1. Introduction
The endoplasmic reticulum (ER) is a complex membrane system that serves as an
essential site for the synthesis, folding, and posttranslational modification of
secreted and membrane proteins [1]. Imbalance in ER caused by certain
physiological or pathological factors leads to the accumulation of a large number
of unfolded or misfolded proteins in cells, resulting in ER stress [2]. To
restore ER homeostasis and reduce the amount of faulty or unfolded proteins in
the reticulum, the unfolded protein response (UPR) is activated [3]. The UPR is
regulated by three proteins, including activating transcription factor 6 (ATF6),
protein kinase R-like ER kinase (PERK) and inositol requiring enzyme 1 (IRE1)
[4]. These three ER transmembrane proteins reversibly bind to the endoplasmic
reticulum partner binding immunoglobulin protein (BiP)/heat shock protein family
A member 5 (HSPA5)/glucose-regulated protein 78 (GRP78) to inhibit activity under
normal physiological conditions; however, HSPA5 dissociates from this protein
when the UPR occurs. These three proteins are activated and initiate a response,
thus regulating transcription and translation, activating protein degradation
pathways and reducing the accumulation of faulty proteins in the ER to restore ER
homeostasis [5]. More intense endoplasmic reticulum stress (ERS) occurs when the
three sensors of the UPR are insufficient to restore unfolded proteins in the ER
to a normal state [6].
As a new mode of death, ferroptosis differs from other programmed types of
death, such as apoptosis, necrosis and autophagy, and its occurrence depends on
the accumulation of iron in cells. Its biochemical, morphological and genetic
characteristics are significantly different from those of other types of
programmed cell death [7, 8]. Morphology is mainly manifested by mitochondrial
agglutination or swelling, increased membrane density, a reduced or disappeared
ridge and outer membrane rupture [9]. When glutathione (GSH) is exhausted in the
cell, the activity of GPX4 is reduced, the GPX4 catalytic reduction reaction
cannot metabolize lipid peroxides and a large amount of reactive oxygen species
(ROS) are generated to induce ferroptosis [10]. Intracellular iron accumulation,
lipid peroxidation and an abnormal antioxidant System Xc- are the key factors
involved in ferroptosis. Ferroptosis is induced by ROS, which are produced by
various sources, such as iron metabolism, the mitochondrial electron transport
chain and the nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (NOX)
protein family.
An increasing number of studies have shown that ferroptosis and ERS are mutually
regulated and that ferroptosis and ERS are involved in the occurrence of various
liver diseases. In this review, the authors summarize the mechanisms by which ERS
interacts with ferroptosis and the roles of these mechanisms in common liver
diseases.
2. Mechanism of ERS
2.1 PERK Pathway
PERK is a kinase of phosphorylated eukaryotic initiation factor 2
(eIF2) and nuclear factor erythroid 2-related factor (Nrf2). When ERS
occurs, PERKs separate from HSPA5s and are activated. Activated PERK
phosphorylates eIF2 and Nrf2. The activation of eIF2 can
cause most protein translation to decrease, and the protein load in the
endoplasmic reticulum is also reduced [1]. The expression of activating
transcription factor 4 (ATF4) is upregulated when the level of phosphorylated
eIF2 reaches a certain level [11]. ATF4 is a major regulator of the
stress response and regulates the expression of genes involved in amino acid
metabolism and redox and protein homeostasis at the transcriptional level. ATF4
enhances mitochondrial respiration and promotes oxidative metabolism by
activating the comprehensive stress-promoting respiratory supercomplex [12]. The
activation of Nrf2 can promote the expression of heme oxygenase-1 (HO-1), ATF3
activating transcription factor 3 (ATF3) and solute carrier family 7 member 11
(SLC7A11), which are other target groups, as well as upregulate ROS and induce
ERS [13].
2.2 The IRE1 Pathway
IRE1 is a type I transmembrane protein that recognizes unfolded
proteins and misfolded proteins in the ER via its N-terminal peptide-binding
domain. IRE1 and GRP78 bind to the downstream X-box-binding protein 1
(XBP1) mRNA transcript after it is bound to a misfolded protein. Spliced X-box
binding protein 1 (XBP1s) is a transcription factor responsible for regulating
the transcription of genes associated with the ER quality control and ERAD
pathways. In addition, regulated IRE1-dependent decay (RIDD), which
degrades or translates mRNA, reduces the protein load in the ER and restores ER
homeostasis [14, 15, 16].
2.3 The ATF6 Pathway
ATF6 is a type II transmembrane protein with two subtypes known as
ATF6 and ATF6. ATF6 is separated from GRP78 and
disrupted by the site-1protease (S1P) and site-2 protease (S2P) when ERS occurs.
The amino-terminal cytoplasmic fragment (ATF6f) is released and translocated to
the nucleus to participate in the expression of proteins, thereby increasing the
folding ability of the ER and restoring ER homeostasis [17, 18]. XBP1 is a target
molecule regulated by ATF6. ATF6 can also upregulate the transcription of XBP1
mRNA, increase the level of XBP1s and activate ERS [19, 20].
