






 , Hai Zhang 5,*
, Hai Zhang 5,*1 Department of Thoracic Surgery, The First Affiliated Hospital of Nanjing Medical University, 210029 Nanjing, Jiangsu, China
2 Institute of Molecular Medicine, Renji Hospital, School of Medicine, Shanghai Jiao Tong University, 200127 Shanghai, China
3 Department of Thoracic Surgery, Zhongshan Hospital, Fudan University, 200032 Shanghai, China
4 Department of Geriatric, Renji Hospital, Shanghai Jiaotong University School of Medicine, 200032 Shanghai, China
5 Department of Pulmonary and Critical Care Medicine, Shanghai Chest Hospital, School of Medicine, Shanghai Jiao Tong University, 200000 Shanghai, China
†These authors contributed equally.
Abstract
Infection is the leading cause of acute lung injury (ALI). Macrophages, which are pivotal innate immune cells, play a critical role in mediating inflammatory processes. Intracellular lipopolysaccharide (LPS) from invasive Gram-negative bacteria can activate the caspase-11 inflammasome, leading to the induction of pyroptosis in macrophages. This process subsequently triggers the release of inflammatory cytokines and damage-associated molecular patterns from pyroptotic macrophages, thereby exacerbating inflammatory progression in ALI. However, the precise regulatory mechanisms governing caspase-11 activation is still unclear. Sentrin-specific proteases (SENPs) have been identified as notable targets for their anti-inflammatory properties. Nevertheless, the specific role of SENPs in macrophage pyroptosis during the pathogenesis of ALI remains unknown.
We used LPS as an endotoxin to induce ALI. We analyzed the expression and location of sentrin-specific protease 1 (SENP1), pulmonary impairment, macrophage infiltration, caspase-11 inflammasome expression and activation, caspase-11 SUMOylation, and inflammatory cytokine secretion.
Upregulated expression of SENP1 in lung tissue and macrophages was observed following LPS stimulation. SENP1 mediates de-SUMOylation and activation of caspase-11 inflammasome in macrophages. Moreover, pharmacological inhibition or genetic deficiency of SENP1 in macrophages significantly improved ALI-related histological damage by reducing the secretion of inflammatory cytokines and suppressing caspase-11-dependent pyroptosis.
Collectively, our findings highlight the involvement of SENP1 in caspase-11 activation and inflammatory progression in macrophages, thereby establishing a scientific foundation for the exploration of novel therapeutic strategies aimed at treating ALI.
Keywords
- sentrin-specific protease 1
- macrophage
- inflammation
- caspase-11
- acute lung injury
Acute lung injury (ALI), and its more severe form, acute respiratory distress syndrome, are respiratory disorders that pose a significant threat to life, with a mortality rate exceeding 40%. Currently, there are no effective treatment methods available [1, 2]. ALI can arise from various pathogenic factors, either through direct damage or indirect injury caused by excessive inflammation [3]. Sepsis, often stemming from infections with Gram-negative bacterial species, constitutes a underlying cause of ALI (6–42%) [4]. Nonpulmonary infections, aspiration of stomach contents, and major trauma accompanied by shock are also typical triggers of ALI. Less frequently, ALI can be associated with acute pancreatitis, blood transfusions, adverse drug reactions, and infections caused by fungi and parasites [5]. Lipopolysaccharide (LPS), a large molecule residing in the outer membrane of Gram-negative bacteria, plays a critical role in the pathogenicity that leads to sepsis. Despite numerous studies aiming to mitigate the progression of ALI following Gram-negative bacterial infection, few have yielded substantial clinical benefits [6, 7].
The pathophysiological mechanism of ALI is believed to be associated with numerous target and effector cells. For example, an excessive inflammatory response promotes the disruption of lung endothelial and epithelial barriers, resulting in increased pulmonary vascular permeability. Excessive neutrophil migration and neutrophil extracellular trap (NET) release have also been associated with the exacerbation of inflammation and tissue injury. Pulmonary macrophages account for 90–95% of lung immune cells at homeostasis, and play a pivotal role in initiating, amplifying, and resolving inflammatory processes [8, 9, 10]. Pyroptosis is an inflammatory form of programmed cell death that is primarily observed in macrophages [11, 12]. Notably, caspase-11, not caspase-1, is the primary mediator of pyroptosis during LPS infection [13]. Upon intracellular LPS stimulation, caspase-11 triggers the cleavage of gasdermin D (GSDMD), a protein responsible for forming pores in the cytoplasmic membrane, leading to membrane rupture and subsequent cell death in macrophages [14]. These pyroptotic macrophages release inflammatory cytokines and other molecular pattern molecules, thereby exacerbating the inflammatory progression in ALI [15, 16]. In addition, caspase-11 is related to NET formation and induces the death of lung epithelial cells [17]. Nonetheless, the regulatory mechanisms governing caspase-11 inflammasome activation and the inflammatory process in macrophages remain incompletely understood.
