1. Introduction
Chronic kidney disease (CKD) is a pathological condition that occurs when the
structure and function of the kidney changes, which in turn can lead to
complications, such as hypertension, edema, and oliguria, as well as high levels
of serum creatinine or blood urea nitrogen [1]. Kidney fibrosis (KF), the most
prevalent histological hallmark of CKD, is a condition marked by an increased
deposition of extracellular matrix (ECM) in the interstitium. KF disrupts kidney
architecture and exacerbates kidney dysfunction [2]. Nowadays, it is widely
accepted that KF involves an epithelial–mesenchymal transition (EMT) of renal
tubular epithelial cells prompted by abnormal activation of myofibroblasts and
subsequent ECM deposition. Transforming growth factor (TGF-)
regulates the most important processes of this complex mechanism. The first phase
of EMT is characterized by both the loss of stability of the various cell–cell
contacts and the disruption of apical–basal polarity. This destabilization is
caused by stimulating tubular epithelial cells through cytokines, such as the
epidermal growth factor (EGF), basic fibroblast growth factor (FGF-2), and
TGF-1. These cytokines are secreted mainly by infiltrating mononuclear
cells, partially reducing epithelial membrane junctions and tight junction
proteins, such as E-cadherin and zonula occludens. In the intermediate phase,
there is a shift towards the expression of new mesenchymal proteins, including
-smooth muscle actin (-SMA) and fibroblast-specific protein 1
(FSP-1) synthesis. In this stage, there is a protein reorganization of the
cytoskeleton and an upregulation of both matrix metalloproteinases (MMP-2 and
MMP-9) and enlarged spindle-shaped myofibroblasts, which show increased capacity
for cell migration and invasion into the tubular interstitium. The observation
that epithelial markers are expressed in interstitial cells in biopsies from End
Stage Renal Disease (ESRD) patients suggests that EMT is involved in KF. The
detection of FSP-1 in the tubular epithelium is another sign of EMT [3]. In human
biopsy samples, there is a correlation between tubular cells presenting EMT
markers, the appearance of interstitial fibrosis, and functional involvement [4].
However, there is a lack of support from in vivo cell-fate tracking
experiments in showing the direct involvement of EMT in contributing to the
myofibroblast population [5]. Recent studies have shown that a partial EMT
(pEMT—wherein tubular epithelial cells acquire one or two phenotypic markers
without leaving the local microenvironment) was induced by Snail1 or Twist-related protein 1 (TWIST1) in
tubular epithelial cells, leading to the onset of fibrosis. Consistent with this,
pEMT has been observed in clinical samples and was found to correlate with the
progression of fibrosis in renal allografts [5]. Inflammatory processes,
beginning with the infiltration and activation of macrophages, are considered to
be major factors leading to fibrotic diseases [6]. The recruitment of macrophages
in the glomerular or tubular interstitium is necessary to promote innate immune
responses and important defensive and disruptive reactions in renal disease. The
subsequent macrophage infiltration leads to kidney structure disruption and
tissue fibrosis development. Glomerular and interstitial macrophages, although
believed to be associated with the development of interstitial fibrosis, play an
important role in stromal remodeling during tissue repair [7, 8]. In addition to
releasing inflammatory cytokines, activated macrophages also secrete matrix
metalloproteinases (MMPs), including high levels of MMP-1, -3, -7, -9, -10, -12,
-14, and -25, along with lower levels of MMP-2, -3, -8, -10, -11, and -12 [9]. An
inflammatory injury in the kidney is also the result of these MMPs contributing
to the degradation of the extracellular matrix [10]. The profibrotic changes in
tubular epithelial cells [11] and the proteolytic activation of osteopontin in
kidney fibrosis led to macrophage recruitment, indicating that MMP-9 causes
fibrosis [12].
2. Matrix Metalloproteinases
Matrix metalloproteinases (MMPs) are zinc-dependent endopeptidase enzymes. They
function to remodel the ECM and are fundamental to tissue development and
homeostasis. Previously, MMPs have been considered to have a negative impact on
human health; today, their role in physiology and general health is being
progressively clarified [11, 12, 13]. In fact, MMPs regulate many physiological
processes. These include angiogenesis, embryogenesis, morphogenesis, and wound
healing, although they may also induce various pathological conditions, similar
to myocardial infarction, fibrotic diseases, osteoarthritis, and cancer.
