies decreased [13, 17]. This led to great interest in evaluating PI for repurposing into antineoplastic drugs. There are numerous activities of PI that lead to anti-cancer effects (Supplementary Table 1), including modulating autophagy.
Among the PI, Nelfinavir (NFV) appears to be the most potent and broadly acting, with effects on angiogenesis, cell invasion, AKT signaling, and apoptosis [17]. NFV also causes significant endoplasmic reticulum (ER) stress [17]. Cells respond to ER stress, in part, by upregulating autophagy. This adaptive response is cytoprotective by reducing the toxicity of accumulated misfolded proteins and protein aggregates. Understanding a cell’s autophagic response to NFV increases knowledge of NFV effects that could be used for chemotherapy.
NFV appears to induce autophagy in several different cancer cell models.
Autophagy was induced in H157 cells, a human non-small cell lung cancer (NSCLC)
cell line, treated with 10 or 20 µM NFV. There was increased LC3-II
protein, increased LC3 puncta by fluorescence microscopy, and vacuoles were noted
on transmission electron microscopy (TEM) [19]. LC3-II protein was also increased
in H157 xenografts in mice treated with NFV [19], suggesting effects on autophagy
occur in vivo as well as in vitro. In another study, autophagy
was induced in TT and MZ-CRC-1 cells, lines used in models of medullary thyroid
cancer. LC3-II protein was increased and p62 protein was decreased after
treatment with 10 µM NFV [20]. Similarly, autophagy was induced in
the human prostate cancer cell lines DU145 and PC-3 [21]. These cells,
transfected to express GFP-LC3, had decreased GFP fluorescence by flow cytometry
after treatment with 20 µM NFV. This result was similar to
Rapamycin, an inducer of autophagic flux. In another study, NFV treatment of
primary human chronic lymphocytic leukemia (CLL) cells also induced autophagy.
Five µM and 10 µM NFV increased LC3 puncta by
immunofluorescence, and decreased p62 protein, similarly to Rapamycin [22].
Lastly, autophagy was induced in mouse embryonic fibroblasts (MEF) knocked out
for Tuberous Sclerosis Complex 2 (TSC2
NFV does not induce autophagy in every cancer cell type; rather, it may inhibit autophagy in some cell types. This was demonstrated in a study of multiple myeloma [24]. Bortezomib, a proteosome inhibitor used in the treatment of multiple myeloma, induced the colocalization of LC3 and Lysosome Associated Membrane Protein 2 (LAMP2), a lysosome marker, in NCI-H929 cells by immunofluorescent microscopy [24]. This effect was mitigated by concomitant treatment with 5 µM NFV. The authors conclude that autophagy was upregulated as a cytoprotective response to Bortezomib stress, and NFV impaired APG maturation, i.e., inhibited autophagy. In another study of ME-180 cells, a cervical cancer cell line, there was increased LC3-II and p62 protein by Western blotting, as well as increased LC3 and p62 puncta by immunofluorescence after 10 µM NFV treatment [25]. The authors conclude that autophagy was induced. While chloroquine (CQ) was used as a positive control for autophagy inhibition, flux analyses were not performed. An alternative interpretation is that with accumulation of both LC3-II and p62 as evidenced by two distinct techniques, NFV inhibited autophagy.
