


1 Laboratory of Integrated Medicine Tumor Immunology, Shanxi University of Chinese Medicine, 030000 Taiyuan, Shanxi, China
2 Department of Pathobiology and Immunology, Hebei University of Chinese Medicine, 050200 Shijiazhuang, Hebei, China
†These authors contributed equally.
Abstract
The Warburg effect, also called aerobic glycolysis, refers to tumor cells that metabolize glucose through glycolysis even in the presence of oxygen. This rapid breakdown of glucose fuels the fast development, growth, and migration of tumor cells. Lactate, the final product of aerobic glycolysis, contributes to an acidic environment within the tumor, promoting the formation of an immunosuppressive microenvironment and accelerating tumor progression by impeding anti-tumor immunity. Numerous studies have confirmed the critical role of aerobic glycolysis in the occurrence and development of hepatocellular carcinoma by influencing tumor cells proliferation, invasion, metastasis, apoptosis, immune escape, angiogenesis, and more. Clinical trials have shown that inhibitors of rate-limiting enzymes in the glycolysis pathway can enhance the effectiveness of sorafenib, a targeted drug for hepatocellular carcinoma, by reducing drug resistance. Additionally, active components of traditional Chinese medicine and specific compound prescriptions are gaining attention for their potential to target and regulate aerobic glycolysis in hepatocellular carcinoma. Therefore, inhibiting the aerobic glycolysis pathway holds promise as a therapeutic strategy for treating liver tumors. This manuscript aims to review the role, research directions, and clinical studies of aerobic glycolysis in hepatocellular carcinoma.
Keywords
- Warburg effect
- hepatocellular carcinoma
- tumor microenvironment
Liver cancer, one of the most common primary malignant cancers, ranks third in cancer-related mortality [1]. Hepatocellular carcinoma (HCC), accounting for 75%–85% of cases, is the predominant type [1]. Currently, surgical resection and radiofrequency ablation remain the mainstay of treatment for early-stage liver cancer, while interventional therapy and radiotherapy are typically used for intermediate and advanced stages. However, due to the high incidence of drug resistance, the prognosis for HCC remains poor [2]. Studies have shown that atezolizumab plus bevacizumab improves recurrence-free and progression-free survival compared to traditional sorafenib treatment [3, 4]. Although immunotherapy has offered new hope for patients, its curative effect is limited, and patient survival times are still short. Therefore, exploring new therapeutic targets for HCC is crucial for developing more effective treatment strategies.
Over the years, many studies have shown that metabolic reprogramming, a significant characteristic of tumors, has gained increasing attention in understanding tumor development. One form of energy metabolism, glycolysis, provides an energy source for tumors [5, 6]. Notably, the liver plays a crucial role in glucose metabolism by regulating glycogen synthesis and breakdown. In the 1920s, Otto Heinrich Warburg made a groundbreaking discovery, observing abnormal glucose metabolism in rat hepatoma cells. Even in the presence of sufficient oxygen, these cells prefer to obtain energy through glycolysis rather than the more efficient mitochondrial oxidative phosphorylation (OXPHOS) pathway, a phenomenon known as the Warburg effect [7]. The main hallmarks of aerobic glycolysis in tumor cells are increased glucose uptake and lactate release. Adenosine triphosphate (ATP) is the main energy source in the body. Despite its inefficiency in ATP production, aerobic glycolysis generates 50% to 70% of ATP in various tumors [8]. Additionally, lactate excretion fosters an acidic tumor microenvironment (TME) linked to tumor progression by facilitating invasion and metastasis [9]. Therefore, targeting tumor aerobic glycolysis holds promise for treating patients with HCC.
Given the key role of aerobic glycolysis in HCC growth, invasion, and treatment. This paper aims to review its characteristics, its relationship with the TME, and the current research and clinical applications of glycolysis in HCC.