Some studies demonstrated that specific increases in MMP could play a role in
arterial remodeling, aneurysm conformation, venous dilatation, and pulmonary
embolism diseases [14, 15]. Many MMPs are secreted as pro-MMPs, requiring activation through
prodomain cleavage, typically by plasmin or other MMPs. Indeed, 24 MMP
genes have been identified in the human genome. Notably, MMP-23A is a
duplicated version of MMP-23B and is likely a pseudogene [16, 17]. Each
MMP contains the following components: (1) an N-terminal signal peptide that
directs the enzyme to the secretory pathway and (2) a prodomain with a conserved
PRCGXPD sequence responsible for the latency of the enzyme bound to a conserved
HEXGHXXGXXH motif [18]. Four twisted -sheets are present in the
hemopexin-like domain’s ‘propeller’, allowing substrate recognition [14]
Together, the hinge region and the hemopexin-like domain break down substrate
proteins for further degradation. The C-terminal domain interacts with Tissue inhibitors of metalloproteinases (TIMPs) and
participates in substrate recognition [19]. MMPs are also able to cleave a range
of non-ECM substrates, such as cell adhesion molecules (cadherins and integrins),
as well as growth factors and their receptors (TGF-), fibroblast growth
factor receptor 1 (FGF-R1) [20]. Tissue inhibitors of metalloproteinases
or TIMPs, which have been identified in vertebrates, control the activities of
MMPs. There are four TIMP members, TIMP-1, -2, -3, and -4, that inhibit all MMPs,
except for TIMP-1, which does not inhibit MMP-14, -16, or -24 [21]. TIMPs can
induce intracellular signaling such as proliferation, angiogenesis, and apoptosis
[22]. Based on substrate and sequence homology, MMPs are classified into six
distinct groups: collagenases (MMP-1, -8, -13, -18), gelatinases (MMP-2, -9),
stromelysins (MMP-3, -10, -11), matrilysins (MMP-7, -26), membrane-bound matrix
MMPs (MMP-14, -15, -16, -17, -24, -25), and the “other MMPs” (MMP-12, -19, -21,
-23a, -23b, -28) [21]. TIMPs are also classified numerically from TIMP-1 to
TIMP-4 [23]. Gelatinases are responsible for cleaving and further degrading
triple-helical fibrillar collagens. They also degrade basement membrane
constituents; for example, MMP-2 (but not MMP-9) can degrade laminin and
fibronectin [24]. Stromelysins degrade extracellular matrix proteins but not
triple-helical fibrillar collagens. Membrane-bound MMPs activate many proteases
and growth factors. All TIMP molecules have 190 amino acids, which are folded
into two distinct domains. These domains comprise a larger N-terminal domain
(approximately 125 amino acid residues) and a smaller C-terminal domain. Three
conserved disulfide bonds stabilize each domain. The N-terminal domain alone can
fold individually and is fully functional for MMP inhibition. While the function
of the C-terminal domain is not fully understood, it has been shown to tightly
bind to the hemopexin domain of latent MMPs [20]. Regulation of MMP activity may
occur through several mechanisms: (1) gene expression, by both transcriptional
and posttranscriptional modifications; (2) compartmentalization that occurs by
incorporating cell-specific and tissue-specific release of extracellular MMPs; or
(3) through the zymogen activation of the pro-enzyme forms of MMPs achieved by
the removal of the 80–84 residue terminal domain; (4) inhibition by TIMPs. While
TIMPs 1–4 all share some sequence similarity, TIMPs 2, 3, and 4 are more
identical than TIMP-1. Furthermore, TIMP-2 is unique in that it can both inhibit
and activate MMPs [25]. All TIMPs inhibit the pro- and active forms of the MMPs,
although with relatively low selectivity, and with which they form tight
non-covalent complexes. TIMP-1 has shown low inhibitory activity against MMP-19
and membrane-bound MMP-14, -16, and -24 while displaying greater potency against
MMP-3 and MMP-7 than TIMP-2 and TIMP-3 [26]. TIMP-2 has an effective inhibitory
activity against all MMPs. Similarly, TIMP-3 inhibits all MMP members while
extending its inhibitory profile to include members of the ADAMs (a disintegrin
and metalloprotease) family of metalloproteinases. MMP-1, -2, -3, -9, -11, -13,
-14, -24, -25, -27, -28 and TIMP-1, -2, -3, -4 are expressed in the kidney [27].