3. Mechanism of Ferroptosis
3.1 Iron Accumulation
Iron is an important trace metal in the body and is involved in biosynthesis,
oxygen transport and the respiratory chain. Cellular iron production is mediated
by transferrin (Tf) and transferrin receptor 1 (TfR1). Intracellular iron can be
transported outside of the cell by the ferroportin (FPN) [21]. Iron overload is
an important cause of ferroptosis. When a large amount of Fe accumulates
in the cell, it induces the Fenton reaction, results in the production of a large
number of ROS, hydroxyl groups and free radicals and activates iron-containing
enzymes. Many known organic compounds, such as carboxylic acids, alcohols and
esters, can be peroxidized by the Fenton reaction and produce corresponding
peroxide products, which ultimately lead to ferroptosis [22, 23, 24].
The iron uptake pathway mediated by Tf and TfR1 is involved in ferroptosis. The
inhibition of the expression of TfR1 can effectively reduce the ferroptosis
induced by erastin. The addition of fe-containing Tf or ferric ammonium citrate
to the cell medium resulted in an increase in the Fe concentration and
induced ferroptosis [25, 26]. Nedd4-like E3 ubiquitin protein ligase (NEDD4L) can
increase the degradation of lactotransferrin (LTF), inhibit intracellular iron
accumulation and reduce ferroptosis [27]. The initial signal of ferroptosis has
not yet been determined; however, ferroptosis must be involved, and the degree of
iron accumulation in the cell directly determines the course of ferroptosis.
3.2 Lipid Peroxidation
The initiation and execution of ferroptosis are regulated by lipids.
Polyunsaturated fatty acids (PUFAs) are essential substrates for fat metabolism
and are catalyzed by coenzyme A (CoA) and 4acyl-CoA synthetase long-chain family
member 4 (Acsl4) to produce polyunsaturated fatty acid membrane phospholipids
(PUFA-PLs) [28]. PUFA-Pls is catalyzed by lipoxygenase (LOX) or cytochrome P450
oxidoreductase (POR) to produce lipid peroxides (LPOs) [8]. LPO can activate
protein kinase II (PKCII) to promote ACSL4 activation and
oxidize additional PUFAs to produce additional LPO [29]. LPO can produce lipids
to freely form lipid ROS through lipid oxidation reactions [30]. Furthermore,
malondialdehyde (MDA) and 4-hydroxynonenal (4-HNE) levels are biomarkers of
ferroptosis [31].
3.3 Antioxidant System Xc- Abnormalities
The oxidation and antioxidant reactions in the cell are in balance under
physiological conditions. When the redox balance system is disrupted, this
scenario causes the accumulation of free radicals and triggers ferroptosis.
System Xc- is a glutamate/cystine reverse transporter. Cystine, glutamate and
glycine are catalyzed by glutamate cysteine ligase (GCL) and glutathione synthase
(GS) to become glutathione (GSH). GSH is an important intracellular antioxidant
that can alleviate oxidative stress by scavenging free radicals in the body [32].
Additionally, GPX4 is a ferroptosis inhibitor that uses GSH as a coenzyme, which
can reduce LPO to nontoxic lipid alcohols and reduce ROS and free radical
accumulation [33]. Ferroptosis can be induced by reduced activity or insufficient
levels of GPX4 [34].
4. The Interaction between Ferroptosis and ERS
4.1 Ferroptosis-Inducing ERS
Ferroptosis and ERS play important roles in cell death. In recent years, studies
have shown that ferroptosis can be accompanied by ERS, which may be closely
related to ROS produced during ferroptosis. ROS can interfere with the activity
of protein folding enzymes, thus resulting in ER protein peroxidation,
dysfunction of ER molecular chaperones and the accumulation of unfolded proteins
in the ER. Eventually, ERS is induced. Additionally, ROS binds to the inositol
receptor 1,4,5-triphosphate (IP3R) in the ER, resulting in high intracellular
Ca accumulation and ultimately ERS [35, 36]. Furthermore, organic extracts
of fine particulate matter (PM2.5) can produce a large amount of ROS by binding
to the aromatic hydrocarbon receptor (AHR) and significantly increase the
expression level of CHOP. CHOP can simultaneously activate the three receptors of
ERS, PERK, IRE1, and ATF6, thereby inducing strong ERS [37]. Iron is also
involved in the occurrence of ERS, and iron activates the
PERK-eIF2-ATF4-CHOP pathway, thereby inducing p53 to upregulate the
expression of apoptosis regulator (PUMA). Ferroptosis inducers that induce
ferroptosis in cells also activate ERS. Ferroptosis inducers can inhibit
cystine-glutamate exchange in cells, increase the expression of ERS-related
protein ChaC glutathione-specific gamma-glutamylcyclotransferase 1 (CHAC1), and
activate ERS. CHAC1 is involved in the ATF4-CHOP pathway in ERS [38]. Artesunate
can be used as an inducer of ferroptosis for the treatment of Burkitt lymphoma
(BL), but artesunate not only induces ferroptosis in BL cells but also induces a
strong UPR. Activation of the ATF4-CHOP-CHAC1 pathway induces an ERS response,
while CHAC1 reduces GSH levels, decreases cell tolerance to ROS and lipid
peroxidation, and increases ferroptosis. Ferrostatin-1 (Fer-1) and desferriamine
not only inhibit ferroptosis but also inhibit ERS [39]. Ferroptosis inducers can
also induce an increase in PUMA expression upregulated by p53 through the
ATF4-CHOP-CHAC1 pathway, thereby causing ERS [38]. In a model of cadmium-induced
hepatocyte damage, cadmium induced ferroptosis in hepatocytes and activated the
PERK-eIF2-ATF4-CHOP pathway to induce ERS, and the iron chelating agent
deferriamine effectively inhibited ferroptosis and ERS [40]. Activation of the
PERK-eIF2-ATF4-CHOP-CHAC1 pathway may be an important factor in the
activation of ERS by ferroptosis inducers. The pathways by which ferroptosis
induces ERS are shown in Fig. 1.