There is a growing consensus suggesting that in HCC, aerobic glycolysis not only exists but is also markedly enhanced [10, 11]. This phenomenon is ubiquitous in HCC and contributes to various aspects of tumor progression. Numerous studies have confirmed that aerobic glycolysis plays a role in tumor cell proliferation, invasion, metastasis, apoptosis, cell cycle, immune escape, angiogenesis, and more [12, 13]. Enhanced aerobic glycolysis is often associated with a poor prognosis for HCC patients [14]. It is well-established that tumor tissues generally exhibit lower glucose levels and higher lactate levels compared to normal tissues [15]. Similar enhancement of aerobic glycolysis has been observed in various other cancers, including cervical cancer [16], breast cancer [17], pancreatic cancer [18], lung cancer [19], gastric cancer [20], and colorectal cancer [21].
While normal cells primarily rely on mitochondrial OXPHOS for energy generation from glucose, tumor cells often depend heavily on glycolysis. However, it is important to note that aerobic glycolysis provides only a portion of the energy needed by most tumor cells [15]. In most normal cells, around 90% of ATP is derived from OXPHOS, with the remaining 10% coming from aerobic glycolysis. While less efficient in ATP production, tumor cells can obtain up to 60% of their ATP through aerobic glycolysis [22]. One study suggested that tumor cells choose the less efficient aerobic glycolysis for two reasons: faster ATP production compared to OXPHOS and the generation of downstream biomacromolecules needed for cell proliferation, achieved by consuming more glucose [23]. This unique ATP production pathway allows tumor cells to take up more glucose, ultimately meeting their specific energy needs during development [6]. In conclusion, the selection of aerobic glycolysis during HCC development appears to be driven by its ability to meet the unique metabolic demands of tumor cells.
Increased glucose uptake, achieved through the glucose transporters (GLUTs) family that facilitates transport from the extracellular space to the cytoplasm, is a well-established characteristic of HCC [24]. GLUTs are frequently upregulated in various cancers, including HCC, colorectal cancer, breast cancer, lung adenocarcinoma, squamous cell carcinoma, ovarian cancer, and glioblastoma, due to mechanisms like gene expression changes, protein relocation, or stability alterations [25]. Specifically, high GLUT1 expression promotes increased glucose uptake, sustains and accelerates aerobic glycolysis, generates substantial lactate, and