Small ubiquitin-like modifier mediated modification (SUMOylation) as a reversible post-translational modification, is essential for diverse biological processes [18, 19], and is readily reversible by sentrin-specific proteases (SENPs) [20, 21]. SENPs regulate certain activities of macrophages including activation, polarization, and apoptosis [22, 23]. However, the role of SENPs in the pyroptosis of macrophages during the ALI process remain unexplored.
In this study, we found that the expression of sentrin-specific protease 1 (SENP1) in macrophages is upregulated in LPS-induced ALI. Knockout (KO) or pharmacological inhibition of SENP1 can protect against the pulmonary damage associated with ALI. Additionally, SENP1 potentiates caspase-11 inflammasome activation and inflammatory cytokine secretion in macrophages stimulated with LPS. These results indicate that SENP1 functions as a positive regulator of caspase-11 activation and the progression of inflammatory processes in LPS-induced ALI, thus providing a novel and promising therapeutic target for ALI treatment.
The C57BL/6 wild-type (WT) mice (6 to 8 weeks old) used in the experiments were obtained from Shanghai SLAC Laboratory Animal Co. Ltd. (Shanghai, China). Lyz2-cre C57BL/6 mice were generated by Cyagen Transgenic Animal Center (Suzhou, China). Lyz2-Cre SENP1 conditional KO (cKO) mice were generated by BRL Medicine Inc. (Shanghai, China). The mice were housed in strict hygienic conditions in a facility designed to prevent pathogen contamination and were provided food and water ad libitum. Approval for conducting animal experiments was obtained from the Institutional Animal Care and Use Committee of the First Affiliated Hospital of Nanjing Medical University (Nanjing, China), ensuring adherence to ethical guidelines and regulations.
RNA sequencing data were obtained from the GSE1871 dataset, and LPS-induced ALI
and control groups were selected. Differentially expressed genes (DEGs) were
analyzed using the R package “edgeR” in R (version 3.17, Boston, MA,
USA). The fold changes were calculated for individual gene expression. Genes
meeting specific cutoff criteria of p
The LPS-induced ALI model was established as previously described [24]. Male C57BL/6 mice were administered an intraperitoneal (IP) injection of LPS at a dose of 10 mg/kg (L2630; Sigma-Aldrich, St. Louis, MO, USA). Momordin Ic (Mc) (HY-N0330; Med Chem Express, Monmouth Junction, NY, USA), a natural pentacyclic triterpenoid, was dissolved in dimethyl sulfoxide (DMSO). For treatment with Mc, mice were administered IP injections of Mc at 10 mg/kg for 20 consecutive days prior to LPS injection and continuing until the end of the experiment. The survival of mice following LPS injection was monitored at 4 h intervals for up to 120 h after administering either Mc or DMSO. The blood and bronchoalveolar lavage fluid (BALF) were collected 0 h, 6 h, 12 h and 24 h after LPS stimulation. The lungs were isolated for histopathologic evaluation after 0 h, 6 h, 12 h and 24 h. SENP1fl/fl and SENP1 cKO mice were administered IP injections of 10 mg/kg LPS. After 24 h, plasma and BALF were collected, and the total lungs were isolated for histopathologic evaluation. Mc dosage was chosen based on previous studies [25, 26, 27]. For sample preparation. Mice were euthanized with carbon dioxide and once unconscious, were subjected to cervical dislocation. Of note, to avoid causing pain, the flow rate of carbon dioxide was displaced at 10-30% of the chamber volume per minute.