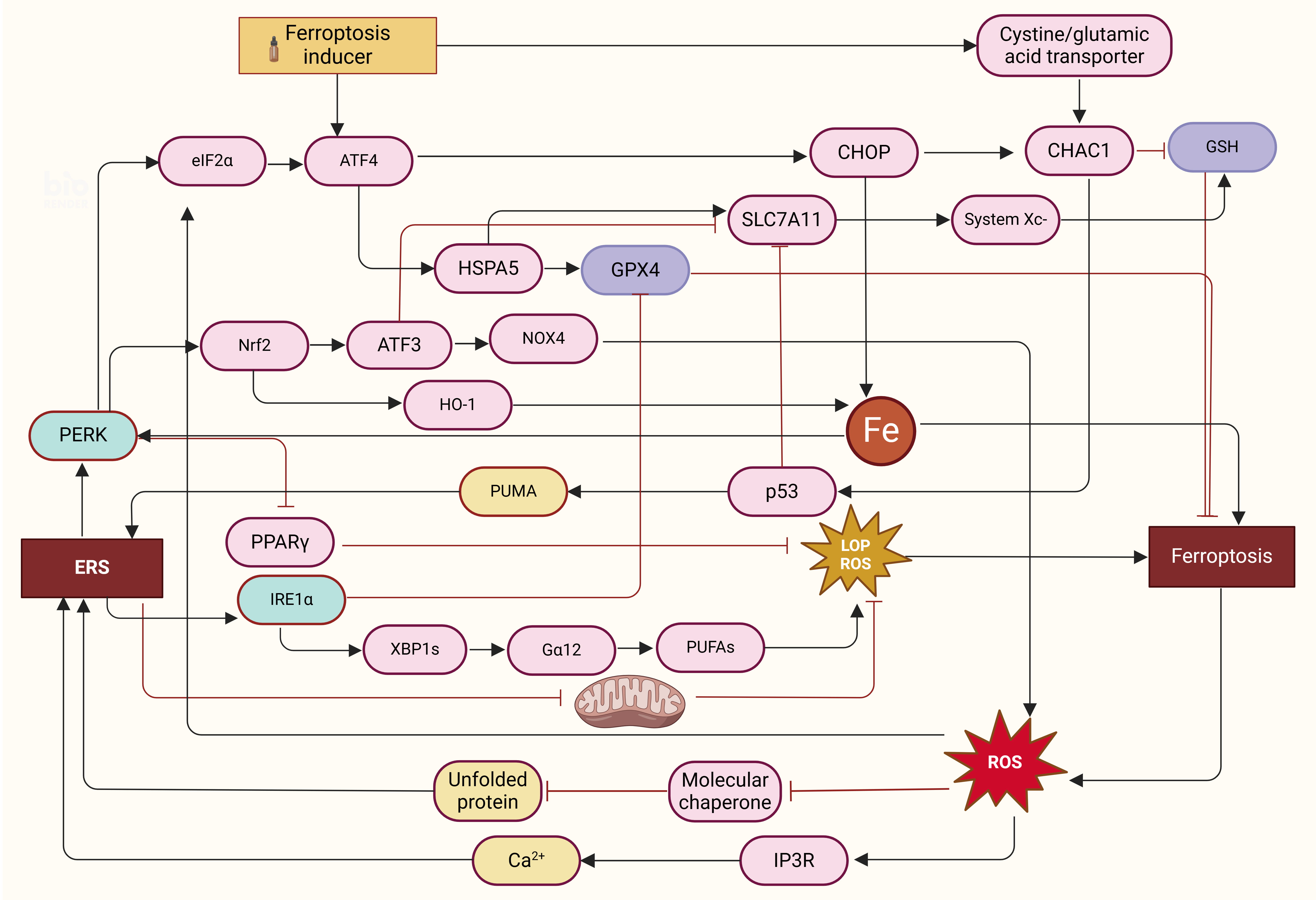 Fig. 1.
Fig. 1.
The mutual regulatory mechanism between ERS and endoplasmic
death. (1) Ferroptosis induces ERS: Ferroptosis can inhibit the chaperone
function of molecules by releasing a large number of ROS, resulting in the
accumulation of a large number of unfolded proteins, and binding with IP3R
releases a large amount of Ca accumulation, resulting in ERS. Excess iron
activation via the PERK-eIF2-ATF4-CHOP pathway induced increased
expression of PUMA. Ferroptosis inducers can inhibit cystine-glutamate exchange
in cells and increase the expression of CHAC1. (2) Ferroptosis induced by ERS:
ERS induces mitochondrial dysfunction and activates
PERK-eIF2-ATF4-CHOP, PERK-Nrf2-HO-1, PERK-PPAR,
PERK-P53-SLC7A11-System Xc-, PERK-Nrf2-ATF3-SLC7A11/NOX4, and
IRE1-XBP1s-G12-PUFAs to induce ferroptosis. (3) ERS
negatively regulates ferroptosis: ERS increases the levels of System Xc- and GSH
and the activities of System Xc- and GPX4 in cells and reduces the production of
lipoperoxide by activating the PERK-ATF4-HSPA5/SLC7A11 pathway. ERS, endoplasmic
reticulum stress; ROS, reactive oxygen species; IP3R, inositol receptor
1,4,5-triphosphate; PUMA, p53 to upregulate the expression of apoptosis; GSH,
glutathione; GPX4, glutathione peroxidase 4; LOP, lipid oxidation product.