influences angiogenesis, migration, and immune escape in HCC [26]. Interestingly, GLUT1 expression has been reported to be negatively correlated with GLUT2 expression. While GLUT2 protein levels were reported to be lower in human HCC compared to normal liver tissue and negatively associated with malignant tumors in previous studies [26], its overexpression has also been documented in HCC samples [27]. Similarly, GLUT3 overexpression has been linked to decreased overall survival in HCC patients [28]. In conclusion, GLUTs play a crucial role in maintaining the energy demands of tumor cells across various biological processes, making them potentially promising targets for anticancer therapies.
It is now understood that three key enzymes limit the rate of aerobic glycolysis
in HCC: hexokinases (HKs), phosphofructokinases (PFKs), and pyruvate kinases
(PKs). Notably, the overexpression of these enzymes has been associated with a
poor prognosis for HCC patients [29]. The first rate-limiting step occurs when
glucose enters tumor cells and is phosphorylated by HKs to form
glucose-6-phosphate (G-6-P) [30]. While there are four HK subtypes (HK1, HK2,
HK3, and HK4), most normal tissues only express HK1. However, high levels of HK2
in HCC tissues are indicative of a poorer prognosis [31]. Next, PFK1 catalyzes
the conversion of fructose-6-phosphate (F-6-P) to fructose-1,6-bisphosphate
(F-1,6-BP) using ATP [32]. Elevated PFK1 expression has been shown to promote
both aerobic glycolysis and tumor cell proliferation [33]. Finally, PKs, the last
rate-limiting enzyme in the aerobic glycolytic process, convert
phosphoenolpyruvate (PEP) to ATP and pyruvate. Pyruvate kinase M2 (PKM2),
specifically, is significantly upregulated in tumor cells and plays a dual role
in both regulating tumor cells metabolism and contributing to poor prognosis
[22]. Its expression is controlled by various signaling pathways and
transcription factors, with HIF-1
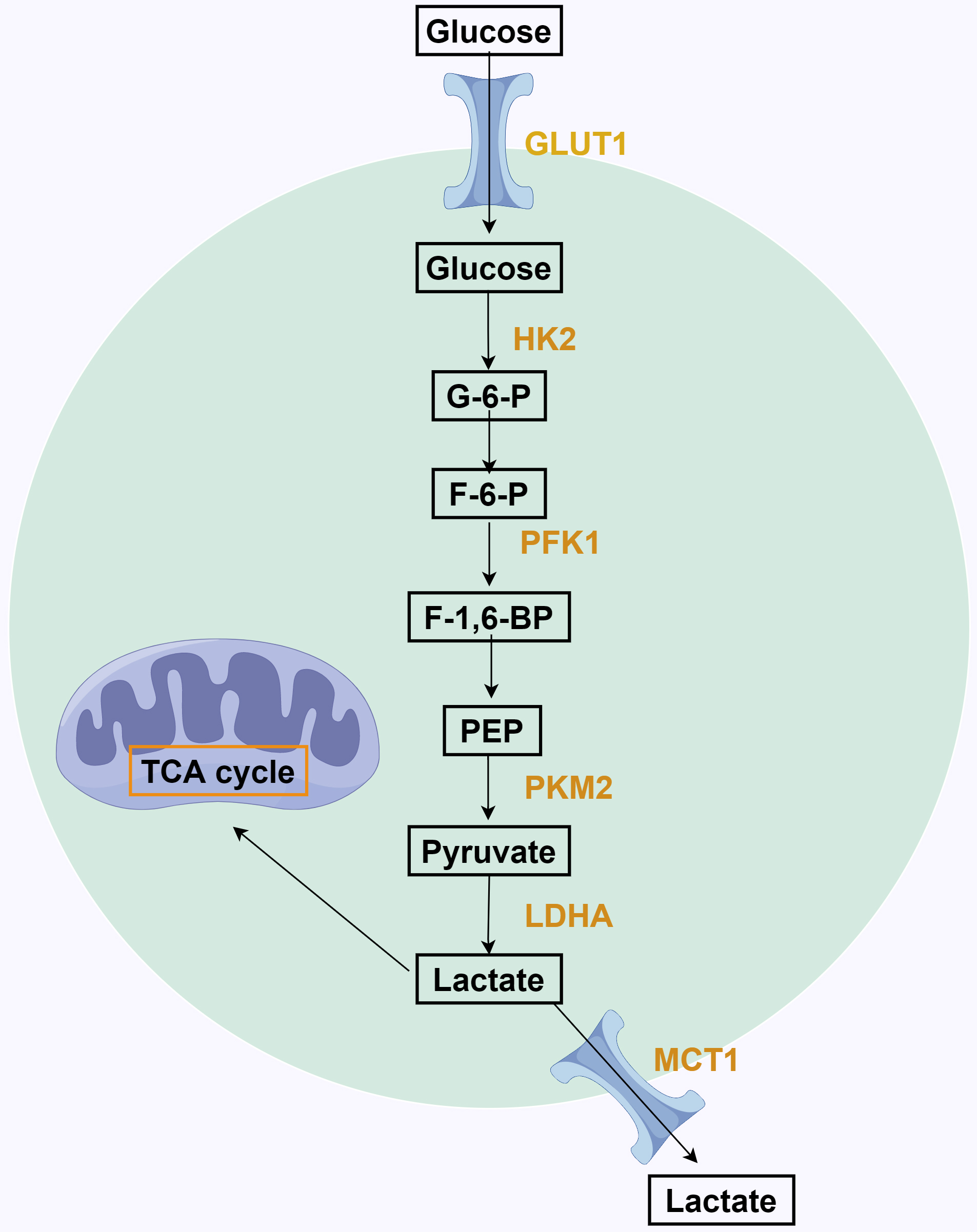 Fig. 1.
Fig. 1.The process of aerobic glycolysis in tumor cells. Glucose entry into tumor cells is catalyzed by hexokinases 2 (HK2) to form Gmur6murp, which is then generated by phosphofructokinases 1 (PFK1) catalysis of Fmur6murp. Then, phosphoenolpyruvate (PEP) produces pyruvate through pyruvate kinase M2 (PKM2), and pyruvate forms lactate under the action of lactate dehydrogenase A (LDHA). Finally, lactate is excreted out of the cell through monocarboxylic acid transporters 1 (MCT1). TCA, tricarboxylic acid; F-1,6-BP, fructose-1,6-bisphosphate; F-6-P, fructose-6-phosphate; G-6-P, glucose-6-phosphate.