Multiple other studies attempt to link the potential chemotherapeutic efficacy of NFV to effects on autophagy but the true effects of NFV on autophagy are difficult to determine based on the studies performed. For example, in a study of four different breast cancer cells lines, T47D, MCF-7, MDA-MB-453, and MDA-MB-435 [26], 26 µM NFV increased LC3-II protein by Western blotting in MDA-MB-453 cells only. The same authors performed a separate study with MDA-MB-453 cells, and found a time-dependent increase in LC3-II protein with the same NFV concentration [27]. In a separate breast cancer model using the MCF-7 breast cancer cell line, treatment with increasing concentrations of NFV (max 6.67 µM) increased LC3-I protein by Western blotting [28]. In another breast cancer model, there was a time-dependent increase in LC3-II after treatment of MDA-MB-231 cells with 25 µM NFV plus 20 µM 2,5-Dimethyl-celecoxib, an ER stress inducer [29]. In a study of non-small cell lung cancer, different concentrations of NFV, 7.5 µM and 25 µM, were packaged into nanoparticles that were used to treat A549 cells, a lung cancer cell line. There was a dose-dependent increase in LC3B protein by Western blotting [30]. In a cervical cancer model using SiHa and HeLa cells, treatment with a combination of 4 µM NFV plus 10 µM Metformin, a widely used type II diabetes medication, led to an increase of APG by TEM, a decrease in the LC3-I/LC3-II ratio, and ATG3, Beclin-1, and ATG7 protein by Western blotting [31]. In a model of pediatric leukemia, neither 10 nor 20 µM NFV treatment of SEM and Molm13 cells, acute lymphocytic leukemia and acute myelocytic leukemia cell lines, respectively, changed LC3-II protein amount by Western blotting [32]. Similarly, no change in LC3-II was detected by Western blotting after 10 µM NFV treatment of H157 or A549 cells in a human lung cancer model [33], nor after 10 µM NFV treatment of CAOV3 cells in an ovarian cancer model [34]. Lastly, in a phase I/II clinical trial of 29 patients with refractory Multiple Myeloma, LC3-II was increased in Peripheral Blood Mononuclear Cells (PBMC) from the patients after 16 weeks of treatment with NFV plus both lenalidomide, an anti-angiogenic agent, and dexamethasone [35]. While the authors of these studies conclude that autophagy was induced in the models, flux assays were not performed to differentiate autophagy induction from inhibition of APG maturation, analysis of LC3-I as opposed to LC3-II was performed, analysis of LC3 without differentiating between LC3-I or LC3-II was performed, or NFV was not studied independently of other drugs. Some studies do have similar findings of increased LC3-II. It is therefore tempting to examine the totality of the data across studies and conclude that NFV consistently induces autophagy. However, it is uncertain that this conclusion can be drawn based on different NFV concentrations, cell lines, and techniques used to measure autophagy.
Saquinavir (SQV) was the first protease inhibitor approved for treatment of HIV,
and was important to the development of combination antiretroviral therapy. One
group examined the antineoplastic effect of SQV on ten different ovarian cancer
cells lines [36]. They found a dose- and time-dependent inhibition of cell growth
and apoptosis with SQV treatment in both chemo-sensitive (A2780) and
chemo-resistant (SKOV3 and CAOV3) lines. To characterize the mechanism underlying
cell death, they examined markers of ER stress, ATF6 and GRP78, and examined
autophagy by presence of APG by electron microscopy, and formation of GFP-LC3
puncta in GFP-LC3 transfected cells after treatment with 50 µM SQV.
They found increased ATF6 and GRP78 suggestive of ER stress, and presence of APG
by EM and an increase in GFP-LC3 puncta. The authors conclude that ER stress
induced autophagy leading to cell death, and therefore SQV may have clinical
application in the treatment of ovarian cancer. While cell death did occur at
lower SQV concentrations, ER stress and autophagy studies were performed with a
high dose of SQV. Ritonavir-boosted SQV serum C
In another study of ovarian cancer, the effects of SQV on cisplatin resistance,
which confers a poor prognosis, was determined [37]. SKOV3 cells were treated
with 10 or 20 µM SQV, and the concentration of cisplatin required to
cause 50% cell death, or IC
Infection with Human Papillomavirus strain 16 (HPV16) increases the risk for development of oropharyngeal and anogenital cancers. HIV PI were shown to decrease cervical dysplasia in cancer and precursor lesions in women with HIV. HPV16 has two oncoproteins, E6 and E7, whose constitutive expression is required to maintain HPV16-positive cancers. One group examined the effects of PI on HPV16 E6/E7 [38]. They used a model of pre-cancerous HPV16 infection: normal immortalized keratinocytes carrying 10-50 extrachromosomal copies of HPVP16 (NIKS16). NIKS16 cells form organoid rafts similar to human skin and maintain infection with HPV16 throughout cell differentiation stages. NIKS16 rafts treated with 5 µM SQV were atrophic, and had decreased cell proliferation relative to control [38]. The authors could not detect E6/E7 by immunofluorescence of the rafts, but they did find MCM7, a protein expressed in the presence of E7. SQV decreased the number of MCM7-positive cells in the rafts, suggesting SQV decreased E7 [38]. E6/E7 inhibit autophagosome fusion with the lysosome, an important step for HPV16 cancer development. The authors examined LC3 and p62 by immunofluorescence of the rafts, and found a small decrease of LC3 and a significant decrease of p62. They conclude that SQV reversed E6/E7 autophagy inhibition. While they did not determine whether the shift in autophagy dynamics by SQV contributed to decreased E6/E7 oncoprotein level, reversal of HPV16 autophagy inhibition is likely important for decreasing HPV16 oncogenic potential.
