





 , Zidun Wang 1,*
, Zidun Wang 1,* , Minglong Chen 1
, Minglong Chen 11 Division of Cardiology, The First Affiliated Hospital of Nanjing Medical University, 211166 Nanjing, Jiangsu, China
2 Department of Cardiovascular Surgery, The First Affiliated Hospital of Nanjing Medical University, 211166 Nanjing, Jiangsu, China
3 Department of Hematology, The First Affiliated Hospital of Nanjing Medical University, 211166 Nanjing, Jiangsu, China
†These authors contributed equally.
Abstract
Background: Ibrutinib could increase the risk of atrial fibrillation (AF) in chronic lymphocytic leukemia (CLL) patients. However, the precise mechanism underlying ibrutinib-induced AF remains incompletely elucidated. Methods: We investigated the proportion of ibrutinib-treated CLL patients with new-onset AF. Optical mapping was conducted to reveal the proarrhythmic effect of ibrutinib on HL-1 cells. Fluorescence staining and western blot were used to compare connexins 43 and 40 expression in ibrutinib-treated and control groups. To identify autophagy phenotypes, we used western blot to detect autophagy-related proteins, transmission electron microscopy to picture autophagosomes, and transfected mCherry-GFP-LC3 virus to label autophagosomes and lysosomes. Hydroxychloroquine as an autophagy inhibitor was administered to rescue ibrutinib-induced Cx43 and Cx40 degradation. Results: About 2.67% of patients developed atrial arrhythmias after ibrutinib administration. HL-1 cells treated with ibrutinib exhibited diminished conduction velocity and a higher incidence of reentry-like arrhythmias compared to controls. Cx43 and Cx40 expression reduced along with autophagy markers increased in HL-1 cells treated with ibrutinib. Inhibiting autophagy upregulated Cx43 and Cx40. Conclusions: The off-target effect of ibrutinib on the PI3K-AKT-mTOR signaling pathway caused connexin degradation and atrial arrhythmia via promoting autophagy. Clinical Trial Registration: ChiCTR2100046062, https://clin.larvol.com/trial-detail/ChiCTR2100046062.
Keywords
- ibrutinib
- atrial fibrillation
- connexins 43
- connexins 40
- autophagy
Bruton’s tyrosine inhibitor ibrutinib is notable efficacy against B-cell malignancies. Nonetheless, pivotal trials, roughly 5–9% of chronic lymphocytic leukemia (CLL) patients developed atrial fibrillation (AF) compared to 0.5–2.4% in controls [1]. A comprehensive investigation based on the VigiBase database (International pharmacovigilance database) included 13,572 patients who were treated with ibrutinib and aimed to characterize adverse cardiovascular effects that were associated with ibrutinib, such as supraventricular arrhythmias, ventricular arrhythmias, and conduction disorders. In total, 957 (7.07%) individuals experienced supraventricular arrhythmia, which accounted for the largest proportion of ibrutinib-associated adverse cardiovascular events [2]. Notably, AF frequently prompted the discontinuation of ibrutinib therapy. The majority of extant studies comprise observational trials wherein AF diagnosis often necessitated hospitalization or other interventions, rather than systematic long-term electrocardiogram (ECG) screening. Consequently, it is conceivable that the true incidence of ibrutinib-associated AF may be underestimated, underscoring the imperative for further research to ascertain a more precise incidence rate [3].
The reason why ibrutinib increases the occurrence of atrial fibrillation remains inadequately elucidated. Existing studies have suggested have reported that structural remodeling via changes in gap junction protein expression could contribute to the development of AF [4]. Gap junction proteins can simultaneously mediate action potential propagation between cardiomyocytes and ensure cardiac electromechanical activity [5, 6]. Within the connexin protein family, comprising 21 isoforms expressed in the human heart [7]. Among these isoforms, Cx43 stands out as the principal isoform and is widely expressed in the ventricle [8], and Cx40 is the predominant connexin protein that is mostly expressed in the atrium and conduction system. Both Cx40 and Cx43 are casually linked to a proarrhythmogenic substrate [9]. Changes in Cx40 and Cx43, including changes in their abundance, subcellular distribution and function, significantly contribute to atrial cardiomyocyte electrical remolding and thereby cause atrial arrhythmia by blocking unidirectional conduction and decreasing conduction velocity [10]. However, the precise impact of ibrutinib on structural alterations within cardiomyocytes, including modifications in gap junction localization and expression, remains unclear.
Autophagy, a catabolic process, involves the sequestration of cellular components within autophagosomes, facilitating the recycling of cellular constituents and preservation of cellular homeostasis [11]. Both autophagy and ubiquitination mechanisms play pivotal roles in the proteolysis of the Cx43 protein [12]. Particularly under conditions of nutrient deprivation, a classical inducer of autophagy, Cx43 can bind to the autophagy-related protein ATG5, which causes the internalization of Cx43 and further enhances its regulatory effect on autophagy [13, 14]. The PI3K-AKT pathway holds paramount importance in cardiac stress responses. Notably, ibrutinib has been reported to modulate this pathway by targeting the alpha-subunit of PI3K and downregulating AKT expression [15]. Furthermore, the off-target effect emerges as a significant mechanism underlying ibrutinib-induced atrial fibrillation. Recent findings by Xiao et al. [16] have identified C-terminal Src kinase (CSK) as a crucial off-target inhibition site of ibrutinib, with CSK knockout mice exhibiting a phenotype akin to atrial fibrillation induced by ibrutinib [16]. Additionally, several studies have implicated the involvement of the PI3K-AKT-mTOR pathway in the off-target effects of ibrutinib [17, 18]. Nevertheless, it remains uncertain whether the off-target impact of ibrutinib on such pathways augments autophagic activity, leading to the degradation of cell gap junction proteins Cx40 and Cx43. Thus, we conducted clinical and experimental investigations to elucidate ibrutinib-induced arrhythmogenesis and the potential role of autophagy-mediated connexin degradation in this pathophysiological process.
A cohort comprising 187 patients diagnosed with CLL administered ibrutinib (single-agent, 420 mg, once daily, continuously) were enrolled in the Chronic Lymphoid Leukemia Center of the First Affiliated Hospital of Nanjing Medical University from March 2019 to March 2020. All of the patients required treatment according to the International Workshop on CLL (iwCLL) criteria [19]. Cardiovascular function was assessed by electrocardiogram and echocardiography in all patients upon admission. Throughout the 12-month follow-up period, electrocardiograms were systematically conducted every three months to monitor the incidence of arrhythmias attributed to ibrutinib.