A large amount of lactate produced by tumor cells not only acidifies the TME but
also promotes tumor angiogenesis, metastasis, drug resistance, and
immunosuppression [38]. The TME, the immediate environment supporting cell
survival, consists of cellular components (T cells, regulatory T cells (Tregs),
natural killer (NK) cells, tumor-associated macrophages (TAMs)) and non-cellular
components (chemokines, cytokines, and other molecules). This acidic TME, also
known as tumor acidosis, results from the accumulation of lactate and H
Recently, the link between aerobic glycolysis and cytokines has gained
increasing attention. Glycolysis promotes the production of the effector molecule
interferon-
In HCC, aerobic glycolysis not only occurs in tumor cells but also extends to
immune cells within the TME [48]. Damaged immune cells with compromised function
contribute to disease progression [49]. T cell function in the TME is intricately
linked to glucose availability, extracellular lactate accumulation, and
interactions with tumor cells. High glucose uptake in tumor cells often leaves T
cells starved, leading to dysfunction [48, 50]. Notably, T cells glucose
transporter GLUT1 is downregulated in tumors, hindering the effector function of
CD4
Unlike normal cells, tumor cells, including HCC cells, primarily rely on aerobic glycolysis for energy production. This phenomenon, known as the Warburg effect, allows HCC cells to thrive and proliferate even under hypoxic conditions [60]. Several key players are involved in this process: two transporter families (GLUTs, and MCTs) and three rate-limiting enzymes (HKs, PFKs, and PKs). In HCC, all of these exhibit varying degrees of upregulation, contributing to enhanced glycolysis. Targeting their overexpression through different mechanisms presents a promising strategy for inhibiting aerobic glycolysis and treating HCC, as summarized in Table 1 (Ref. [61, 62, 63, 64, 65, 66, 67, 68, 69, 70, 71, 72, 73, 74, 75, 76, 77, 78, 79, 80, 81, 82, 83, 84]).
| Intervention means | Target | Role | Reference |
| BAY-876 | GLUT1 | antagonist of GLUT1 receptor | [61] |
| Benzamides and Rapafucins | GLUT1 | inhibit the expression of GLUT1 | [62] |
| WZB117 | GLUT1 | a GLUT1 inhibitor | [63] |
| Napabucasin | GLUT1 | reduces the expression of CD47 and GLUT1 | [64] |
| Sirolimus combined with Huai Er | PI3K/Akt/mTOR-HIF-1 |
inhibits the expression of LDHA and GLUT1 | [65] |
| Down-regulation of BTF3 | GLUT1 | inhibits the expression of GLUT1 | [66] |
| STF-31 | GLUT1 | directly binds to GLUT1 | [67] |
| 2-DG | HK2 | inhibits the expression of HK2 | [68] |
| 3-BrPA | HK2 | inhibits the expression of HK2 | [69, 70] |
| NaBu | HK2 and c-Myc | inhibits the expression of HK2 | [71] |
| miR-202 | HK2 | inhibits the expression of HK2 | [72] |
| Metformin | HIF-1 |
inhibit the expression of PFK1 | [73] |
| Vitamin K |
PFK1 and HKs | inhibits the expression of HKs and PFK1 | [74] |
| TRIM35 | PKM2 | blockades the phosphorylation levels of PKM2 Y105 | [75] |
| SETD5 | PKM2 | inhibits the expression of PKM2 | [76] |
| CaMKK |
PKM2 and PI3K/AKT | inhibits the expression of PKM2 | [77] |
| TKP | PKM2 | inhibits the expression of PKM2 | [78] |
| MiR-342-3p | MCT1 | inhibits the expression of MCT1 | [79] |
| Syrosingopine | MCT1 and MCT4 | inhibits the expression of MCT1 and MCT4 | [80] |
| AZD3965 | MCT1 | inhibits the expression of MCT1 | [81, 82, 83] |
| AR-C155858 | MCT1 | inhibits the expression of MCT1 | [84] |
Abbreviations: BTF3, basic transcription factor 3; 2-DG, 2-Deoxyglucose; 3-BrPA,
3-bromopyruvate; NaBu, sodium butyrate; TRIM35, Tripartite motif-containing
protein 35; SETD5, SET domain-containing 5; CaMKK
GLUT1, a crucial protein for glucose uptake in tumor cells, is upregulated in
HCC [85]. BAY-876, a GLUT1 antagonist, significantly inhibited tumor growth in
HCC models [61]. Direct treatment of HCC tumors with microcrystalline BAY-876
reduced glucose uptake by the cancer cells [61]. Meanwhile, several other
strategies target GLUT1 to inhibit tumor growth. Benzamides and rapafucins can
interfere with GLUT1 expression, preventing or reducing tumor cell proliferation