The distribution of MMPs and TIMPs in human kidney tissue and their localization
at the subcellular level and immunohistochemical study are available at
https://www.proteinatlas.org/[28].
Matrix Metalloproteinases in Kidney Fibrosis
The accumulation of proteins in the extracellular matrix and an increased
activity of myofibroblasts cause the development and progression of renal
fibrosis. In this process, MMPs and TIMPs play a very important role, and this is
demonstrated, for example, by the role that MMP-2 and MMP-9 play in particular.
The latter causes the EMT of tubular cells. It favors the activation of resident
fibroblasts, endothelial–mesenchymal transition (EndoMT), and
pericyte–myofibroblast transdifferentiation [29]. Many cells, including
mesangial, endothelial, epithelial, collecting duct tubular cells, macrophages,
and fibroblasts, express MMP-2 and MMP-9, which could directly induce the entire
course of renal tubular cell EMT, as demonstrated by in vitro studies
[30, 31]. MMPs are usually responsible for the degradation of mature ECM proteins,
such as collagens and fibronectin. TIMPs decrease MMP activity and reduce ECM
degradation rates. The accumulation of collagen types I, III, and IV in the
glomeruli, interstitial tissue, and vessels causes progressive renal scarring.
TGF-1 induces increased MMP-9 expression and decreased TIMP-1
expression, altering ECM homeostasis and leading to kidney fibrosis. This process
has been recognized as an essential component of EMT activation and permits the
migration and invasion of newly transformed mesenchymal cells in the interstitial
space, resulting in fibrosis progression through extracellular matrix deposition
[32]. However, it should be noted that some investigators have reported an
increased expression of TIMP-1 by TGF-1. One study observed the
induction of TIMP-1 by TGF-1 via the proximal promoter activator protein
1 (AP1) site while having the opposite effect on MMP-1 expression [33]. Another
study investigating animal models of intestinal fibrosis observed increased
phosphorylation of Smad-2 and Smad-3 proteins upon treatment of myofibroblasts
with TGF-1 and increased TIMP-1 levels [34]. Akool et al. [35]
also noted TIMP-1 expression induced by nitric oxide (NO) in mesangial cells—NO
exerted this effect through the TGF-1/Smad-2 signaling pathway. Yang and
co-workers underlined the importance of MMPs in disrupting the tubular basement
membrane and demonstrated that the tubular basement membrane integrity was
preserved due to the indirect reduction of MMP-9 activity in mice with
tissue-type plasminogen activator (t-PA) deficiency. This reduction caused a
decrease in tubular cell EMT and kidney fibrosis in obstructive nephropathy.
Furthermore, Fernandez-Patron and Leung [29] reported that MMP-2 could
reduce inflammation because it antagonizes phospholipase A2 (PLA2). Additionally,
MMP-2 may facilitate the activation of the MMP-1, MMP-9, and MMP-13 zymogens.