HL-1 cells obtained from Dr. William Claycomb (Louisiana State University, New
Orleans, LA, USA) were validated by STR profiling and tested negative for mycoplasma and cultured in Claycomb medium (51800C, Sigma Aldrich, St. Louis, MO,
USA) supplemented with penicillin‒streptomycin solution (C0222, Beyotime,
Shanghai, China), 10% fetal bovine serum (F2442, Sigma Aldrich), 2 mM
L-glutamine (G7513, Sigma Aldrich), and 0.1 mM norepinephrine (A0937,
Sigma Aldrich) [20]. Prior to cell seeding, culture flasks were coated with
gelatin containing 0.02 mg/mL Fibronectin (F1141, Sigma Aldrich) and
incubated at 37 °C for one hour. Subsequently, cells were passaged at a
ratio of 1:4 upon reaching full confluence. The detachment of cells from flasks
was facilitated by the addition of trypsin (0.25%)/EDTA (Gibco, Waltham, MA, USA) and
incubation at 37 °C for 5 minutes, followed by termination of the
digestion process through dilution with culture medium. After centrifugation at
500
We used western blotting (WB) to measure the expression levels of autophagy marker proteins, including Beclin-1 (1:2000, Abcam, Cambridge, UK, ab207612), Atg5 (1:1000, CST, Boston, MA, USA, 2630s), LC3B (1:500, Abmart Shanghai Co, Shanghai, China, T55992), and P62 (1:1000, CST, 88588s), as well as Cx43 (1:2000, Abcam, ab217676) and Cx40 (1:2000, Abcam, ab183648). The levels of phosphorylated-Bruton’s tyrosine kinase (p-BTK) (1:2000, Abcam, ab68217), BTK (1:1000, CST, 8547s), mTOR (1:1000, CST, 2972s), p-mTOR (1:1000, CST, 5536s), PI3K (1:3000, CST, 4255s), p-PI3K (1:3000, CST, 4249s), Akt (1:2000, CST, 4685s) and p-Akt (1:2000, CST, 4060T) were also measured. In brief, HL-1 cells were cultured with varying concentrations of ibrutinib for 72 hours. Protein extraction from the cells was performed using radioimmunoprecipitation assay lysis buffer (RIPA), and quantification was conducted utilizing a BCA Protein Assay Kit (23225, Thermo Fisher, Waltham, MA, USA). Subsequently, 30 µg of protein from each experimental group was separated via SDS-polyacrylamide gel electrophoresis (SDS-PAGE), followed by transfer onto polyvinylidene difluoride membranes (Millipore, Boston, MA, USA). Membranes were then blocked with 5% bovine serum albumin at room temperature for two hours and subsequently incubated with primary antibodies overnight at 4 °C. The following day, membranes were washed thrice with Tris-buffered saline with Tween 20 (TBST) for 5 minutes each and then incubated with secondary antibodies for 2 hours. Visualization of proteins was achieved using the FD bio-pico enhanced chemiluminescence kit HRP (FD8030, FD bio, Hangzhou, Zhejiang, China), and protein expression levels were quantified using ImageJ software (NIH, Bethesda, MD, USA).
Following the plating of an equivalent number of HL-1 cells in a six-well plate, we adhered to the protocol outlined in the instruction manual of the Mouse Bruton’s Tyrosine Kinase Activity Assay Kit (FY-EM12093-0, Wuhan, Hubei, China) to lyse the HL-1 cells treated with varying concentrations of ibrutinib and extract the resulting supernatant for BTK activity assessment. Subsequently, the absorbance value was determined at a wavelength of 450 nm, and a standard curve was generated to calculate the active concentration of mouse BTK in the sample based on the optical density (OD) values
The effects of ibrutinib on HL-1 cell viability were assessed using the Calcein-AM/propidium iodide (PI) kit (Beyotime). Cells were treated with varying concentrations of ibrutinib (0, 0.01, 0.1, 1, 10, or 100 µM) for 72 hours. Subsequently, cells were subjected to three washes with phosphate-buffered saline (PBS) and then incubated with 1x washing buffer containing Calcein-AM and propidium iodide in the dark at 37 °C for 30 minutes. Following incubation, the cells were visualized using a fluorescence microscope (Carl Zeiss, Oberkochen, canton Batten-Würburg, Germany) under dark conditions. Red fluorescence indicated dead cells, whereas green fluorescence indicated viable cells. The percentage of positive cells was quantified using Image-Pro software (version 7.0.1, NIH, Bethesda, MD, USA).
To visualize cell morphology using a Zeiss Axio Observer microscope, live cells were stained with wheat germ agglutinin (1 mg/mL, Sigma Aldrich) diluted to a ratio of 1:40 in Hanks’ balanced salt solution (HBSS) supplemented with 1% bovine serum albumin. The staining process lasted for 10 minutes.
HL-1 cells were cultured on gelatin-coated coverslips, washed twice with
PBS, and fixed with methanol at room temperature for
20 minutes. Subsequently, the cells were washed with PBS for 5 minutes and
incubated with a solution comprising PBS, 0.2% Triton X-100, and 10% goat serum
for 1 hour at room temperature. HL-1 cells were then subjected to primary
antibody incubation against Cx43 (Abcam, ab217676), Cx40 (Abcam, ab183648),
HL-1 cells were initially fixed with 2.5% glutaraldehyde for a duration of 1.5 hours, followed by detachment using a cell scraper. The cells underwent centrifugation at 1500 rpm for 5 minutes and were then subjected to fixation in 1% aqueous uranyl acetate for a period of 1 hour. Post-fixation, the HL-1 cells were subjected to dehydration using a series of graded acetone solutions (ranging from 30% to 100%) subsequent to thorough washing with distilled water. Ultrathin sections were prepared, impregnated, and embedded onto copper grids utilizing an appropriate kit. Finally, the prepared samples were examined utilizing a transmission electron microscope (JEM-1400Flash, Shanghai J&R Instrument Technology Co., Shanghai, China).
Total RNA was extracted from HL-1 cells cultured with either ibrutinib or the
control condition. A total of 500 ng of total RNA was reverse-transcribed into
cDNA with the Hifair 1st Strand cDNA Synthesis Super Mix kit (11119ES60,
Yeasen Biotechnology, Shanghai, China). cDNA was quantified with specific primers
for Cx43 and Cx40 using the Hieff UNICON power qPCR SYBR Green Master Mix kit
(Yeasen Biotechnology).