[62]. WZB117, another GLUT1 inhibitor, suppresses tumor growth in mouse models by
downregulating GLUT1 [63]. Similarly, knocking down GLUT1 expression restrains
aerobic glycolysis and inhibits prostate cancer growth [86]. Importantly,
combining GLUT1 inhibitors with conventional therapies significantly enhances
their efficacy [87, 88, 89]. Interestingly, signal transducer and activator of
transcription 3 (STAT3) directly regulates the transcription of both CD47 and
GLUT1 (SLC2A1). HCC cells and patients typically show high SLC2A1 expression,
while SLC2A2 expression is comparatively low [90]. Napabucasin, a STAT3
inhibitor, reduces tumor growth by decreasing CD47 and GLUT1 expression in
subcutaneous and orthotopic mouse models [64]. Additionally, combining sirolimus
and Huai Er can inhibit aerobic glycolysis by downregulating LDHA and GLUT1
through reduced HIF-1
HK2 inhibition offers a promising strategy for treating human liver cancer by hindering tumor cells aerobic glycolysis and inducing cell death [93]. 2-Deoxyglucose (2-DG) is a widely studied HK2 inhibitor that has been reported to inhibit glycolysis by inhibiting hexokinase [68]. 2-DG, similar to glucose, catalyzes the production of 2-deoxy-D-glucose-6-phosphate (2-DG-6P) through HK2, which inhibits the activity of HK2 and indirectly inhibits aerobic glycolysis, unlike G-6-P, which is produced by glucose [94]. In rat HCC models, 2-DG was shown to reduce PKM2 and LDHA expression, leading to decreased aerobic glycolysis and tumor cell death [95]. Combining 2-DG with sorafenib demonstrated superior antitumor effects compared to sorafenib alone, suggesting its potential for synergistic action with other anticancer drugs [96]. Besides, this combination synergistically inhibited the development of sorafenib-resistant HCC cells by targeting ATP production [97]. Mitochondrial fusion protein mitofusin-1(MFN1) downregulation, linked to metastasis and poor HCC prognosis, is significantly inhibited by 2-DG treatment, demonstrating its potential in combating metastasis [98]. Moreover, 2-DG fusion nanoparticles such as (2-DG)-encapsulated poly (lactic-co-glycolic acid) (PLGA) nanoparticles (2DG-PLGA-NPs) exhibit potent antitumor effects by enhancing T cells transport, cytotoxicity, and antitumor immunity, leading to HCC growth suppression in mice [99]. 2-DG also demonstrates strong cytotoxicity in prostate cancer cell lines. Phase II clinical trials using 45 mg/kg of 2-DG showed promising results in inhibiting tumor cell metabolism and achieving antitumor effects in patients with prostate cancer and other solid tumors [100]. 3-bromopyruvate (3-BrPA), another inhibitor of HK2, is also often used to inhibit aerobic glycolysis [69]. The combination of 3-BrPA and l,3-bis(2-chloroethyl)-1-nitrosourea (BCNU) was found to significantly reduce the production of ATP and the excretion of lactate in HCC cell lines, including SMMC-7721 and HepG2 cells [70]. Other studies have found that 3-BrPA can promote the apoptosis of Hep3B and SK-Hep1 cells by inducing endoplasmic reticulum stress [101]. Further study demonstrated that 3-BrPA inhibited hypoxia-induced mitochondrial stability and promoted tumor cell apoptosis, leading to its ability to inhibit tumor progression in an MH3-induced HCC mouse model [102]. Both 3-BrPA and sodium citrate (SCT) inhibited tumor progression in mice with transplanted tumors by inhibiting aerobic glycolysis and inducing tumor cells apoptosis [103]. 3-BrPA has shown strong anti-tumor effects by inhibiting aerobic glycolysis and reducing ATP production in other cancer treatments, including malignant peripheral nerve sheath tumors, pancreatic cancer, and breast cancer [104, 105, 106]. Similar conclusions support the view that HK2 knockdown, when combined with metformin or sorafenib, significantly increases HCC cell death and slows tumor growth compared to metformin or sorafenib alone [93]. Sodium butyrate (NaBu) has been reported to inhibit HK2 expression through the c-Myc signal pathway, reducing aerobic glycolysis of HCC cells and enhancing the anti-tumor effect of sorafenib [71]. Besides, miR-202 can suppress aerobic glycolysis and cell proliferation by regulating HK2 expression in HCC [72].