Lopinavir (LPV) is a unique PI in that it was not formulated for use
independently from Ritonavir (RTV, discussed below) because it has poor
bioavailability. One group identified Lopinavir as a potential chemotherapeutic
agent through its effects on autophagy in a model of metastatic gastric cancer
[39]. When non-cancerous cells detach from the extracellular matrix (ECM), they
undergo a type of programmed cell death termed anoikis. Cancer cells detaching
from the ECM and intravasating into the vasculature must be anoikis resistant to
survive and establish metastatic disease. In certain prostate and liver cancer
models, autophagy was shown to promote metastasis by inhibiting anoikis. ATG4B,
which is essential for the lipidation of LC3 (Fig. 1), is expressed highly in
various cancer types. Inhibiting ATG4B may induce anoikis, thus inhibiting
metastatic processes, but ATG4B inhibitors are not currently clinically viable
options. Using AGS cells, a human gastric cancer cell line, the authors
demonstrate that high levels of ELAVL1, an RNA-binding protein, suppress
circSPECC1 levels, a specific sequence of circular RNA, which binds to
and negatively regulates ATG4B [39]. The effect of ELAVL1 on circSPECC1
led to increased ATG4B levels, thereby increasing autophagy as shown by LC3 and
p62 Western blotting and fluorescence microscopy of an mCherry-GFP-LC3 reporter.
This interaction conferred anoikis resistance to the cells. Molecular docking and
virtual screening studies identified LPV as a molecule that could disrupt the
interaction between ELAVL1 and circSPECC1. After treatment with 25
µM or 50 µM LPV, circSPECC1 levels were
restored, and correspondingly, ATG4B levels and autophagy were decreased. LPV
increased anoikis in vitro, and decreased the incidence of pulmonary
metastasis after tail vein injection of GC in a nude mouse model [39]. This study
introduces LPV as a viable chemotherapeutic agent for metastatic gastric cancer.
One limitation of the study is the concentration of LPV used. The serum C
Having determined that a nitric oxide (NO)-releasing derivative of SQV had
anti-cancer properties, one group generated a similar molecule from Lopinavir
(LPV-NO) to examine its efficacy in glioblastoma and melanoma models in two
separate studies [40, 41]. LN-229 and U-251 cells, human glioblastoma cell lines
[40], and B16, B16F, mouse melanoma cells lines, and A375 cells, a human melanoma
cell line [41], were treated with a range of LPV and LPV-NO concentrations. In
both models, the IC
Indinavir has been suggested to block angiogenesis and tumor cell invasion, but
not inhibit proliferation or induce apoptosis in cancer cells in vitro [58]. To improve Indinavir’s antineoplastic properties, one group developed an
analog, CH05-10 [42]. Human lung cancer cells, A549, were treated with CH05-10,
and apoptosis, ER stress, and autophagy were assessed. CH05-10 decreased cell
viability, inhibited proliferation, caused G
Originally used as treatment in PWH to inhibit HIV protease, Ritonavir (RTV) is
now used in current regimens to increase, or boost serum levels of other protease
inhibitors. This is because RTV is a potent inhibitor of Cytochrome P450 (CYP3A).