The HL-1 cells were stained with freshly prepared FluoVolt TM membrane potential
dye (F10488, Thermo Fisher, Waltham, MA, USA). The medium was removed, and the
cells were washed with physiological buffer and then incubated with prepared dye
loading buffer containing Component B (100
HL-1 cells were seeded onto glass coverslips and subsequently washed with Hanks’ balanced salt solution (HBSS) prior to loading with 3 µM Fluo-4 AM (Invitrogen F14201). Following loading, the HL-1 cells were incubated in darkness at room temperature for 60 minutes. Upon completion of the incubation period, Fluo-4-loaded HL-1 cells were washed with HBSS and subjected to imaging using a Dhyana 400BSI V2 camera coupled with an Olympus IX71 inverted microscope (IX71-RC, Olympus, Tokyo, Japan). Videos capturing spontaneous calcium transients were recorded. For statistical analysis, twenty-five cells from both the control and ibrutinib-treated groups were included. Fiji software (Plot Z-axis profile, NIH, Bethesda, MD, USA) was employed to measure the average pixel intensity (F) in randomly selected regions, serving as a reflection of calcium activity. Relative changes in fluorescence were determined utilizing the formula: (F-F0)/F0, where F0 represents the fluorescence intensity at time 0. Arrhythmia incidence and transient calcium flux amplitude were assessed using calcium graphs.
We downloaded the relevant transcriptome [16] and proteomics data [23] of the left atrial appendage tissues from mice treated with Ibrutinib in previous studies. Initially, we conducted a statistical analysis to identify differentially expressed proteins and genes in the Ibrutinib intervention group compared to the control group. Subsequently, we performed consensus Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis on the respective KEGG enrichment results derived from the differentially expressed proteomes and transcriptomes. A Venn diagram was then generated using the ggVennDiagram R package (https://cran.r-project.org/web/packages/ggVennDiagram/index.html) to illustrate the overlapping pathways. Furthermore, based on the consensus KEGG differential genes, Gene Ontology (GO) pathway enrichment analyses were carried out employing the clusterProfiler 4.6.2 package (https://bioconductor.org/packages/release/bioc/html/clusterProfiler.html), with an adjusted p value threshold of less than 0.05 utilized as the cut-off criterion.
The characteristics of the patients enrolled in the study were delineated in
terms of the median (interquartile range), mean
To ascertain the incidence of ibrutinib-induced atrial fibrillation in clinical practice, a cohort comprising one hundred eighty-seven chronic lymphocytic leukemia (CLL) patients undergoing ibrutinib therapy was recruited for our study (registered under clinical trial number: ChiCTR2100046062). Prior to treatment initiation, baseline electrocardiograms were conducted for all 187 patients, revealing sinus rhythm in each case (Supplementary Fig. 1B). Additionally, transthoracic echocardiograms demonstrated normal myocardial systolic function without evidence of valvular disease. Throughout a median follow-up period of 12 months post-treatment, five patients developed new-onset atrial arrhythmia, as discerned from electrocardiograms (Supplementary Fig. 1C). Detailed characteristics of these five patients are presented in Supplementary Fig. 1A.
We exposed HL-1 cells to increasing concentrations (0, 0.01, 0.1, 1, 10, and 100 µM) of ibrutinib to investigate its impact on BTK activity and cell viability. Significant inhibition of BTK was observed with ibrutinib treatment at doses exceeding 0.01 µM, as depicted in Fig. 1A. Furthermore, the inhibition effects of ibrutinib on BTK were found to manifest in a dose-dependent manner. Specifically, treatment with 1 µM ibrutinib resulted in a reduction of BTK activity to 46.6% of the control group (Fig. 1C), mirroring the suppression observed at concentrations of 0.1 µM and 10 µM (Supplementary Fig. 2). To assess the potential cytotoxicity of various concentrations of ibrutinib on HL-1 cells, we quantified cell viability using calcein-AM/PI staining. Our findings indicate that treatment with 0.01 µM to 10 µM ibrutinib elicited negligible cardiotoxic effects on HL-1 cells. Only the 100 µM dose resulted in a reduction of HL-1 cell viability by 4.2% (Fig. 1B,D). Based on the outcomes of these experiments, we identified 1 µM as an appropriate concentration of ibrutinib, demonstrating efficacy without inducing cytotoxicity.
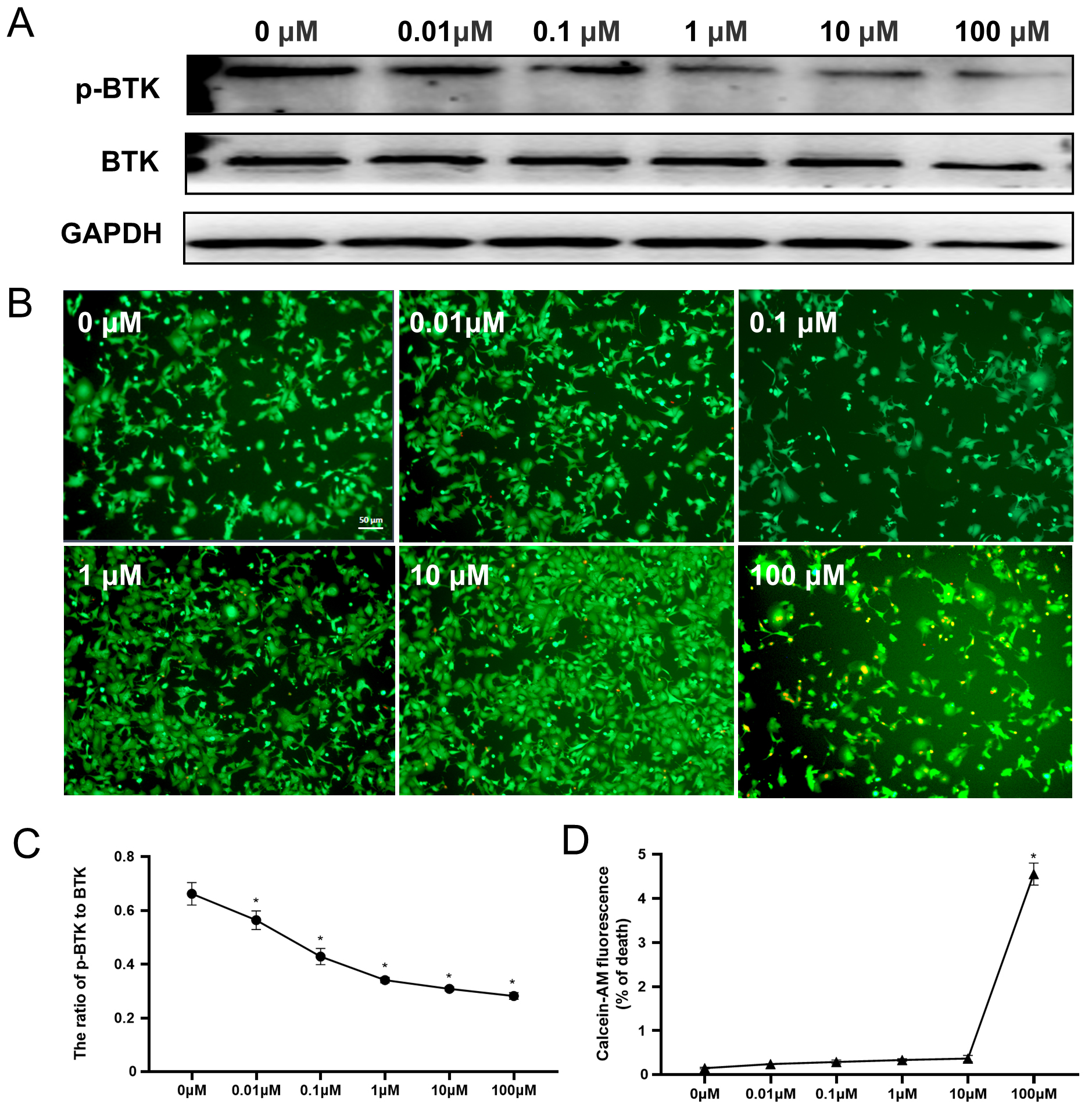 Fig. 1.