4.2 ERS Induces Ferroptosis
Ferroptosis is regulated by ERS, and under certain conditions, ERS can promote
ferroptosis. The PERK-eIF2-ATF4-CHOP pathway is not only involved in
ferroptosis-induced ERS but also involved in ferroptosis induced by ERS. Xu
et al. [41] reported that the levels of iron ions and phospholipids in
the intestinal epithelial cells of the ulcerative colitis (UC) group were
significantly greater than those in the control group, and the observed
mitochondrial atrophy was consistent with the morphological characteristics of
ferroptosis. These findings suggested that ferroptosis may be closely related to
UC. However, the activation of PERK-eIF2-ATF4-CHOP pathway was also
observed during this process. Combining PERK inhibitors with RSL3 (a ferroptosis
inducer) did not inhibit PERK-eIF2-ATF4-CHOP and ERS activation but
also reduced ferroptosis in cells. ERS was also found to activate the
PERK/eIF2 signalling pathway and induce mitochondrial dysfunction to
regulate ROS production and promote ferroptosis in cells induced by whole-smoke
condensates [42]. Peroxisome proliferator-activated receptor (PPAR) is involved in the regulation of lipid metabolism and blood
glucose. PPAR is also involved in ferroptosis induced by ERS. When ERS
occurs, the PERK signalling pathway is activated, the expression of
PPAR is inhibited, the level of PPAR decreases, and the level
of lipid peroxide increases, promoting ferroptosis [43, 44]. Additionally, PERK
can upregulate p53 to inhibit the transcription of SLC7A11, decreasing the
activity of System Xc- and promoting ferroptosis [45]. In exploring novel
therapeutic strategies for lung cancer, Fu et al. [46] also reported
that biomineralized liposomes (LDMs) containing dihydroartemisinin (DHA) and
pH-responsive calcium phosphate (CaP) can drive and enhance ferroptosis by
inducing ERS. Mechanistically, CaP causes a sharp increase in the intracellular
Ca concentration, triggering intense ERS, followed by mitochondrial
dysfunction, ROS accumulation, and ferroptosis. This is followed by a surge of
Ca entering the cell through iron pathways on the cell membrane, driving
the “Ca burst-ER stress-ferroptosis” cycle. Further experiments revealed
that the process by which ERS promotes ferroptosis is also the process by which
ROS and LPO accumulation trigger cell swelling and cell membrane destruction.
Nrf2 is also involved in ERS-induced ferroptosis. Nrf2 is an upstream element
involved in the PERK-Nrf2-HO-1 cascade in ERS. Activated Nrf2 activates target
genes, including HO-1, and increases HO-1 expression. HO-1 metabolizes
heme to Fe and induces ferroptosis by regulating the increase in unstable
iron content and ROS production in the iron pool [13, 47]. ATF3 is an ERS-induced
transcription factor that can regulate the transcription of target genes
according to the cellular environment [48]. When activated, Nrf2 can bind to the
ATF3 promoter and upregulate the expression of ATF3 through ROS, inhibiting the
expression of SLC7A11 and resulting in reduced activity of System Xc-, a lack of
intracellular GSH, the promotion of lipid peroxidation, and the induction of
intracellular ferroptosis [49]. ATF3 can also upregulate the expression of NADPH
oxidase 4 (NOX4), activate NADPH oxidase, promote the production of superoxide
and ROS, and promote iron-mediated cell death [50]. Therefore, ATF3 plays an
important role in the regulation of ferroptosis by ERS. IRE1 is also
involved in the regulation of ferroptosis. On the one hand, IRE1 can
inhibit GPX4 expression and promote ferroptosis; on the other hand,
IRE1 can promote the transcription of XBP1s and activate G12
to promote iron peroxide-mediated death in PUFAs [51, 52]. The pathways by which
ferroptosis is induced by ERS are shown in Fig. 1.
4.3 ERS Inhibits Ferroptosis
ERS and ferroptosis coparticipate in cell death. Under certain conditions, ERS
can inhibit ferroptosis and participate in cancer cell resistance. In cancer cell
ferroptosis induced by ferroptosis inducers, the ERS response is simultaneously
activated, and the PERK-eIF2-ATF4 pathway inhibits ferroptosis by
upregulating the expression of HSPA5, System Xc-, and other molecules [53]. When
pancreatic ductal adenocarcinoma (PDAC) cells were treated with a certain dose of
erastin, the transcriptional translation level of the ER chaperone HSPA5 was
significantly greater than that in the control group. The PERK and
eIF2-ATF4 pathways were also activated in these cells, the expression
of ATF4 and the molecule CHOP was upregulated, and the expression of the ATF4
target gene HSPA5 was also upregulated. Erastin-induced lipid peroxidation can be
significantly enhanced by inhibiting HSPA5 expression via RNA interference (RNAi)
or knocking out the ATF4 gene, leading to ferroptosis. The above results
indicate that HSPA5 can inhibit ferroptosis in PDAC cells, which might be related
to the inhibition of GPX4 degradation by HSPA5 to improve the intracellular
antioxidant capacity [54]. As an anticancer drug, dihydroartemisinin can induce
ferroptosis in glioma cells, and the activation of PERK-ATF4-HSPA5 pathway was
also found when studying the related mechanism; additionally, upregulated HSPA5
increased the activity of GPX4 and reduced the production of lipoperoxide [53].