One group examined its possible efficacy in treating glioblastoma [43].
Patient-derived glioblastoma cells (pGBM) were treated with 30 µM
RTV. The mean serum C
Darunavir is the first-line preferred protease inhibitor in ART regimens [60].
As for all PI in modern regimens, Darunavir is boosted with either low-dose RTV,
or with cobicistat, a non-antiretroviral inhibitor of Cytochrome P450 (CYP3A).
Precursor analogs of Darunavir, RDD-19 and RDD-142, which had been developed
originally for potential use as antiretrovirals, were tested for efficacy as
antitumor agents against hepatocellular carcinoma [44]. While Darunavir itself
did not cause any cytotoxicity, RDD-19 and RDD-142 reduced viability and induced
apoptosis in HepG2 cells, a hepatocellular carcinoma cell line, in a
dose-dependent manner. These findings were associated with a dose-dependent
increase of ER stress markers as well as inhibition of the proteosome. They also
performed Western blotting for LC3 and Beclin-1, and found significantly
increased amounts of LC3-II relative to control, but no change in Beclin-1.
Additional studies were not performed to further evaluate the effects of the
compounds on autophagy. The authors conclude that while the IC
The first FDA approved medication to treat HIV infection was a reverse transcriptase (RT) inhibitor (RTI). While RTI inhibit viral reverse transcriptase with different mechanisms, they all cause chain termination during viral replication (Table 1). These remain the mainstay of HIV treatment [60]. Similar to PI, the incidence of cancers in PWH decreased with the institution of RTI in HIV treatment regimens [61, 62]. As was also found for PI, RTI may mediate chemotherapeutic effects by several mechanisms (Supplementary Table 1), including autophagy modulation.
Efavirenz (EFV) is a non-nucleoside reverse transcriptase inhibitor that was an important component of ART regimens for almost two decades. Many studies showed that EFV can inhibit cell proliferation and promote cell death in various carcinoma and sarcoma cell lines [63, 64, 65, 66, 67, 68, 69], and a small clinical trial showed EFV may have some efficacy in treating metastatic castration-resistant prostate cancer [70]. In a recent publication, 20 µM EFV and 20 µM SPV122.2, a stereoisomer of EFV, induced significant DNA damage, chromatin reorganization, lamin B1 breakdown with micronuclei formation, and ultimately, decreased proliferation and significant apoptosis of PC3 cells, a prostate carcinoma cell line [45]. As LC3-II plays a role in lamin B1 degradation [71, 72], the authors examined whether autophagy contributes to EFV/SPV122.2 induced nuclear changes. They saw increased LC3 positive puncta by immunofluorescent microscopy, as well as increased LC3-II, p62, and ATG7 by Western blotting. While these studies do not exclude the possibility that APG maturation was inhibited, pharmacologic inhibition of autophagy with 3-MA mitigated the effects of EFV and SPV122.2 on nuclear architecture, and restored viability of the malignant cells, suggesting EFV and SPV122.2 induced autophagy which contributed to the effects on the nucleus [45]. Daily dosing of EFV leads to an average serum level range between 3.17 and 12.67 µM [73]. Due to major interpersonal EFV pharmacokinetic variability, 20–40% of people can have plasma levels that reach as high as 30–50 µM [74, 75, 76]. Their data suggest that activating autophagy by EFV may have chemotherapeutic benefit in prostate cancer.