Fig. 1.Efficacy and toxicity of ibrutinib in HL-1 cells.
(A,C) Western blotting of the P-BTK and BTK levels after treatment with
different doses (0, 0.01, 0.1, 1, 10, or 100 µM) of ibrutinib and
statistical analysis. (B) Calcein and PI staining of cells treated with ibrutinib
at various concentrations. Red fluorescence indicates dead cells, while green
fluorescence indicates live cells. (D) Quantitative analysis of the dead cell
ratio (red cells/green cells). All data are expressed as
Mean
Given the prevalence of atrial fibrillation (AF) as the foremost atrial rhythm
disorder associated with ibrutinib, our investigation aimed to assess whether
ibrutinib could exacerbate susceptibility to atrial arrhythmia.
Immunofluorescence staining for the atrial cardiomyocyte marker protein ANP
(Supplementary Fig. 3) was employed to confirm the atrial myocardial
properties of HL-1 cells. Subsequently, optical mapping was utilized to evaluate
the arrhythmogenic effects on transient Ca
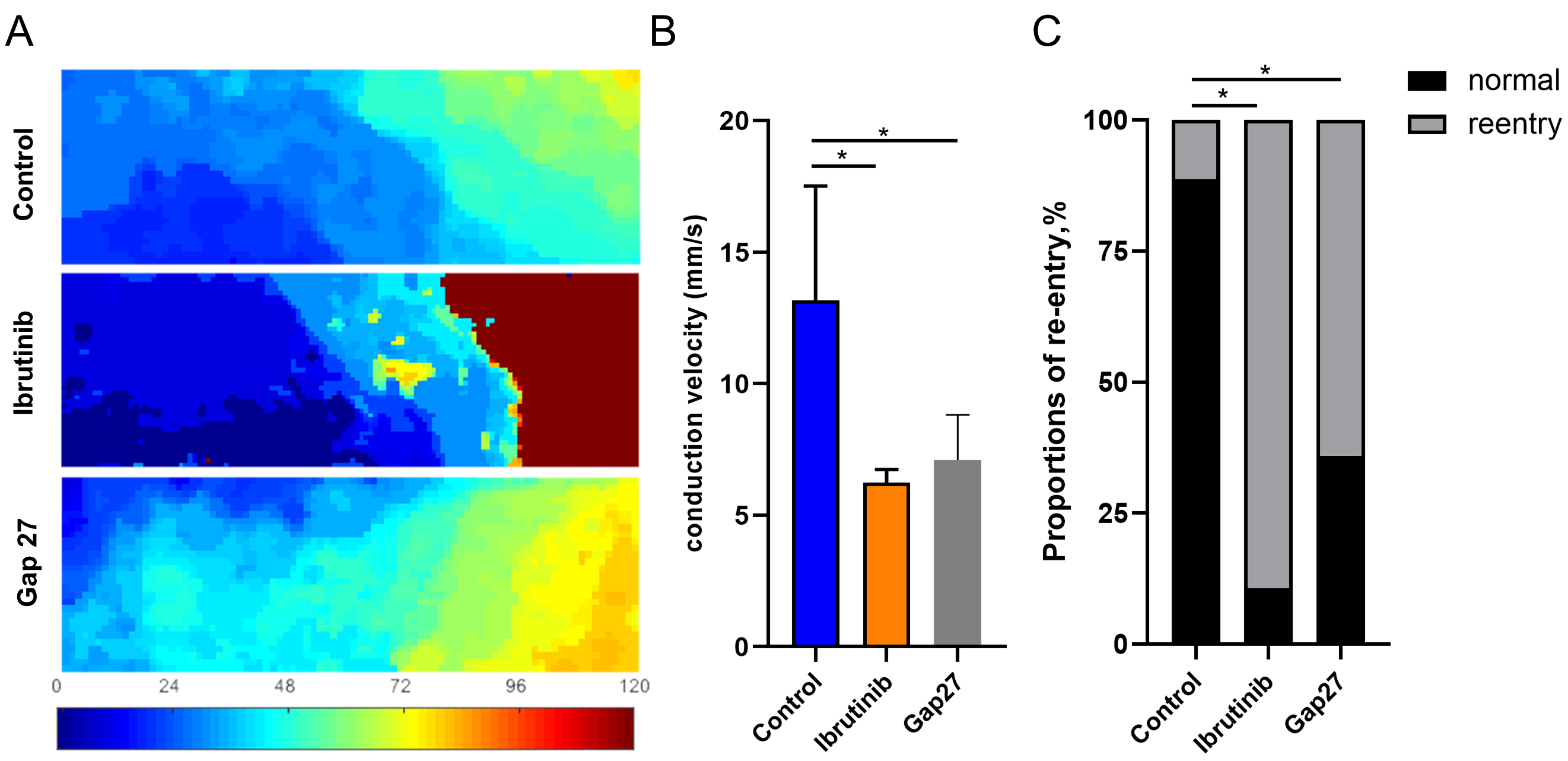 Fig. 2.