ATF4 is highly expressed in injured cardiomyocytes induced by Sorafenib (SOR),
and ATF4 can enhance the expression of SLC7A11, the active regulatory subunit of
System Xc-, through transcriptional regulation, increase the activity of System
Xc-, inhibit ferroptosis, and promote the survival of cardiomyocytes [55].
Harding et al. [56] reported that wild-type mouse fibroblasts were prone
to amino acid depletion when the ATF4 gene was knocked out. In the
absence of exogenous amino acid supplementation, peroxides rapidly accumulate in
cells, resulting in cell death. Ferroptosis inducers such as artesunate and
dihydroartemisinin can induce ferroptosis in cells and negatively regulate
ferroptosis, and ERS is activated. The expression of HSPA5 and SLC7A11 increased
under the regulation of ATF4, which increased the levels of System Xc- and GSH
and the activities of System Xc- and GPX4 in cells and reduced the occurrence of
ferroptosis [53]. This finding suggested that the expression level of ATF4 may be
closely related to the sensitivity of cells to oxidative stress and that ATF4 may
be another important molecule involved in the regulation of ferroptosis by ERS.
The pathways by which ERS inhibits ferroptosis are shown in Fig. 1.
5. The Role of ERS and Ferroptosis Cross-Talk in Common Liver Diseases
5.1 Non-Alcoholic Fatty Liver Disease (NAFLD)
The disease progression of non-alcoholic fatty liver disease (NAFLD) encompasses
a range of diseases from simple steatosis to non-alcoholic hepatitis (NASH),
which can progress to cirrhosis and hepatocellular carcinoma (HCC), a metabolic
syndrome of liver damage with an unknown pathogenesis. However, ferroptosis and
ERS are closely related to the occurrence of NAFLD. IRE1/XBP1 plays an
important role in lipid regulation in hepatocytes. IRE1/XBP1 can
directly upregulate the regulation of sterol regulatory element-binding
protein-1c (SREBP-1c), which is involved in fatty acid synthesis. When
IRE1/XBP1 is continuously activated, it leads to the accumulation of
triglycerides and cholesterol in the liver, resulting in NAFLD [57]. GRP78 is an
ER molecular chaperone, and the expression of ATF6, an ERS receptor, is
significantly increased in the cells of mice fed a high-fat diet. Aerobic
exercise can reduce the expression levels of GRP78 and ATF6 and reduce ERS, thus
improving the symptoms related to NAFLD [58]. PERK is also activated in ERS
through the PERK-eIF2-ATF4 pathway to increase the intracellular CHOP
level, activate the CHOP pathway, and induce cell apoptosis; at the same time,
CHOP can induce PPAR expression and promote lipid accumulation in NAFLD
[59]. In NAFLD cells, the increased expression level of lysophosphatidylcholine
acyltransferase (LPCAT) increases the content of intracellular amino acids, which
are one of the substrates of PUFAs. The increase in the content of amino acids
can significantly increase the level of lipid peroxidation and induce
ferroptosis. Although GPX4 can inhibit ferroptosis, the overexpression of GPX4 in
liver cells can activate enolase3 (ENO3) and increase the content of
intracellular lipids to induce ferroptosis [60]. Ferroptosis and ERS participate
in the occurrence of NAFLD and can regulate each other. Sodium arsenite (NaAsO2)
is a carcinogen that can cause immune inflammation, fibrosis, and even cancer in
liver cells. Ferroptosis in a NaASO2-induced NAFLD rat model involved ERS. After
NaAsO2 treatment, the iron content significantly increased, MDA, ACSL4, and
5-hydroxy eicosatetraenoic acid (5HETE) expression increased, GSH expression
decreased, and linear membrane rupture and ridge reduction were observed in rat
hepatocytes. When liver cells were pretreated with Fer-1 or ACSL4 inhibitors,
GPX4 levels were significantly restored, mitochondrial structure and morphology
were significantly improved, and ferroptosis was inhibited. In rat hepatocytes
treated with NaAsO2, the expression of IRE1, one of the receptors of
ERS, was upregulated, which activated ERS, and IRE1 promoted NAFLD and
ferroptosis in hepatocytes by upregulating 5-HETE, ACSL4, and MDA and inhibiting
GPX4 expression [51]. The ferroptosis and ERS pathways in NAFLD are shown in Fig. 2A.