In a study of colon cancer, EFV was used in combination with Metformin and Fluoxetine, a common anti-depressant, to generate excess ROS as a means to induce cancer cell death [46]. There was a significant increase in ROS production in HCT116 cells, a colon cancer cell line, after treatment with 1.5 µM EFV plus Metformin and Fluoxetine relative to any of the three drugs alone. This combination also reduced mitochondrial membrane potential, induced cell cycle arrest, and caused significant cell death. To identify the underlying mechanisms of toxicity, Western blotting was performed for markers of DNA damage, apoptosis, necroptosis, and of p62 to assess autophagy. Most markers were increased relative to control, including p62. However, the increase in p62 was small and not statistically significant. They conclude that this drug combination had profound antitumor effects due to ROS induced DNA damage, and upregulation of apoptosis, necroptosis and autophagy. However, an increase in p62 suggests autophagy may be inhibited. In addition, excess p62 stabilizes nuclear factor erythroid 2-related factor 2 (NRF2) [77], a major transcription factor involved in the defense against oxidative and electrophilic stresses. SQSTM1, the gene for p62, is a target of NRF2. [78]. The increased p62 may be a result of a positive feed-back loop established with activation of the NRF2 anti-oxidant system rather than resulting from a change in autophagy. As there were no direct studies of autophagy, it remains unclear what effect EFV or the 3-drug combination may have on autophagy in this model.
Dapivirine is a unique antiretroviral, developed as an active ingredient in
microbicides and vaginal rings to prevent HIV acquisition in women. One study
showed Dapivirine to have potential therapeutic benefit in a glioblastoma model
[47]. U87 cells, a human glioblastoma cell line, were treated with a range of
Dapivirine concentrations, and cell viability and apoptosis were examined.
Dapivirine decreased cell proliferation, and increased apoptosis with an
IC
Zalcitabine (ddC) did not achieve great success in the treatment of HIV as it
was inconveniently dosed, less potent than other NRTI, and had overlapping
toxicities with RTI, limiting its ability to be used in combination with other
RTI. One study examined the effect of ddC on mitochondrial function and autophagy
in a pancreatic cancer model. Treatment of two human pancreatic ductal
adenocarcinoma (PDAC) cell lines, PANC1 and Capan2, and of ex vivo
primary human cells with 20 µM ddC led to significant mitochondrial
dysfunction with reduced mtDNA copy number, increased ROS, decreased oxygen
consumption, and reduced ATP production. ddC also induced cell death [48]. mtDNA
replication requires Transcriptional Factor A, Mitochondrial (TFAM), which can
also activate the DNA damage sensing pathway. ddC treatment reduced TFAM, and
increased cytosolic mtDNA, ultimately leading to increased phosphorylation of
Stimulator of Interferon Response CGAMP Interactor 1 (STING1) [48], a cytosolic
DNA sensor linked to the innate immune response and autophagy. The authors used a
novel autophagic flux reporter, GFP-LC3-RFP-LC3
The guanosine analog Abacavir (ABC) was once a preferred backbone agent for PWH,
and is still a first-line preferred and alternative agent for children with HIV
[60]. One study examined ABC as a potential chemotherapeutic agent for
medulloblastoma (MB) [49], a common pediatric brain tumor with a low 5-year
survival rate. MB is thought to arise, in part, from abnormal neuronal or glial
cell differentiation. ABC had been previously shown to induce cell
differentiation [83]. The authors used ABC in an attempt to induce cell
differentiation and reduce cancer-forming capacity. Three human medulloblastoma
cell lines, DAOY, MEB-Med8a, and D238-Med, were treated with 250 µM,
40 µM, and 240 µM ABC, respectively, after which they
found significantly more double-strand DNA breaks, reduced cell viability,
increased apoptosis, and decreased clonogenic survival. This was associated with
a significant increase in the “autophagy activity factor (AAF)” which they
derived from the mean fluorescence intensities of treated and control cells using
a commercially available dye kit for measuring autophagic flux. The authors
conclude that the increased AAF is evidence of increased autophagy [49], but it
is not clear if the appropriate controls were used in order to analyze autophagic
flux properly. In addition, the concentrations of ABC used represent an
inhibitory concentration by which 90% of clonogenic cells were killed
(IC