Fig. 2.Ibrutinib altered the conduction direction and velocity in cell
monolayers. (A) Representative conduction velocity map of HL-1 cells monolayer
with ibrutinib or Gap27 intervention. (B) Statistical analysis of the conduction
velocity along the vector. n = 15 dishes for each group. (C) Statistical analysis
of the proportion of re-entry or normal conduction events in the two groups, with
an 89.33% re-entry occurrence rate in the ibrutinib group (n = 134/150), 64% in
the Gap27 group (n = 96/150) and a 11.33% re-entry occurrence rate in the
control group (n = 17/150), n = 150 videos of different fields of view captured
on 15 dishes. *p
Optical mapping analysis revealed a pronounced reduction in conduction velocity and an elevated incidence of re-entry in the ibrutinib-treated group. Electrical conduction among atrial cardiomyocytes is intricately linked to gap junction proteins such as Connexin 43 and 40. Moreover, we conducted an integrated analysis of transcriptomic sequencing [16] and proteomics [23] data from previous studies involving intraperitoneal injection of ibrutinib (25–30 mg/kg) in C57BL/6J mice. This analysis revealed enrichment of numerous differentially expressed proteins and genes (Supplementary Fig. 6A) in pathways related to ECM-receptor interaction KEGG and extracellular matrix structural constituent GO, both of which encompass connexins (Supplementary Fig. 6B–D). This underscores the pivotal role of connexins in the aberrant electrical conduction between cardiomyocytes induced by ibrutinib. According to qRT-PCR, ibrutinib treatment did not induce significant changes in the transcription levels of Connexin 43 and connexin 40 (Fig. 3A). However, Western blot analysis demonstrated a significant reduction in the protein levels of both Connexin 43 and Connexin 40 following ibrutinib treatment compared to the control group (Fig. 3B, C). Furthermore, immunofluorescence analysis was employed to evaluate the localization and abundance of Cx40 and Cx43. Consistently, a notable decrease in the fluorescence intensity of Cx40 and Cx43 was observed in the ibrutinib-treated group, without evident translocation (Fig. 3D,E). Our experimental findings indicate that while ibrutinib does not impact the expression of Cx43 or Cx40 at the transcriptional level, it significantly diminishes the expression of the functional protein units. These results align with the outcomes of our KEGG and GO analyses, which highlighted enrichment of the autophagic pathway and phagosome pathway (Supplementary Fig. 6B–D), suggesting that post-transcriptionally, the protein levels of Cx43 and Cx40 may be attenuated through degradation processes.
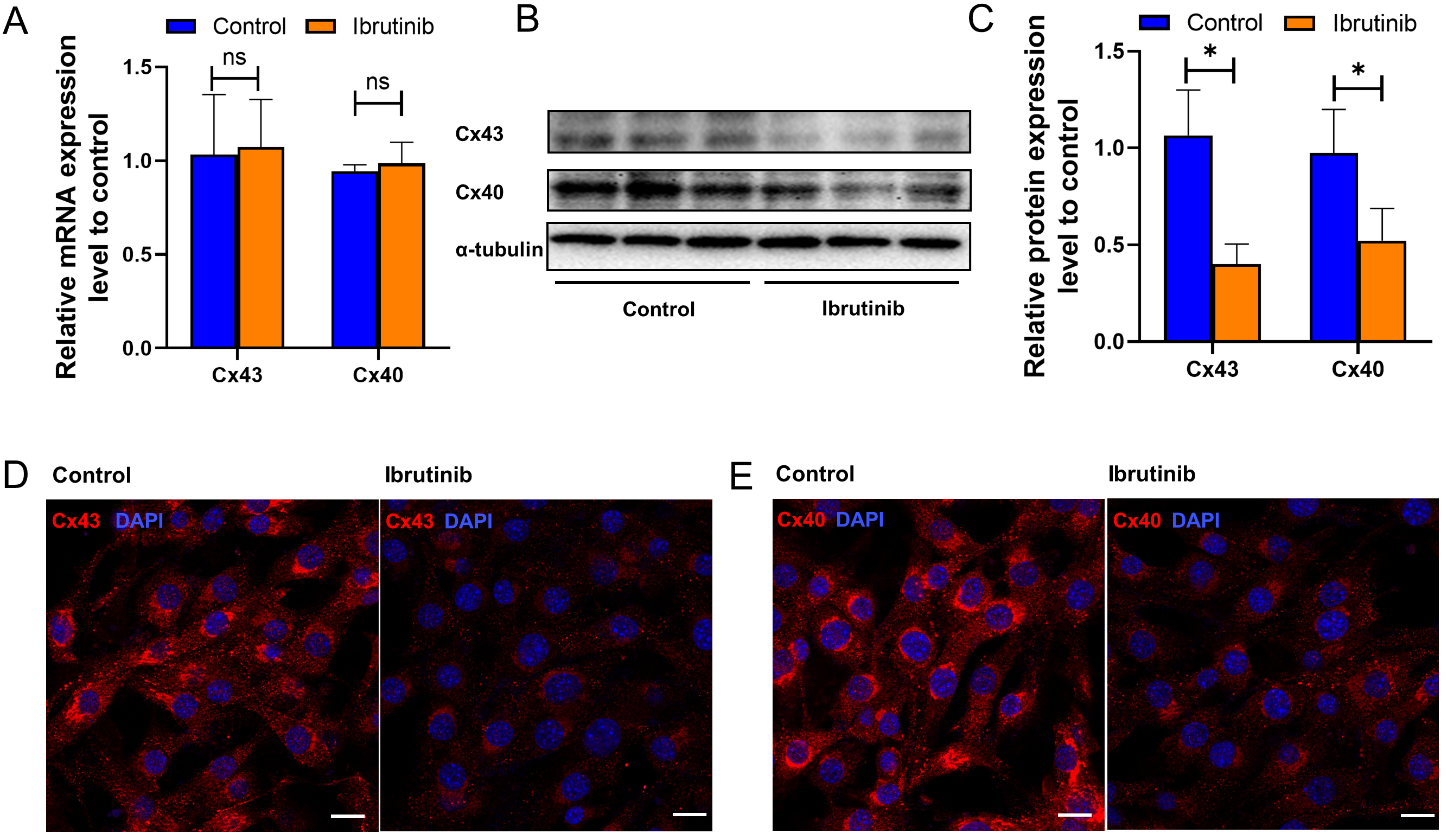 Fig. 3.