We validated all cell lines by Short Tandem Repeat (STR) profiling and confirmed negative for mycoplasma. The RAW264.7 cell line was purchased from the Shanghai Academy of Sciences (Shanghai, China). To obtain bone marrow-derived macrophages (BMDMs), the mouse femur and tibia were isolated, and bone marrow cells were collected by repeated washing of the bone marrow cavity. The cells were differentiated into macrophages through treatment with recombinant macrophage colony-stimulating factor (25 ng/mL) for 7 days. Flow cytometry analysis was used to identify and validate the purity of the BMDMs. Cells were primed with Pam3CysSerLys4 (6 h, 1 µg/mL, tlrl-pms; Invivogen US, San Diego, CA, USA), and ultrapure LPS (1 µg/mL, 24 h) was transfected using the DOTAP Liposomal Transfection Reagent (11202375001; Roche, Indianapolis, IN, USA). FLAG-tagged caspase-11, His-tagged SENP1, His-tagged SENP1 mutant C603S, and HA-tagged SUMO plasmids were established by adding the coding sequences on the plasmid pHAGE-CMV-MCSIzsGreen. Lipofectamine 2000 (11668019; Invitrogen, Carlsbad, CA, USA) was utilized to perform transfection with plasmid DNA in RAW264.7 cells. To identify the purity of the BMDMs. Cells were incubated with PE rabbit anti-mouseF4/80 (565410, BD Bioscience, CA, USA) for 30 min at 4°C in the dark. Then, cells were washed and subjected to flow cytometry. Data were analyzed by Beckman CytoFlex S (Brea, CA, USA).
To identify the purity of the BMDMs. Cells were incubated with PE rabbit anti-mouseF4/80 (565410, BD Bioscience, CA, USA) for 30 min at 4 °C in the dark. Then, cells were washed and subjected to flow cytometry. Data were analyzed by Beckman CytoFlex S (Brea, CA, USA).
The fixed lungs were embedded and transversely sectioned and stained with hematoxylin and eosin (H&E) for the observation of lung injury. The quantification of lung injury was conducted following a previously described method [28]. A pathologist was assigned to grade lung injuries in a blinded fashion. To summarize, histological analysis was performed to assess the thickness of alveolar wall/hyaline membrane formation, hemorrhage, neutrophil infiltration in air spaces, and alveolar congestion. Each category of pulmonary injury was assigned a grade ranging from 0 to 4, and the score was determined by summing the scores from each individual category.
The initial step involved weighing the freshly harvested lung to determine its wet weight. Subsequently, the lung tissues were subjected to a 24 h drying process at 80 °C, and post-drying, the tissues underwent another weighing to ascertain their dry weight. The wet/dry (W/D) weight ratio was employed as a metric for assessing pulmonary edema.
The procedure involved dissection of the mouse trachea, and a 20 G catheter was used for its subsequent cannulation. Then the lungs were subjected to three lavages with 1.5 mL ice-cold phosphate-buffered saline (PBS). Following this, BALF was obtained and centrifuged to obtain the supernatant. The protein concentration was determined using the QuantiPro™ BCA Assay Kit (Sigma-Aldrich).
The process began with retrieval of the mouse lungs, followed by thorough
rinsing with PBS and subsequent homogenization. The resulting homogenates were
subjected to centrifugation at 400
Tissue sections were incubated overnight with the following antibodies: F4/80 (1:200, ab6640; Abcam, Cambridge, MA, USA), GSDMD (1:200, AF4013; Affinity Biosciences, Cincinnati, OH, USA), and SENP1 (1:200, ab236094; Abcam). Following this incubation, the slides underwent a washing step and then were incubated in the dark for 1 h at room temperature with secondary antibodies conjugated to fluorochromes. DNA staining for 1 min was performed using DAPI. Images were captured using a fluorescence microscope (Zeiss, Oberkochen, Germany).
Lung tissues or cells were lysed using Radio-Immunoprecipitation Assay (RIPA) lysis buffer obtained from Sigma-Aldrich. After determining the protein concentration, each sample (40 µg) was loaded on gels and subsequently electrotransferred to polyvinylidene fluoride (PVDF) membranes. After blocking, the membranes were incubated overnight with SENP1 antibody (ab236094; Abcam), caspase-11 antibody (ab180673; dilution 1:200; Abcam), GSDMD antibody (ab209845; dilution 1:200; Abcam), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) antibody (ab9485; dilution 1:200; Abcam). Following a series of washes, membranes were incubated with secondary antibody (ab6721; dilution 1:2000; Abcam). Then a standard enhanced chemiluminescence reagent from Millipore (Burlington, MA, USA) was used to visualize the bands. Quantification of blots was performed in a blinded fashion by using ImageJ software, version 1.8.0 (National Institutes of Health, USA).