Tenofovir is a NRTI (Table 1) and a major component of contemporary ART, being one of two NRTI recommended in first- and second line regimens. It is also a component of Pre-exposure Prophylaxis (PrEP) as well as Pose-Exposure Prophylaxis (PEP), preventing HIV infection in individuals at risk for HIV acquisition. It was recently examined as a possible chemotherapeutic agent for breast cancer [50]. Breast cancer was induced in Sprague Dawley rats with 7,12-dimethylbenz(a) anthracene (DMBA) given twice weekly for four weeks. During this four-week period, rats were also given 25 mg/kg Tenofovir (TF25), 50 mg/kg Tenofovir (TF50), or Doxorubicin (DOX)+TF50. TF25 and TF50 conferred a survival advantage, and mammary gland tissue weight was significantly decreased after all three treatments. Markers of oxidative stress, cell proliferation, and apoptosis were all decreased after the three treatments. Enzyme-linked immunosorbent assay (ELISA) for Beclin-1 and LC3 were performed on supernatants obtained from homogenized breast tissue, and the three treatments decreased Beclin-1, and increased LC3 relative to DMBA alone. The authors conclude that TF offers therapeutic benefit, even more so when combined with DOX, as a result of decreasing breast tissue weight, decreasing oxidative stress, apoptosis and cell proliferation, as well as increasing autophagy [50]. However, the ELISA measured total LC3, and no other autophagy analysis was performed. Tenofovir may be beneficial in the treatment of breast cancer, but the effects on autophagy are not fully characterized.
Etravirine (ETR) is a NNRTI (Table 1) that has a flexible conformation, enabling
it to bind RT effectively even in the presence of RT mutations. At the time of
its FDA approval, ETR offered HIV treatment efficacy for those in whom resistance
to other NNRTI had developed. ETR was shown to have anti-cancer effects in an
ovarian cancer model, possibly by impacting autophagic degradation of Anterior
gradient protein 2 homolog (AGR2) [51]. AGR2, thought to play a role in protein
folding, is expressed highly in many cancers, promoting angiogenesis, enhancing
metastasis and tumor progression. Its expression is associated with
chemoresistance and a poor prognosis. The authors treated two ovarian cancer cell
lines, A2780 and A2780-ADR, with ETR ranging from 1.25 µM to 10
µM, and performed LC3-II Western blotting. They found a
dose-dependent increase in LC3-II, corresponding to a dose-dependent decrease of
AGR2. By immunofluorescence, LC3 was increased after 10 µM ETR and
this also corresponded with decreased AGR2. When autophagy was inhibited with
wortmannin, a PI3K inhibitor, CQ, or ATG7 siRNA knockdown, the effects of ETR in
AGR2 were reversed, suggesting that ETR decreased AGR2 level by
autophagy-mediated degradation. ETR also decreased cell proliferation, viability
and migration in vitro, and when combined with Paclitaxel, a common
chemotherapy agent, decreased cancer progression and metastasis in a xenograft
mouse model. The authors conclude that ETR is a promising chemotherapeutic agent
for its ability to upregulate autophagic degradation of AGR2 [51]. Of note, the
median serum C
Entry Inhibitors (Table 1) were important to the evolution of successful ART regimens, as they offered a novel mechanism of action against HIV in treatment-experienced patients in whom resistance to PI and RTI had developed. Members of this class of medications have diverse mechanisms of actions, all preventing HIV from entering cells. They too have shown potential to act as chemotherapeutic agents (Supplementary Table 1). One group studied how endothelial cells contribute to prostate cancer metastasis, incorporating Maraviroc into their animal model [52]. Maraviroc is a C-C chemokine receptor type 5 (CCR5) antagonist, blocking HIV gp120 from binding CCR5, one of two cell surface co-receptors needed for HIV to enter a cell. Using a coculture of C4-2 cells, a metastatic prostate cancer cell line, and human umbilical vein endothelial cells (HUVEC), the authors show that HUVEC enhanced prostate cancer cell invasion by decreasing androgen receptor (AR) activity in a C-C chemokine ligand type 5 (CCL5)- dependent manner. AR signaling inhibits autophagy, which degrades paxillin, an important component of focal adhesion sites where cells anchor their actin cytoskeletons to the ECM. Paxillin accumulates during autophagy inhibition, which can increase cell mobility. In their model, CCL5 repression of AR led to increased autophagy (as evidenced by increased LC3-II and decreased p62 on Western blotting), reduced paxillin, and subsequently increased cell migration. When either a CCL5 neutralization antibody or CQ was added to the co-culture, autophagy was inhibited (as shown by accumulation of LC3-II on Western blotting), paxillin was increased, and cell mobilization was decreased. In their mouse model in which prostate cancer was induced by injection of C4-2 plus HUVEC cells, mice treated with either Maraviroc or CQ had decreased metastatic foci compared to control mice, and mice treated with both Maraviroc and CQ had no metastatic disease. This supported their conclusion that CCL5, CCLR5 and/or autophagy are potential drug targets to inhibit endothelial cell promotion of prostate cancer metastases [52].