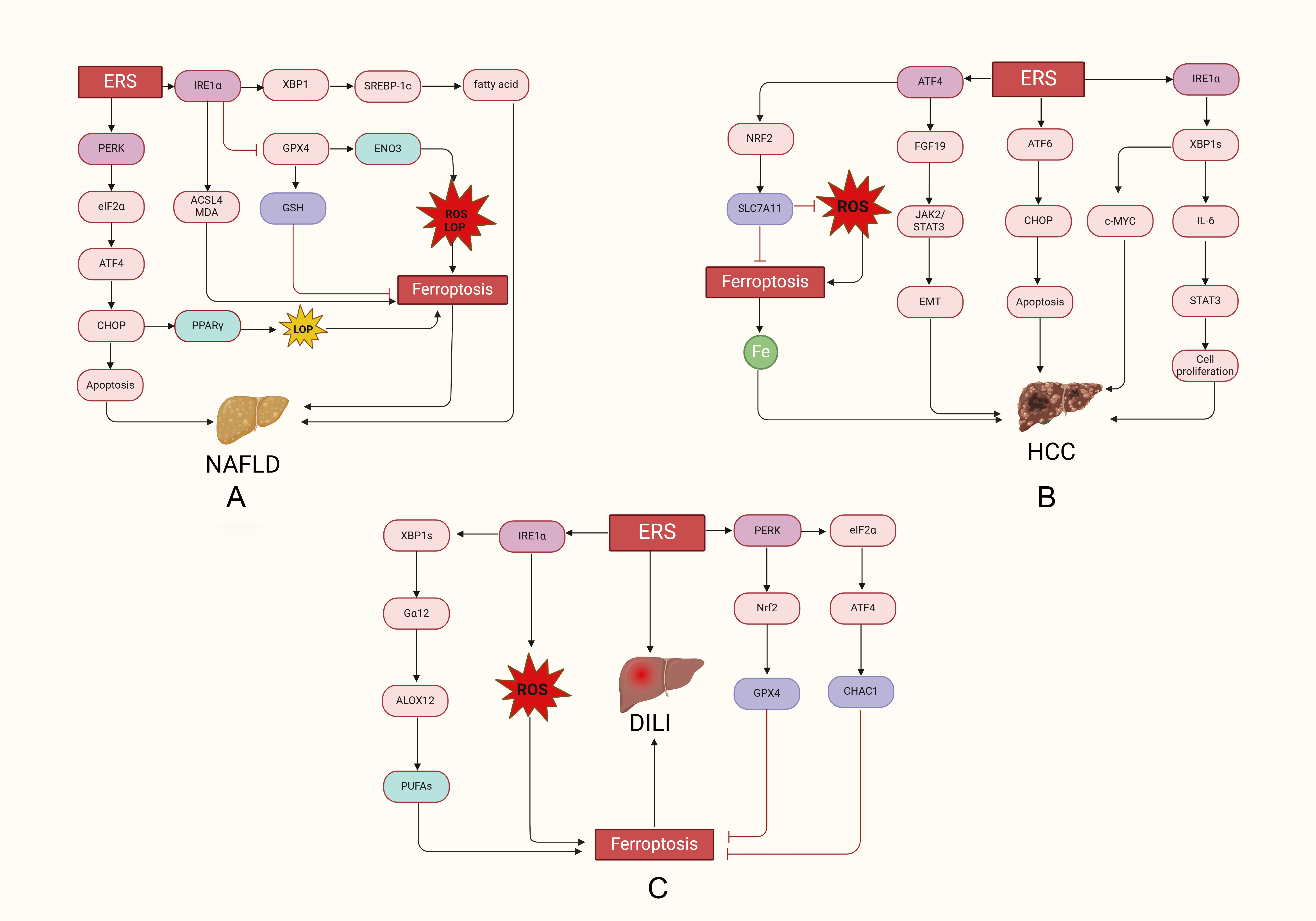 Fig. 2.
Fig. 2.
Ferroptosis and the regulation of ERS in liver disease. (A)
In NAFLD, ERS increased the expression of IRE1, PERK, ACSL4 and GRP78
related proteins, and IRE1 promoted ferroptosis in hepatocytes by
upregulating 5-HETE, ACSL4 and MDA and inhibiting GPX4 expression. (B) In HCC,
ERS promotes the growth and proliferation of cancer cells through the
ATF4-FGF19-JAK2/STAT3-EMT, IRE1-XBP1s-IL-6-STAT3, and
IRE1-XBP1s-c-MYC pathways and inhibits ferroptosis through the
PERK-ATF4-Nrf2-SLC7A11 pathway. (C) In DILI, ERS induces ferroptosis in
hepatocytes by activating IRE1-XBP1-G12-ALOX12-PUFAs and PERK
eIF2-ATF4-CHAC1, but ERS inhibits ferroptosis in hepatocytes via PERK
Nrf2-GPX4. NAFLD, non-alcoholic fatty liver disease; DILI, Drug-induced liver
injury; HCC, hepatocellular carcinoma; PERK, protein kinase R-like ER kinase;
ACSL4, 4acyl-CoA synthetase long-chain family member 4; GRP78, glucose-regulated
protein 78; MDA, malondialdehyde.