PFK1 constitutes the majority of PFKs isoforms within the human liver [107].
HIF-1
The high expression of PKM2 in HCC tissues suggests a poor prognosis [110]. Some
studies have shown that PKM2 knockdown suppresses the migration of HCC cells and
prevents the development of tumors in HCC xenografted mice [111]. Both in
vivo and in vitro, guanosine triphosphate binding protein 4 (GTPBP4)
induces PKM2 expression by modifying protein ubiquitination, promoting the
development of aerobic glycolysis and liver cancer [112]. Similarly, heat shock
protein 90 (HSP90) binds to PKM2, increasing its abundance and enhancing the
effect of aerobic glycolysis, thereby promoting HCC cells development [13].
Overwhelming evidence currently supports that inhibiting PKM2 expression is an
effective treatment strategy for HCC. Tripartite motif-containing protein 35
(TRIM35) has been reported to block the Y105 phosphorylation of PKM2, thereby
inhibiting aerobic glycolysis in HCC cells and achieving antitumor effects [75].
Similarly, down-regulation of SET domain-containing 5 (SETD5) suppressed PKM2
expression, inhibiting aerobic glycolysis in HCC cells and tumor growth in
xenografted mice [76]. Additionally, calcium ions and calmodulins stimulate
protein kinase kinases
It has been reported established that MCT1, a key transporter of lactate
excreted from cells, significantly promotes tumor cell death when inhibited,
causing lactate accumulation within the cells [37]. Additionally, autophagy
induces MCT1 expression by activating the Wnt/
Recent studies have increasingly shown that traditional Chinese medicine
monomers and their extracts can inhibit aerobic glycolysis and exhibit strong
anti-tumor effects, as summarized in Table 2 (Ref. [112, 115, 116, 117, 118, 119, 120, 121, 122, 123, 124, 125]). Notably,
dihydroartemisinin (DHA) has emerged as a potential anti-tumor agent by directly
targeting aerobic glycolysis metabolism. Several studies, including our previous
work, have suggested that DHA inhibits tumor growth by targeting glycolysis
metabolism. For example, we demonstrated that DHA suppresses the Warburg effect
in HepG2215 cells and HCC mice by reducing SLC2A1 levels through YAP1 inhibition
[115]. Similarly, DHA effectively suppressed aerobic glycolysis and ESCC
progression by downregulating PKM2 expression in esophageal squamous cell
carcinoma (ESCC) and ESCC cells [126]. Furthermore, DHA inhibited leukemia cell
K562 proliferation by suppressing GLUT1 and PKM2 levels, thereby regulating
glucose uptake and inhibiting aerobic glycolysis [127]. In LNCaP cells, DHA
reduced Akt/mTOR and HIF-1
| Intervention means | Target | Role | Reference |
| DHA | GLUT1 | inhibits the expression of YAP1 to reduce the expression levels of SLC2A1 | [115] |
| Ginsenoside Rg3 | PI3K/Akt/HK2 | inhibits the expression of HK2 | [116] |
| Ginsenoside CK | AKT/mTOR/c-Myc | inhibits the expression of HK2 and PKM2 | [117] |
| QUE | AKT/mTOR | inhibits the expression of HK2 | [118] |
| Astragaloside | miR-2b | inhibits the expression of HK2 | [119] |
| Oroxylin A | HIF-1 |
inhibits the expression of PDK1, LDHA and HK2 | [120] |
| Chrysin | HK2 | inhibits the expression of HK2 | [121] |
| Genistein | GLUT1 and HK2 | inhibits the expression of HK2 and GLUT1 | [122] |
| Rosmarinic acid | GLUT1 and HK2 | inhibits the expression of HK2 and GLUT1 | [123] |
| Dauricine | HK2 and PKM2 | reduces the expression of HK2 and PKM2 | [124] |
| Shikonin | PKM2 | reduces the expression of PKM2 | [112] |
| PB2 | PKM2 | reduces the expression of PKM2 | [125] |
Abbreviations: DHA, dihydroartemisinin; Ginsenoside CK, ginsenoside Compound K; QUE, Quercetin; PB2, proanthocyanidin B2.