Autophagy is required for maintaining cell, and by extension, tissue and organism homeostasis. A decline in or dysregulation of autophagic activity contributes to aging, as well as to age-dependent pathologies, including cancer. Tumorigenesis and autophagy have a complex and incompletely understood relationship, but their interrelationship affords great opportunity for chemotherapy development. Repurposing antiretroviral drugs would be a more time efficient and less costly mechanism to bring “new” chemotherapeutic agents to market. The positive effects of antiretroviral drugs on cancer cells in in vitro and pre-clinical studies have led to many clinical trials of antiretrovirals to reposition them as adjunctive chemotherapeutic agents in people with a variety of malignancies (Supplementary Table 2). The impacts of antiretroviral drugs on autophagy likely contribute to their anti-cancer properties.
The studies reviewed highlight that antiretroviral drugs have varied,
cell-specific effects on autophagy (Table 2). Effects on autophagy are not
antiretroviral drug class specific, nor is there one specific effect by
individual drugs. NFV best underscores this point, as NFV can induce [19, 20, 21, 22, 23],
inhibit [24, 25], or have no effect [32, 33, 34] on autophagy depending on the model
examined. The choice of antiretroviral drug to be used as part of a chemotherapy
regimen will need to be tailored to the specific cancer depending on the desired
effect on autophagy of the antiretroviral drug. To provide clinical benefit
through autophagy modulation, the effect on autophagy would need to potentiate
cytotoxicity or cell death. While many studies demonstrate increased cell death
after antiretroviral drug treatment [19, 20, 21, 23, 24, 25, 36, 42, 45, 47, 48, 49], not all
studies link changes in autophagy to cell death [25, 26, 27, 47, 49], nor did all
antiretroviral drugs induce cell death despite possibly altering autophagy [20, 22, 24, 28, 37]. For example, SQV increased the IC
There are several potential mechanisms underlying the effects of antiretroviral drugs on autophagy. ER stress is a well-known inducer of autophagy. Autophagy is upregulated to mitigate damage from toxic, misfolded proteins and protein aggregates. A few studies show increased markers of ER stress with clear evidence of autophagy induction after NFV treatment [19, 22, 23]. Other studies demonstrate presence of ER stress after antiretroviral treatment [26, 27, 28, 29, 32, 36, 42, 43, 44, 50], and while data may suggest autophagy was induced in these models, it is unclear whether autophagy is consistently induced with ER stress [86]. The specific molecular factors involved in the ER stress response and downstream effects on autophagy and apoptosis in the presence of antiretroviral drugs are important to identify for possible chemotherapeutic potential. Other mechanisms by which antiretroviral drugs may alter autophagy include mitochondrial damage and dysfunction [20, 46, 48], DNA damage and nuclear structural abnormalities [23, 45, 46, 49], oxidative stress [26, 31, 50], and possibly calpain inhibition [24].