BMDM cells were seeded and cultivated in 96-well plates. Following priming and stimulation, each well received 90 µL glucose medium without fetal bovine serum, along with the addition of 10 µL Cell Counting Kit-8 assay reagent (40203ES60; Yeasen Biotechnology, Shanghai, China). The cells were subsequently incubated for 24 h. After incubation, absorbance values were determined at 450 nm using a multifunctional enzyme marker.
Following the priming and stimulation steps, the cell culture medium was aspirated. In accordance with the assay kit’s established protocol, Lactate Dehydrogenase (LDH) Assay Buffer (40209ES76; Yeasen Biotechnology) was added to the samples and maintained at 37 °C for 1 h in a light-free environment. Absorbance readings were taken at a wavelength of 450 nm.
RNA extraction was carried out from BMDMs and lung tissue employing TRIzol
reagent (9108; Takara Bio USA, San Jose, CA, USA). Subsequently, cDNA was
produced using the Reverse Transcription System (A2791; Promega, Madison, WI,
USA). Validated primers for SENP1, TNF-
| Gene | Primer sequence |
| GAPDH | F: AGTCCCTGCCCTTTGTACACA |
| R: CCGAGGGCCTCACTAAACC | |
| IL-1 |
F: AGGCTTCCTTGTGCAAGTGT |
| R: TGAGTGACACTGCCTTCCTG | |
| IL-6 | F: CCGGAGAGGAGACTTCACAG |
| R: ACAGTGCATCATCGCTGTTC | |
| TNF- |
F: GGCTGCCCCGACTACGT |
| R: AGGTTGACTTTCTCCTGGTATGAGA | |
| SENP1 | F: CCTAGCTGGACTGCAGAA |
| R: CACCACTGGCCAGAATGATGA |
All sequences are in 5′-3′ direction. GAPDH, glyceraldehyde-3-phosphate dehydrogenase; IL, interleukin;
TNF-
The co-immunoprecipitation (co-IP) procedure was conducted following previously established protocols [29]. Briefly, RIPA buffer (87787; Thermo Fisher Scientific, Waltham, MA, USA) was used to lyse the cells. The lysates underwent a 2 h incubation with protein A/G-agarose for preclearing and then were incubated overnight with specific antibodies. Subsequently, protein A/G agarose beads were added. Finally, loading buffer was used to wash and mix the beads, followed by immunoblot analysis.
The results are shown as the mean
To investigate the role of SENPs in ALI, we first revisited the results of a previous Affymetrix microarray study that profiled lung gene expression following LPS stimulation [30]. Our reanalysis aimed to identify the expression pattern of SENPs in the lung during ALI. We discovered that among the SENP family members, only SENP1 expression was upregulated by LPS treatment (Supplementary Table 1), suggesting that SENP1 might be involved in ALI progression. To validate this finding, we employed an LPS-induced mouse model of ALI. The results of enhanced lung injury (Fig. 1A,B), total protein concentration in BALF (Fig. 1C), and pulmonary edema (Fig. 1D) after LPS stimulation confirmed the successful establishment of the LPS-induced ALI model. Next, lung tissues were collected to measure the expression of SENP1 at both the mRNA and protein levels. Our results confirmed that SENP1 mRNA expression was increased in the lungs following LPS injection (Fig. 1E). Consistent with this increased transcription, immunofluorescence and western blot analysis revealed the significant upregulation of SENP1 protein expression in response to LPS injection (Fig. 1F–H).