MMP-2 performs many pathophysiological functions, creating a close association
between fibrosis and inflammation. In fact, many studies report that it is
produced in many nephron sites (such as the glomerular mesangial cells, renal
tubular epithelial cells, and interstitial cells) and can play different roles
during the progression of CKD. The rapid progression of CKD is also due to the
ability of MMP-2 to degrade the basement membrane, alter the glomerular
filtration membrane, activate TGF-1, and promote the phenotypic
transformation of tubular epithelial cells [36]. Deficient MMP-2 activity leads
to renal tubular atrophy, fibrosis, and excessive ECM deposition in the basement
membrane and interstitium, a sign of advanced chronic kidney disease. This not
only contributes to ECM degradation and the development of chronic inflammation
but also leads to insufficient degradation of endothelin-1, adrenomedullin, and
calcitonin gene-related peptides by MMP-2, exacerbating the intrarenal arteriolar
spasms and sclerosis [30]. MMP-7 may also contribute to renal fibrosis mainly
through EMT transition, TGF- signaling, and ECM accumulation [31]. MMP-7
mediates EMT through three distinct pathways: Through the first mechanism, MMP-7
causes ECM destruction by cleaving collagen type IV and laminin. Secondly, MMP-7
mediates E-cadherin degradation, leading to a disruption of tubular epithelial
integrity. The last process induces the expression of the Fas ligand (FasL) in
interstitial fibroblasts, leading to apoptosis [31]. MMP-7 expression in
the kidney is also upregulated by age, and its level correlates with renal
healing [37]. The thickening of the basement membrane and mesangial expansion
precede the onset of renal scarring and are favored by MMP-7. The latter also
enzymatically cleaves numerous extracellular matrix proteins such as type IV
collagen, laminin, fibronectin, proteoglycans, and entactin. Interestingly, in a
rat model of age-associated renal interstitial fibrosis, MMP-7 expression is
significantly elevated in fibrotic samples, whereas non-fibrotic samples do not
show this expression profile. Furthermore, the expression of MMP-7 strongly
correlates with the fibrosis degree [38].
Zhang et al. [39] investigated the role of TIMP-1 in fibrosis using a
transgenic TIMP-1 mouse model and observed that intercellular adhesion molecule-1 (ICAM-1) expression was increased
in these mice partly due to MMP-9 inhibition. They also noted that the increase
in ICAM-1 level led to increased renal macrophage infiltration, kidney collagen
levels, and renal fibrosis, which correlated with the age of the transgenic
animal. Zhang et al. [39] suggested that an imbalance between TIMPs/MMPs
and inflammation could lead to fibrosis. A recent study utilizing TGF-1
transgenic mice also demonstrated that renal fibrosis was correlated with TIMP-1
expression, and using an anti-TIMP1 neutralizing antibody ameliorated
glomerulosclerosis and tubulointerstitial fibrosis [40]. They also observed
increased TIMP1 levels in human kidneys with focal segmental glomerulosclerosis,
suggesting a correlation between TIMP-1 and fibrosis. Other groups have also
noted a correlation between TIMP-1 and renal fibrosis: Increased expression of
TIMP-1 mRNAs in glomerular resident cells and tubular epithelial cells have been
associated with interstitial injury [41]; a reduction in TIMP-1 mRNA levels was
associated with improved symptoms of nephritis in a murine model of lupus
nephritis [42]; decrease in TIMP-1 expression was also accompanied by a reduction
in renal collagen expression and glomerulosclerosis in diabetic rats treated with
the soluble guanylate cyclase activator cinaciguat [43].
3. MMP Gene Polymorphisms and Kidney Disease
Gene polymorphisms can influence both the expression and activity of MMP-9. Two
functional polymorphisms affecting the transcriptional activity of the
MMP-9 gene were identified in the promoter region, namely the
C-1562T (rs3918242) and microsatellite (CA) (rs3222264) polymorphisms [44]. In particular, the microsatellite
(CA)n region located near the -90 positions functions as a binding site
for a specific DNA regulatory protein and also promotes the unwinding of the DNA
helix and the transcription process by allowing the DNA to adopt a Z structure.