5.2 Hepatocellular Carcinoma (HCC)
HCC is a malignant tumor that occurs in hepatocytes or intrahepatic bile duct
epithelial cells and has a poor prognosis, and it is urgent to explore the
pathogenesis of HCC [61]. Recent studies have shown that HCC can develop from
NAFLD and viral hepatitis B [62, 63]. The team of Wu [64] reported that ERS
was activated in hepatitis B virus (HBV)-induced HCC cells. HBV significantly
increased the expression of ERS-related proteins BiP and ATF4, and the secretion
of fibroblast growth factor 19 (FGF19) increased under the regulation of ATF4,
which activated Janus kinase 2 (JAK2)/activator of transcription 3 (STAT3)
pathway and leaded to epithelial-mesenchymal transition (EMT) in HCC cells. EMT
is closely related to the occurrence of HCC, and when EMT is inhibited, it can
inhibit the formation of hepatic vessels [65, 66]. IRE1 is also involved
in the development of HCC. IRE1 is overexpressed in HCC cells, and a
large amount of IRE1 accumulates in cells to stimulate RNase activity
and catalyze the formation of XBP1s. XBP1s increased IL-6 expression through
signal transduction, induced STAT3 activation, and promoted the proliferation of
HCC cells. Treatment of HCC with toluene can significantly improve the
value-added effect of IL-6-STAT3 on HCC [67]. c-MYC is closely related to the
occurrence of cancer and has a strong carcinogenic effect, participating in the
drug resistance of cancer cells. Knockout of IRE1 can inhibit
the IRE1-XBP1s-C-MYC pathway, which can significantly improve the
resistance of HCC to sorafenib [60, 61, 62, 63, 64, 65, 66, 67, 68, 69]. In addition, ATF6 and its target gene
(CHOP apoptosis gene) are involved in the development of HCC [70].
ATF6/CHOP is significantly activated in hepatocytes treated with cytochrome P450
2E1 (CYP2E1), which inhibits the apoptotic pathway of hepatocytes and promotes
the occurrence and development of HCC [71]. Moreover, single nucleotide
polymorphisms (SNPs) in ATF6 significantly increase the risk of HCC
[72]. Studies have shown that long-term iron overload in cells can cause cell
damage, which eventually manifests as HCC [73]. Excessive accumulation of iron in
HCC can promote the proliferation of cancer cells and promote the growth of
tumors [74]. Moreover, studies have shown that the expression of GPX4, a
regulator of ferroptosis, is significantly increased in HCC, which may be related
to the reduction in oxidative stress in cancer cells caused by GPX4 [75]. Both
ERS and ferroptosis regulators are involved in the development of HCC, and
inducing ferroptosis in HCC is a promising therapeutic strategy. Thus, there is
mutual regulation between ERS and ferroptosis, and the same is true in HCC. ATF4
is a transcription factor activated by ERS, and ATF4 and Nrf2 can coupregulate
the expression of SLC7A11 in HCC, reduce oxidative stress, and inhibit
ferroptosis [76]. The ferroptosis and ERS pathways in HCC are shown in Fig. 2B.
5.3 Drug-Induced Liver Injury (DILI)
Drug-induced liver injury (DILI) refers to liver damage caused by the drug
itself or its metabolites, as well as due to the supersensitivity or tolerance of
special constitutions to the drug during the use of the drugs, which can lead to
liver failure. Moreover, liver injury caused by acetaminophen (APAP) is more
common [77]. APAP is catalyzed by cytochrome P450 2E1 (CYP2E1) to produce
N-acetyl-p-benzoquinoneimine (NAPQI), which is a highly toxic product. An
excessive concentration of APAP in vivo increases GSH consumption and
NAPQ and ROS levels, and this process leads to ferroptosis and ERS, ultimately
leading to hepatocyte death [78, 79]. ERS is involved in drug-induced liver
damage, and Galpha12 (G12) is associated with cell activity.
Apap-induced acute liver damage in patients with significantly increased
G12 expression, downregulated GPX4 expression, lipid peroxide
accumulation and typical ferroptosis may also be observed. An increase in ERS was
positively correlated with the expression of G12 and trans-activated
G12 through the IRE1-XBP1 pathway. Moreover, G12
can promote the peroxidation of PUFAs and induce ferroptosis in hepatocytes by
inducing arachidonate 12-lipoxygenase (ALOX12) [52]. These results indicate that
ERS can promote ferroptosis in hepatocytes by increasing the expression of
G12 and that G12 is expected to be a new target for the
treatment of DILI. Xu and his team [80] reported the protective effect of ERS in
mouse hepatocytes with APAP-induced acute liver injury, which promoted
ferroptosis in hepatocytes through the activation of
PERK-eIF2-ATF4-CHAC1. The morphology of mitochondria is a
characteristic feature of ferroptosis, and a large amount of ROS, iron and lipid
peroxides can accumulate in a liver injury model induced by carbon tetrachloride
(CCl). Similarly, the expression levels of SLC7A11 and GPX4 were
significantly lower in these cells than in control cells. However, bicyclol can
improve ferroptosis by directly increasing the activity of other enzymes. This
effect can also be achieved by activating another branch of PERK known as the
Nrf2-GPX4 axis [81]. In addition, CCl can lead to decreased transcription
levels of antioxidant enzymes, including GSH and superoxide dismutase (SOD), as
well as increased ROS levels and activation of the ERS-related protein
IRE1-. The production of excessive ROS further promotes the occurrence
of ERS. P. umbrosa can significantly improve ferroptosis in hepatocytes
and ERS [82]. The ferroptosis and ERS pathways involved in DILI are shown in Fig. 2C.