Fig. 3.Ibrutinib decreased Connexin43 and Connexin40 expression. (A)
Quantitative real-time polymerase chain reaction (qRT-PCR) analysis of the gap
junction proteins Gja1 (encodes Connexin43) and Gja5 (encodes Connexin40). The
Messenger RNA (mRNA) levels of Gja1 and Gja5 were not statistically significantly different
between the ibrutinib and control groups. (B) Representative Western blot of
Connexin 40 and Connexin 43.
A prior investigation noted elevated levels of the autophagy protein microtubule associated protein 1 light chain 3 beta (LC3B) in tumor cells of certain advanced CLL patients following standard ibrutinib treatment [24]. Furthermore, studies have demonstrated that ibrutinib can inhibit the PI3K/Akt/mTOR signaling pathway, thereby inducing autophagic cell death and mitigating cerebral ischemia/reperfusion injury [25].
Herein, representative transmission electron microscopy (TEM) images of HL-1 cells exhibited characteristic autophagic bodies with bilayer membrane structures and single-membrane autolysosomes in the ibrutinib-treated group (Fig. 4A). To further quantify autophagic levels, mCherry-GFP-LC3 viral transfection was employed to label autophagosomes and autolysosomes, facilitating assessment of the strength of the autophagic flux (Fig. 4B). The mCherry-GFP-LC3 protein participates in autophagosome formation, emitting yellow fluorescence owing to concurrent mCherry and GFP expression. Upon fusion of autophagosomes with lysosomes, the mCherry-GFP-LC3 protein emits solely red fluorescence due to GFP quenching in the acidic environment. Our findings revealed a significant increase in the numbers of intracellular green and red spots in the ibrutinib-treated group (Fig. 4C). Discrimination between autophagosomes and autophagolysosomes was achieved, with analysis indicating elevated vesicle counts following ibrutinib treatment (Fig. 4D).
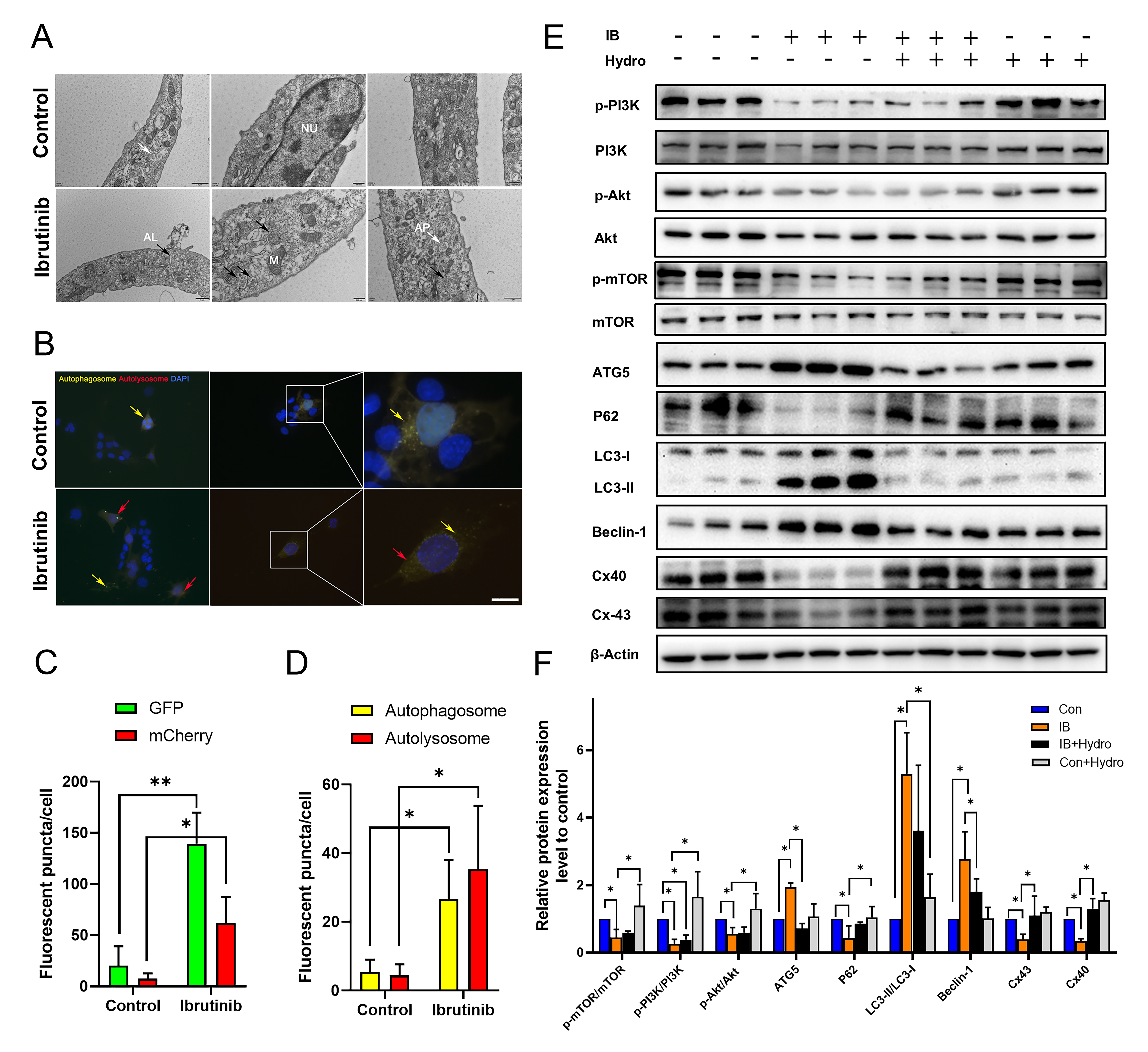 Fig. 4.