The longest repeat alleles of the microsatellite were correlated with the highest
activity, as demonstrated in vitro. The single nucleotide polymorphism
C-1562T prevents the nuclear repressor protein from binding to the
MMP-9 gene promoter region, resulting in increased MMP-9 expression, as
observed in some in vitro studies [45, 46]. A large Japanese cohort study
has highlighted that the MMP-9-1562T/279R/668Q haplotype reduced the
risk of CKD compared with the MMP-9-1562C/279R/668R. Furthermore, the
MMP-9-1562TT/279RR/668QQ genotype combination has been linked to a
decreased CKD risk compared with the major combination of homozygous allele
MMP-9 -1562CC/279RR/668RR. The association of these genotypes supports
the idea that they play a protective role in kidney disease and could cause an
increase in MMP-9 expression [47]. Earlier studies have also shown that the
CC genotype for the -1562 C/T (rs3918242) polymorphism in the
promoter region causes a significant increase in serum MMP-9 in hemodialysis (HD)
patients compared with the CT genotype [48]. Conversely, in
nephrolithiasis, the -1562 TT (rs3918242) genotype, but not the
Q279R (rs17576) single nucleotide polymorphism (SNP) of the
MMP-9 gene, was associated with a higher risk of lithiasis [49]. It has
been shown, in a 3-year study of HD patients, that the 2G/2G homozygous
genotype for MMP-1 was associated with increased mortality in contrast to the
6A/6A homozygous genotype for MMP-3 in the same population [48]. These
findings suggest that investigating the genetic polymorphisms in MMP genes might
help identify high-risk populations that can benefit from a targeted treatment
approach.
4. MMP-9 Signaling
Identifying the molecular components involved in the signal transduction
pathways activated by MMPs may allow interventions to be developed by modifying
their activity. However, there have only been a few studies investigating the
signaling pathways involved in the cellular actions of MMP-9, with several
molecules identified as playing a role in MMP-9 signaling, as discussed in the
following subsections.
4.1 p38 Mitogen-Activated Protein Kinase and NF-kB
In addition to its proteolytic action on the ECM, MMP-9 activates
interleukin-1 [50] and plays a role in cell signaling. Notably, a study
by Al-Sadi et al. [51] reported that MMP-9, at clinically relevant
concentrations, rapidly upregulated p38 mitogen-activated protein kinase (MAPK)
levels in Caco-2 cells. This, in turn, led to an increase in myosin light-chain
kinase gene and protein levels. However, no details on the mechanism through
which MMP-9 caused activation of the MAPK were given. Furthermore, it was noted
that MMP-9 did not activate the extracellular-signal-regulated protein kinases
(ERK)1/2 and c-jun N-terminal kinase (JNK) members of the MAPK family [51]. In a
subsequent report, the same authors observed that p38 activation led to the
activation and subsequent nuclear translocation of the proinflammatory
transcription factor nuclear factor kappa B (NF-B) [52]. Notably, the
p38 isoform may regulate MMP-9 expression [53]. While these studies
were conducted in non-renal cells, it is possible that MMP-9 could have similar
effects in renal cells. In such a scenario, the p38/NF-B axis would
lead to inflammation and increase MMP-9 levels, creating a positive feedback loop
that could intensify the inflammatory milieu.
4.2 Collagen Degradation Products
MMP-9 action on the ECM leads to the production of collagen fragments, such as
the tripeptide N-acetyl Pro–Gly–Pro (Ac-PGP), a neutrophil chemoattractant
[54]. AcPGP also binds to neutrophils to induce further production of MMP-9
via an ERK1/2–MAPK-dependent pathway [22], creating a positive feedback
loop. AcPGP binding appears to be on the CXC motif chemokine receptor 1 and CXC
motif chemokine receptor 2 (CXCR1 and CXCR2) receptors on neutrophils and may
interact with these receptors wherever they are present in other cells, including
renal cells [55].
4.3 Notch Signaling
Zhao et al. [56] reported the induction of Endo-MT in human kidney
glomerular endothelial cells (HKGECs) by recombinant MMP-9 (rMMP-9). It was
observed that rMMP-9 caused a decrease in Notch1 receptor levels while
simultaneously increasing the levels of the Notch intracellular domain (NICD)
[56]. The Notch signaling pathway consists of the Notch receptors together with
several ligands, although in the canonical signaling pathway, Notch is cleaved to
form the NCID, which is then transported to the nucleus to regulate transcription
of its target genes [57]. Zhao et al. [58] also demonstrated that using
a Notch pathway inhibitor, a -secretase inhibitor (GSI), inhibited the
rMMP-9-induced morphological changes and decreased NICD levels in HKGECs. In a
following study performed by the same group, Zhao et al. [58] used
primary mouse renal peritubular endothelial cells (MRPECs) to observe Endo-MT
following induction by MMP-9. Once again, rMMP-9 caused an increase in NICD,
whose levels were decreased by GSI. Furthermore, in MRPECs isolated from MMP-9
knockout (KO) mice and subjected to TGF-1, higher levels of Notch1 and
lower levels of NICD were observed. Finally, Zhao et al. [58] performed
unilateral ureteral obstruction (UUO) of the kidneys in MMP-9 KO and wild-type
mice to induce fibrosis. Zhao et al. [58] noted that the MMP-9 deficient UUO
kidneys had less fibrosis, and levels of hairy/enhancer-of-split related with
YRPW motif 1 (Hey-1), a downstream target gene in the Notch signaling pathway,
decreased, implicating Notch signaling in the MMP-9 mechanism of action.