6. The Application of ERS and Ferroptosis in the Treatment of Liver
Disease
Compared with that in normal cells, the metabolism of iron in cancer cells is
significantly increased, and the demand for iron is also increased, so cancer
cells have a higher susceptibility to ferroptosis. Sorafenib is a systemic
therapy for the treatment of advanced HCC, and the development of drug resistance
is the main reason for its limited use. Metallothionein (MT)-1G is an
Nrf2-dependent protein that can reduce the levels of GSH and lipid peroxidation
in HCC, inhibiting ferroptosis in cells. The upregulation of MT-1G expression is
related to the resistance of hepatocellular carcinoma cells to sorafenib [83].
Additionally, ERS participates in sorafenib resistance by upregulating pyruvate
kinase subtype M2 (PKM2) through the microRNA-188-5p (miR-188-5p)/heterogeneous
nuclear ribonucleoprotein A2B1 (hnRNPA2B1) pathway [84]. These findings
demonstrate that both ferroptosis and ER are involved in sorafenib resistance.
The Nrf2, as the junction of ferroptosis and ER stress, may effectively improve
sorafenib resistance and enhance its anticancer efficacy. Currently, there is a
lack of effective drugs for NAFLD treatment, but acacetin can effectively improve
NAFLD by leveraging the crosstalk between ERS and ferroptosis. Acacetin can
reduce the expression of MDA, GSH, ATF6, and CHOP in high-fat diet-fed mice,
inhibiting ferroptosis and ERS. Acacetin can directly inhibit ferroptosis and
indirectly inhibit the expression of ACSL4, a promoter of ferroptosis, by
inhibiting the ATF4-CHOP axis in ERS. Therefore, acacetin has a strong ability to
inhibit lipid accumulation in liver cells and is expected to become a candidate
drug for the treatment of NAFLD in the future [85]. ERS and ferroptosis promote
each other and participate in the occurrence and development of drug-induced
liver injury. Salidroside inhibited the cationic transport regulator CHAC1 and
the PERK-eIF2-ATF4 pathways in mouse models, reducing GSH degradation
and the intracellular iron content, inhibiting ferroptosis, and preventing
APAP-related liver damage caused by ERS. Increased transcription of the PERK
activators CCT020312 and ATF4 can reduce the inhibitory effect of salidroside on
CHAC1. Additionally, the activation of AMP-activated protein kinase
(AMPK)/sirtuin-1 (SIRT1) signal transduction plays an important role in the
inhibition of ferroptosis and ERS by salidroside, and when SIRT1 is inhibited,
the protective effect of salidroside on ferroptosis and ERS can be weakened [80].
In fact, many studies have shown that daglizin and irisin can inhibit ferroptosis
and ERS by activating SIRT1, reducing ROS and MDA production, reducing the
intracellular iron content, and inhibiting the PERK-eIF2-ATF4 axis
[86, 87]. These findings suggest that the activation of SIRT1 may be a valuable
target for evaluating DILI treatment.
7. Summarize
According to recent studies, ferroptosis can induce ERS by activating the
PERK-eIF2-ATF4-CHOP pathway and upregulating PUMA and ROS, while ERS
can negatively regulate ferroptosis by activating the PERK-ATF4-HSPA5/System Xc-,
PERK-Nrf2-GPX4, and other pathways. However, ERS can also induce ferroptosis
through PERK-eIF2-ATF4-CHOP, PERK-Nrf2-HO-1, and other pathways.
Additionally, cross-talk between ferroptosis and ERS also plays an important role
in diseases. In NAFLD, HCC and DILI, ferroptosis characteristics are observed,
such as increased ROS and lipid peroxides, as well as iron accumulation, while in
NAFLD, ERS manifests as increased expression of IRE1, PERK, and GRP78.
In HCC, the expression of ERS-related proteins such as IRE1, XBP1s,
ATF6, and CHOP was upregulated, and ferroptosis was inhibited by
PERK-ATF4-Nrf2-SLC7A11. In DILI, the activation of the
IRE1-XBP1-G12-ALOX12 pathway promotes ferroptosis, but ERS
inhibits ferroptosis through the PERK-Nrf2-GPX4 pathway. Understanding the
circumstances under which the ability of ERS to induce ferroptosis is greater
than that of ERS to negatively regulate ferroptosis may lead to the
identification of new therapeutic targets for overcoming cell drug resistance.
Moreover, the targeting of ferroptosis plays an important role in preventing
diseases mediated by ROS, lipid peroxidation and inflammatory infiltration. The
involvement of the ERS also provides additional options and directions for the
treatment of related diseases. The combination of ERS and ferroptosis inhibitors
may achieve improved efficacy. However, the mechanism of crosstalk related to ERS
and ferroptosis has still not been studied in detail, and the crosstalk between
ERS and ferroptosis has not yet undergone clinical translation. A further
elucidation of the crosstalk mechanism between ERS and ferroptosis for clinical
application will be beneficial for the treatment of related diseases.
Author Contributions
MH, YW, WL and XW conceived and designed the study. MH, YW,
and XW drafted the manuscript. MH and YW revised the paper. WL edited the
article. All authors read and approved the final manuscript. All authors have
participated sufficiently in the work and agreed to be accountable for all
aspects of the work.
Ethics Approval and Consent to Participate
Not applicable.
Acknowledgment
Not applicable.
Funding
This work was supported by the National Natural Science Foundation of China
(82100894, 82100630).
Conflict of Interest
The authors declare no conflict of interest.