Fig. 4.Ibrutinib promotes Cx40 and Cx43 downregulation through
autophagy. (A) Representative transmission electron microscope (TEM) images of
HL1 cells treated with or without 1 µM ibrutinib. Nu, nucleus; Mt,
mitochondrion; AP, autophagosome; AL, autolysosome. Bar shown on the bottom of
the corresponding images. (B) Cells transfected with mCherry-GFP-LC3 virus for 24
hours before ibrutinib treatment for 24 hours were analyzed under a fluorescence
microscope. Nuclei were stained with DAPI. Scale bar 50 µm. Yellow arrows,
autophagosome and Red arrows, autolysosome. (C) Calculation of mCherry-GFP-LC3
puncta/cell with GFP puncta (green) and mCherry puncta (red), n = 50 cells for
each group. (D) Analysis of mCherry-GFP-LC3 puncta/cell with yellow puncta
(autophagosome) and red puncta (autolysosome), n = 50 cells for each group. (E)
Representative Western blots of p-mTOR, mTOR, ATG5, LC3II/I, Beclin-1, P62,
Additionally, western blotting assays were employed to assess the expression of proteins involved in the upstream autophagy-related PI3K/Akt/mTOR pathway, alongside autophagosome markers LC3I, LC3II, Beclin-1, and p62. Our results delineated the activation of autophagic flux from initiation to culmination stages in the ibrutinib-treated group. Exposure to ibrutinib suppressed PI3K/AKT/mTOR activity by inhibiting its phosphorylated expression, consequently activating autophagic flux. This activation was evidenced by the upregulation of ATG5 and Beclin-1, an elevated LC3II/LC3I ratio, and downregulation of p62 (Fig. 4E,F). Furthermore, hydroxychloroquine, an autophagy inhibitor, effectively counteracted the degradative effects of ibrutinib on Cx43 and Cx40 proteins by reversing the autophagic process (Fig. 4E,F). Collectively, these findings suggest that ibrutinib augments the autophagic degradation of connexins through inhibition of the PI3K-Akt-mTOR pathway.
The high prevalence of atrial fibrillation associated with ibrutinib poses challenges to its clinical utility. Our investigation revealed an incidence of atrial fibrillation at 2.7%, based on data collected at the Hematology Research Center of Jiangsu Province People’s Hospital. Notably, this incidence rate slightly diverged from that reported in Jennifer R. Brown’s clinical trial [26], which stood at 6.5%. Furthermore, ibrutinib administration correlates with a heightened risk of bleeding, thereby complicating the use of anticoagulants and presenting challenges in managing ibrutinib-induced atrial fibrillation [27]. Given the incomplete understanding of ibrutinib’s proarrhythmic mechanisms, treatment decisions are hindered by a dearth of clinical experience and experimental evidence. Hence, there is a pressing need for numerous studies to unravel the underlying mechanisms.
Connexin proteins serve as principal mediators in facilitating the transmission of electric and metabolic signals among cardiomyocytes, thereby being indispensable for the maintenance of cardiac function. Various mutations identified in cardiac Cx40 and Cx43, including heterozygous missense mutations in GJA5 (G38D, P88S, A96S, and M163V) and GJA1 (c.932delC), have been associated with atrial fibrillation [28]. Notably, Leybaert et al. [29] reported that decreased connexin density at cellular poles, attributable to redistribution towards lateral margins, fosters conduction abnormalities, suggesting a potential substrate for atrial fibrillation within Cx43 remodeling. Given the pivotal role of both aberrant protein expression levels and lateralization of Cx43 and Cx40 in structurally predisposing arrhythmia [7], an evaluation of conduction associated with connexin proteins in ibrutinib-related atrial fibrillation is warranted. The HL-1 cell line, derived from atrial cardiomyocytes, was employed in our investigation to assess conduction at the cellular level. Acknowledging that the propagation of action potentials through gap junctions in a 2D homogeneous monolayer mirrors that in complex 3D models [30], the arrhythmogenic impact of ibrutinib was evaluated using optical mapping with a high spatiotemporal resolution, facilitating visualization of propagation trajectories. Intriguingly, our study observed a notable delay in conduction within HL-1 cell monolayers, accompanied by the emergence of reentrant-like waves. However, the differences in transient calcium fluxes between the control and intervention groups did not attain statistical significance. These findings suggest that the deceleration of conduction attributable to abnormal cell connections may constitute a pivotal factor in the pathogenesis of ibrutinib-induced atrial fibrillation.
Our research identified no disparity in the transcriptional expression of Cx43 and Cx40. However, notably diminished protein expression levels observed in the ibrutinib-treated group suggest a potential interference of the drug with post-transcriptional modification or degradation processes of connexin proteins. Autophagy, a crucial degradation mechanism in eukaryotes, plays a pivotal role in maintaining normal physiological functions. Various stressors, including hypoxia, nutrient deprivation, and cytotoxic agents, can induce autophagy [31]. Prior studies have demonstrated the ability of apelin-13 to counteract Ang II-induced Cx43 downregulation by mitigating increased autophagy [32]. Connexins can undergo degradation via autophagy-mediated formation of autophagic lysosomes [33]. Lu et al. [34] observed myocardial ischemia-reperfusion injury-induced Cx43 remodeling through autophagy. Our investigation revealed heightened degradation of Cx43 and Cx40 alongside augmented autophagic flux following ibrutinib treatment. Several reports have also highlighted ibrutinib’s role as an inducer of autophagy in antitumor therapy, evidenced in conditions such as glioblastoma [35] and skin cancer [36]. Consequently, our endeavor aimed to elucidate the potential mechanistic linkages among ibrutinib, Cx43, autophagy, and atrial fibrillation. Ibrutinib, a well-established small-molecule inhibitor targeting Bruton’s tyrosine kinase (BTK), exerts its effects by selectively and irreversibly binding to BTK, thereby inhibiting its enzymatic activity and disrupting pro-survival signals stemming from B cell receptor (BCR) inactivation. Notably, ibrutinib can impede downstream molecules within the BCR pathway, including PI3K, AKT, and Erk [37]. Of particular relevance, the PI3K/Akt/mTOR pathway serves as a critical regulator of autophagy [38]. Confirmation of the PI3K/Akt/mTOR pathway’s activity in HL-1 cells treated with 1 µM ibrutinib was established in our present study.
In our investigation, the autophagy inhibitor hydroxychloroquine demonstrated the capacity to mitigate the decline in Cx43 and Cx40 expression, thereby ameliorating aberrant conduction in HL-1 cell monolayers. Furthermore, ibrutinib exhibited inhibitory effects on skin cancer cell proliferation, a phenomenon notably potentiated in the presence of 3MA, an autophagy inhibitor [36]. Likewise, the augmentation of ibrutinib’s anticancer activity in glioblastoma was observed upon autophagy inhibition by 3MA in vivo [35]. Moreover, the autophagy flux was inhibited in CLL cells via post-transcriptional modulation by chidamide, which could induce cytotoxicity [39]. Ibrutinib, as a pioneering therapeutic agent for CLL treatment, is widely prescribed and well-tolerated, manifesting rapid and enduring responses. Nonetheless, atrial fibrillation, a prevalent adverse effect, often necessitates therapy discontinuation in clinical trials [40]. The precise mechanism underlying ibrutinib’s off-target cardiac effects remains incompletely elucidated. However, our findings offer a novel hypothesis: ibrutinib induces connexin degradation, thereby elevating the incidence of re-entry phenomena in vitro. Considering our study alongside prior investigations, the potential therapeutic synergy of ibrutinib in combination with autophagy inhibitors in CLL patients warrants consideration. Such a combination regimen may yield enhanced tumor cytotoxicity and a reduced occurrence of ibrutinib-induced atrial fibrillation.