Some studies have shown that ginsenoside Rg3 combined with sorafenib can
significantly inhibit the PI3K/Akt signaling pathway and the expression of HK2 in
HCC cells [116]. Similarly, ginsenoside Compound K (CK) effectively inhibits the
AKT/mTOR/c-Myc pathway and HK2 and PKM2 expression, leading to aerobic glycolysis
inhibition in HepG2 and Huh7 cells and inducing apoptosis [117]. Quercetin (QUE),
a bioactive flavonoid, inhibits tumor growth in HCC xenograft mice by suppressing
the AKT/mTOR pathway and HK2 expression [118]. Recent studies have found that
astragaloside suppresses HCC cell proliferation by upregulating miR-2b and
inhibiting HK2 expression [119]. Additionally, oroxylin A inhibits
HIF-1
Aerobic glycolysis plays a pivotal role throughout HCC development, supplying a significant amount of ATP for both initiation and progression of the disease. By targeting key factors within this pathway, such as GLUT1, HK2, PFK1, PKM2, and MCT1, alongside their regulatory signaling pathways, we can effectively inhibit aerobic glycolysis and impede tumor growth. Consequently, transporters and key enzymes involved in aerobic glycolysis emerge as promising candidates for targeted drug development in tumor therapy.
Our comprehensive literature analysis suggests three highly promising research directions for HCC treatment. The tumor microenvironment plays a crucial role in tumorigenesis. As tumor cells rely on glycolysis for energy, they produce large amounts of lactate that acidify the TME, suppressing immune cell response and promoting tumor progression. Restoring this acidified TME to a normal pH and inhibiting lactate transporter MCT1 in tumor cells (preventing intracellular lactate from entering TME) represent two powerful treatment options. Additionally, inhibiting GLUT1 reduces glucose uptake, hindering glycolysis at its source and starving tumor cells of energy for growth. Finally, suppressing HK2 expression directly decreases the rate of aerobic glycolysis, inhibiting tumor cell growth and development. Collectively, these three approaches offer effective means to inhibit aerobic glycolysis and achieve successful HCC treatment.
While the potential of targeting aerobic glycolysis for HCC treatment shows promise, most supporting evidence currently comes from animal and cell experiments, with limited clinical data available. We acknowledge that our review primarily focused on preclinical studies and lacked in-depth analysis of clinical trials investigating aerobic glycolysis inhibition. Moving forward, we will concentrate on exploring the ongoing clinical advancements in this promising therapeutic approach. In-depth research and development of targeted aerobic glycolysis therapies hold the potential to offer new treatment options for HCC patients. Combining these novel drugs with existing chemotherapy regimens could further enhance therapeutic outcomes. However, it is crucial to consider that inhibiting aerobic glycolysis may also negatively impact the antitumor activity of immune cells. Therefore, significant research efforts are still warranted to optimize this approach and ensure its safe and effective application in HCC patients.
YY, YGao, and YX performed the research, analyzed the literature data, and wrote the original draft. XS designed the research study, reviewed this paper, and supervised the progress of the research. JL and YGong analyzed the collected literature and made charts. YZ, DW, and ZL performed the acquisition and analysis of data. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This research was financially supported by the National Natural Science Foundation of China (grant number 82274315) and the Foundation for High-levels Talents of Shanxi University of Chinese Medicine (grant number 2023RC03).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.