Interestingly, it is worth mentioning that a complex interaction between Notch
and NF-B exists, which could activate the NF-B pathway (Fig. 1) [59].
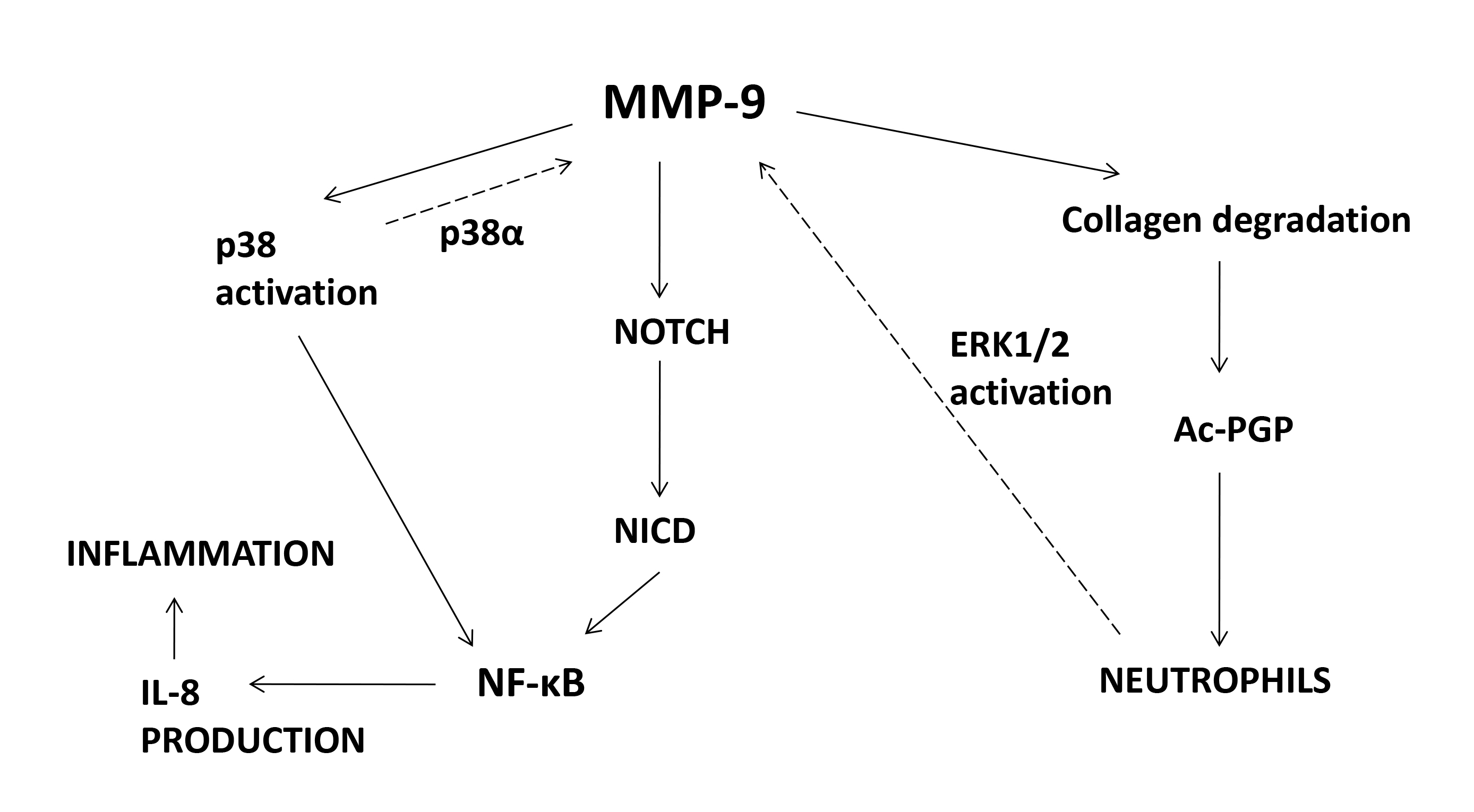 Fig. 1.