In our current investigation, we have uncovered a notable finding regarding the potential of ibrutinib to heighten susceptibility to atrial fibrillation in patients with CLL. Subsequent research has elucidated an association between ibrutinib-induced conduction abnormalities and the downregulation of Connexin 40 and 43. Moreover, heightened autophagic activity has been identified as a contributing factor to the decreased expression of Connexin 40 and 43. Notably, intervention through autophagy inhibition in HL-1 cells resulted in a substantial reversal of the degradation process. These findings suggest that targeting autophagy holds promise as a viable approach to mitigate ibrutinib-related atrial arrhythmia (Fig. 5).
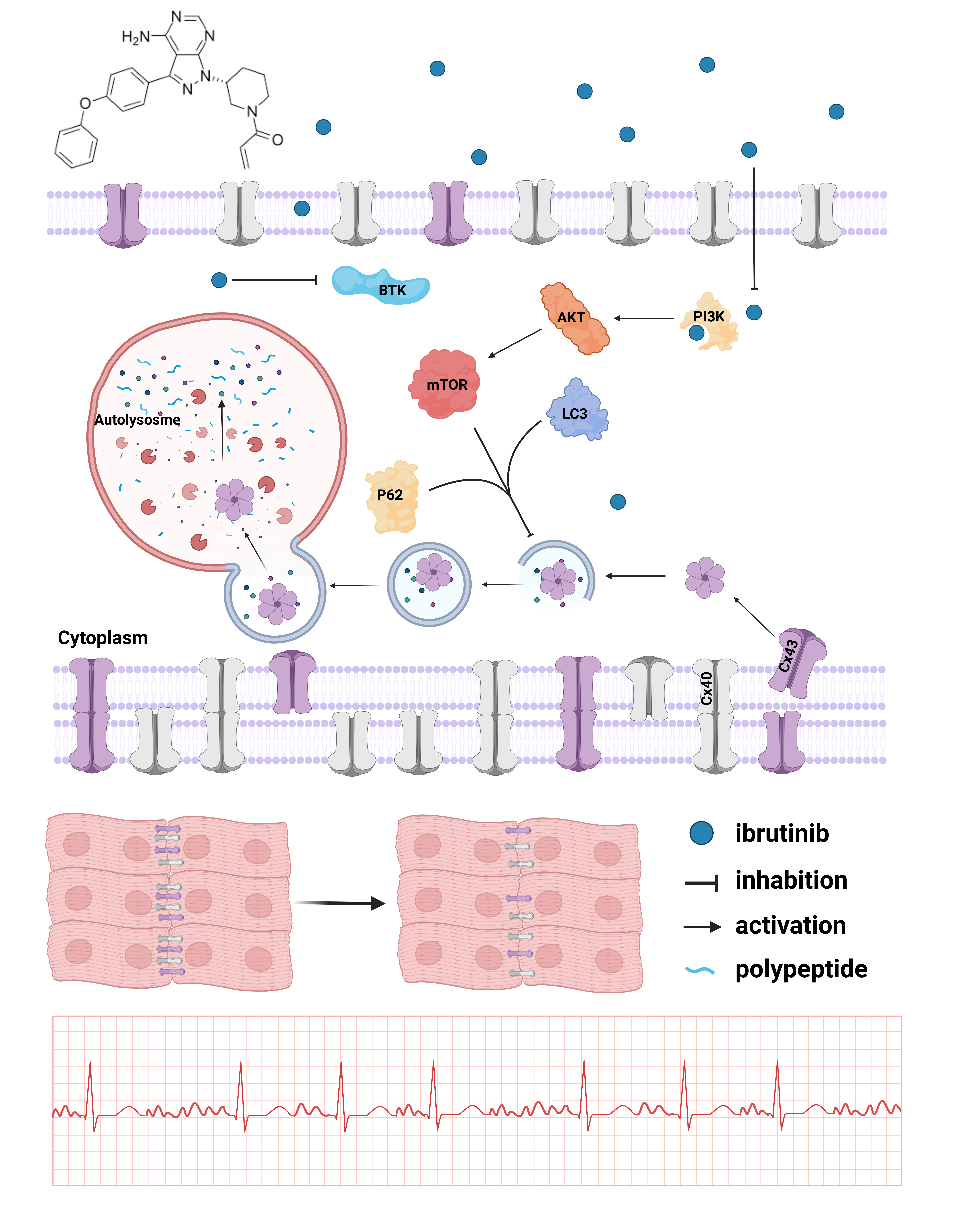 Fig. 5.
Fig. 5.Schematic diagram. Showing ibrutinib decreases Cx43 and Cx40 expression by inducing autophagy via the PI3K-AKT-mTOR signaling pathway in HL-1 cells. As a result, the decreased expression of Cx43 and Cx40 increases susceptibility to atrial fibrillation. (Created with https://www.biorender.com/).
The study’s supporting data are available on request from the corresponding author.
ZW, CC, and MC designed the research. HQ and BZ performed the research. YJ, HZ and CW collected and analyzed the clinical data of CLL patients. HQ and ZL analyzed the experiment data and drew figures and supplementary figures. HQ and CW wrote the manuscript. All authors have participated sufficiently in the work to take public responsibility for appropriate portions of the content and agreed to be accountable for all aspects of the work in ensuring that questions related to its accuracy or integrity. All authors read and approved the final manuscript. All authors contributed to editorial changes in the manuscript.
The clinical study was established, according to the ethical guidelines of the Helsinki Declaration and was approved by the Human Ethics Committee of The First Affiliated Hospital of Nanjing Medical University (2021-SR-006). Written informed consent was obtained from individual or guardian participants.
We would like to thank Dongxia Ji, Meng Sun, and Sanli Li (Feiyuebio, Wuhan, China) for working overtime during the Chinese Spring Festival to provide us with the ELISA kit.
This work was supported by a grant from the National Nature Science Foundation of China [grant number: 82100338 to Cheng Wang].
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.