There are two major limitations to many studies reviewed herein. First, autophagic flux assays or multiple complimentary autophagy assays were frequently not performed to assess autophagy dynamics. While there are no fixed criteria for determining the status of autophagy in every experimental context, there are regularly updated guidelines that detail acceptable assays as well provide as an extensive framework with which to interpret experimental results [79]. Autophagy is a highly dynamic, multi-step process that can be positively and/or negatively impacted at several steps. It is important to perform techniques that allow for the distinction of changes in specific steps in autophagy. For example, the accumulation of GFP-LC3 puncta by fluorescent microscopy represents either increased APG formation, or inhibition of APG maturation. The experimental result is the same, yet the step in autophagy that is affected is different. The effect of autophagy on cell death and ultimately on human diseases mandates the use of precise tools such as in [39, 48] to measure and correctly interpret autophagic flux changes in response to stimuli, especially in the context of repositioning antiretroviral drugs as cancer treatment. Second, the concentrations of antiretroviral drugs used in many studies exceeds achievable human serum or CSF concentrations, at least with the current antiretroviral formulations. Some antiretroviral drugs have considerable variability in steady-state serum concentrations both inter- and intra-individually, however, use of high drug concentrations in in vitro studies may lead to detection of artifact. Drugs should be used in concentrations reflective of serum or CSF levels, depending on the tissue of interest, to model human pharmacokinetics and pharmacodynamics better. In addition, an antiretroviral drug progressing through the chemotherapy pipeline based on data generated from concentrations exceeding current formulations would have to undergo reformulation and new clinical safety trials.
A single molecular pathway does not contribute solely to either aging or to cancer biology. There are several hallmarks of each, many of which overlap, including autophagy [2]. Decreased autophagic activity is highly correlated with both aging and tumor progression. As autophagy contributes to many cell processes including mitochondrial and other organelle homeostasis, ROS neutralization, degradation of toxic aggregates, and metabolism, age-associated decline in autophagy likely dysregulates these processes too, thereby contributing to cancer development. It is not known to what degree these other cell processes are impacted by autophagy in the context of aging, nor the extent of their contributions to tumorigenesis. Also, age-related changes to autophagy may promote cancer development in both cell-autonomous and non-autonomous manners. Autophagy likely shapes the microenvironment of tissue as much as it does the intracellular environment of cells. There is currently little understanding of the impacts of non-autonomous age-related autophagy changes to cancer cells and the development or progression thereof. This review has addressed only macroautophagy, but the other forms of autophagy, such as chaperone-mediated autophagy, also decrease with advancing age [87, 88]. As knowledge related to macroautophagy in the context of aging and cancer increases, understanding of other types of autophagy and how they intersect with macroautophagy, cancer, and aging should also expand. Research opportunities, and thus cancer treatment opportunities, will continue to increase when effects of antiretroviral drugs are examined within all of these contexts. From the clinical and drug development perspective, antiretrovirals provide a research advantage over novel molecules because the safety profiles of antiretroviral drugs in aging persons have been determined.
As the population grows and ages, the need for new chemotherapy agents will continue to increase. Repurposing approved drugs that could effect cancer cell death or increase chemosensitivity by autophagy modulation is both time-saving and cost-effective. A more complete understanding of the effects of antiretroviral drugs on autophagy in cancer models will increase the opportunity to reposition antiretrovirals into the antineoplastic arsenal, positively impacting the morbidity and mortality, as well as the financial burden of care for people with cancer.
LC, JMB and JWB conceptualized the review. LC performed the literature searches. LC, JMB, and GM analyzed and interpretated the reviewed studies. The original draft was prepared by LC, JMB, and GM. All authors contributed to editorial changes of the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Fig. 1 was designed with the assistance of Biorender.com.
Support is provided by NIH grants T32GM00728846, F30DA053118 (J.M.B.), K08MH130210 (L.C.), and R01DA044584, R01DA048609, R01DA056261, and R01DA041931 (J.W.B.).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.