Fig. 1.
Signaling molecules affected by MMP-9. MMP-9, matrix
metalloprotease-9; Ac-PGP, N-acetyl–proline–glycine–proline; NICD, Notch
intracellular domain; IL-8, interleukin-8; NF-B, nuclear
factor-B; ERK1/2, extracellular signal-regulated protein kinase 1/2;
p38, p38 mitogen-activated protein kinase.
5.MMP-9 as a Promising Therapeutic Target
Several studies have indirectly demonstrated the involvement of MMP-9 in renal
fibrosis through the inhibition of t-PA (as t-PA is an inducer of MMP-9) and
through knockout mice. To determine the role of MMP-9 in vivo, direct
inhibition of MMP-9 activity is the recommended approach [60]. Indeed, a study
[60] has shown that inhibiting the activity of the MMPs early, particularly of
MMPs-2, 3, and 9, seems to protect against Alport’s disease in mice deficient in
the 3(IV) chain of type IV collagen. The study showed that late-stage
inhibition of MMP activity results in more rapid disease progression, which is
associated with interstitial fibrosis and early mortality. Some macrophage
phenotypes may contribute to fibrosis, and evidence suggests that neutralizing
proinflammatory macrophages may prevent renal fibrosis [61]. In vitro
studies have shown that MMP-9 cleaves osteopontin, inducing migration of
macrophages, and that MMP-9 from both tubular epithelial cells (TECs) and
macrophages induces EMT in tubular cells [12]. In the same study, a unilateral
ureteral obstruction murine model of renal fibrosis was used to show that TECs
were the main source of MMP-9 in the early stages of UUO, whereas TECs,
macrophages, and myofibroblasts were the main sources in the later stages. Using
a murine renal tubular epithelial cell line, a study investigated the effects of
incubating these cells in an activated macrophage-conditioned medium (AMCM)
containing MMP-9 secreted by Lipopolysaccharide (LPS)-stimulated macrophages. They noted that the AMCM
could induce EMT in tubular cells, and this effect was inhibited in the presence
of MMP-9 inhibitors and by immunoprecipitation of MMP-9 from the AMCM [11]. A
recent study also demonstrated how diosmin has a potential multicomponent
molecular mechanism of action in treating renal fibrosis. The study indicates
that caspase 3, MMP-9, annexin A5, and heat shock protein 90-alpha family class A
member 1 could be important direct targets of diosmin and that the MAPK, Ras,
phosphatidylinositol-3 kinase (PI3K) Protein kinase B (Akt), FoxO, and hypoxia
inducible factor-1 (HIF) signaling pathways could play a major role in its
mechanism of action. Further studies on diosmin in treating renal fibrosis should
be performed to demonstrate its efficacy [62]. However, as evidence for the role
of MMP-9 in renal fibrosis increases, understanding its secretion, regulation,
and mechanism of action is necessary to develop therapeutics to regulate its
action.
Control of Secretion and Expression
Bellosta et al. [63] reported that the hydroxyl methyl glutaryl
coenzyme A (HMG-CoA) reductase inhibitor (statin), Fluvastatin, could reduce
MMP-9 activity by up to 50% in human macrophages. Furthermore, they hypothesized
that the statin interfered with the prenylation of small Ras-like guanine
nucleotide-binding proteins (GTPases) that regulate membrane traffic involving
endocytic and exocytic processes, leading to reduced MMP-9 secretion by
macrophages. Other studies have also reported similar effects of various statins
on MMP-9 secretion by different cell types and macrophages [64, 65, 66]. However,
other workers have observed that under certain conditions, some statins may
increase MMP-9 levels in macrophages [67]. Surprisingly, one report attributes
the beneficial effects of a hydrophilic statin, rosuvastatin, on preventing an
increase in MMP-2 and a decrease in MMP-9 in hypertensive stroke-prone rats [68].
Confocal microscopic analysis of kidney sections from Zucker diabetic rat kidneys
showed increased MMP-9 in the glomeruli, particularly in the parietal epithelial
cells (PECs) [69]. Albumin administration of cultured primary PECs resulted in a
dose-dependent increase in MMP-9 in the culture medium, together with increased
phosphorylation (activation) of extracellular-signal-regulated kinases (ERKs)1/2 of the mitogen-activated protein kinase (MAPK) family. Other kinases
investigated included Akt and p38 MAPK, which showed no increase in their
phosphorylation status. Furthermore, using the inhibitor U0126, which inhibits
the upstream kinases, MAPK kinase1/2 (MEK1/2), of the ERK1/2 kinases, resulted
in lower MMP-9 levels. Many factors positively regulate MMP-9 gene expression
including transcription factors E-26 (Ets), NF-B, polyomavirus
enhancer-binding protein 3 (PEA3), activator protein 1 (AP-1), specificity
protein 1 (Sp-1) and serum amyloid A activating factor (SAF)-1 [70], all of which
may be considered as viable targets for MMP-9 regulation, together with the
signal transduction pathways that modulate their activity. However, more research
must be undertaken to delineate these pathways in renal cells and determine the
conditions under which they are activated.
6. Conclusions
Renal fibrosis is caused by excessive accumulation of the extracellular matrix
(ECM), and its pathogenesis is a progressive process that leads to end-stage
renal disease. The accumulation of ECM and uncontrolled matrix degradation play
an important role, mainly involving MMPs and TIMPs, which are particularly
important in the development and progression of renal glomerulosclerosis and
tubular interstitial fibrosis. MMPs have traditionally been considered
antifibrotic factors due to their proteolytic activity and extracellular matrix
degradation. However, a decrease in MMP proteolytic activity or an increase in
MMP tissue inhibitors (TIMPs) is thought to be responsible for extracellular
matrix accumulation and fibrosis [27]. In particular, in this review, we have
highlighted the important role that MMP-9 plays in up-regulating pathways
involved in renal fibrosis [58]. Therefore, further studies are needed to
demonstrate that inhibition of MMP-9 pathways could represent a therapeutic
strategy for treating renal fibrosis in chronic kidney disease. In addition, it
would be interesting to understand the intracellular signaling pathways involved
in the action of MMPs, particularly the role of MMPs and TIMPs in the kidney, to
elucidate the underlying mechanisms and develop biomarkers and therapeutic
targets for renal fibrosis.
Author Contributions
ALR, RS and MA designed the study. ALR, RS, TF, GCr, AB, GCo, DB, YB, NI, DC, AM
and MA performed the search and analyzed the data. ALR, RS, AM and MA wrote the
manuscript. All authors contributed to editorial changes in the manuscript. All
authors read and approved the final manuscript. All authors have participated
sufficiently in the work and agreed to be accountable for all aspects of the
work.
Ethics Approval and Consent to Participate
Not applicable.
Acknowledgment
Not applicable.
Funding
This research received no external funding.
Conflict of Interest
The authors declare no conflict of interest. Raffaele Serra is serving as one of
the Editorial Board members of this journal. We declare that Raffaele Serra had
no involvement in the peer review of this article and has no access to
information regarding its peer review. Full responsibility for the editorial
process for this article was delegated to Amancio Carnero Moya.


 , Teresa Faga 3, Giuseppina Crugliano 1, Angelica Bonelli 1, Giuseppe Coppolino 1, Davide Bolignano 2, Yuri Battaglia 4,5, Nicola Ielapi 6, Davide Costa 7, Ashour Michael 1, Michele Andreucci 1
, Teresa Faga 3, Giuseppina Crugliano 1, Angelica Bonelli 1, Giuseppe Coppolino 1, Davide Bolignano 2, Yuri Battaglia 4,5, Nicola Ielapi 6, Davide Costa 7, Ashour Michael 1, Michele Andreucci 1