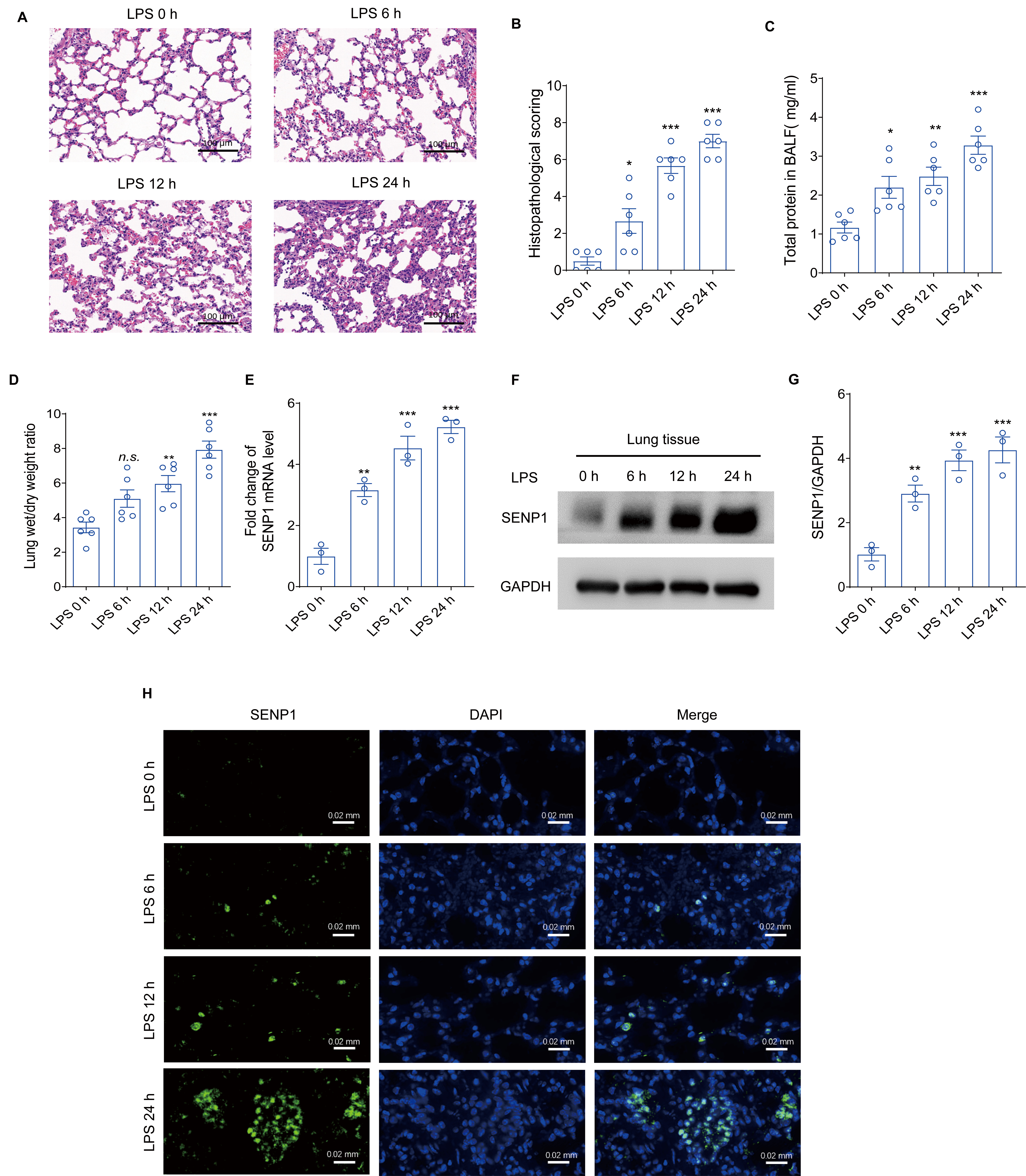 Fig. 1.
Fig. 1.
Sentrin-specific protease 1 (SENP1) expression was elevated in
the lungs following lipopolysaccharide (LPS) stimulation. (A) Representative
images of lung sections stained with hematoxylin and eosin (H&E) post-LPS
stimulation (20
To determine whether SENP1 contributes to the progression of ALI, we employed a
chemical inhibitor to suppress SENP1 function. Mc, a naturally occurring
pentacyclic triterpenoid compound, was chosen due to its documented ability to
inhibit SENP1 activity [25, 27]. Then we examined the effects of Mc on
LPS-induced ALI. The dosage of Mc was chosen based on a previous study [25]. Our
findings revealed that pre-treatment with Mc effectively mitigated the
pathological alterations induced by LPS, as evidenced by the reduced
histopathological scores of lung tissue, total protein levels, and pulmonary W/D
weight ratio (Fig. 2A–E). Furthermore, we assessed the survival rates of various
groups up to 120 h post-LPS stimulation. Remarkably, LPS+DMSO mice exhibited a
significant decline in survival rates, whereas Mc-treated mice showed a notable
increase (Fig. 2F). Given that a severe inflammatory response is a hallmark of
LPS-induced ALI, we further examined the impact of Mc on the systemic
inflammatory response. ALI induction led to a pronounced inflammatory response in
both plasma and BALF. However, in the presence of Mc, there was a significant
reduction in the levels of TNF-
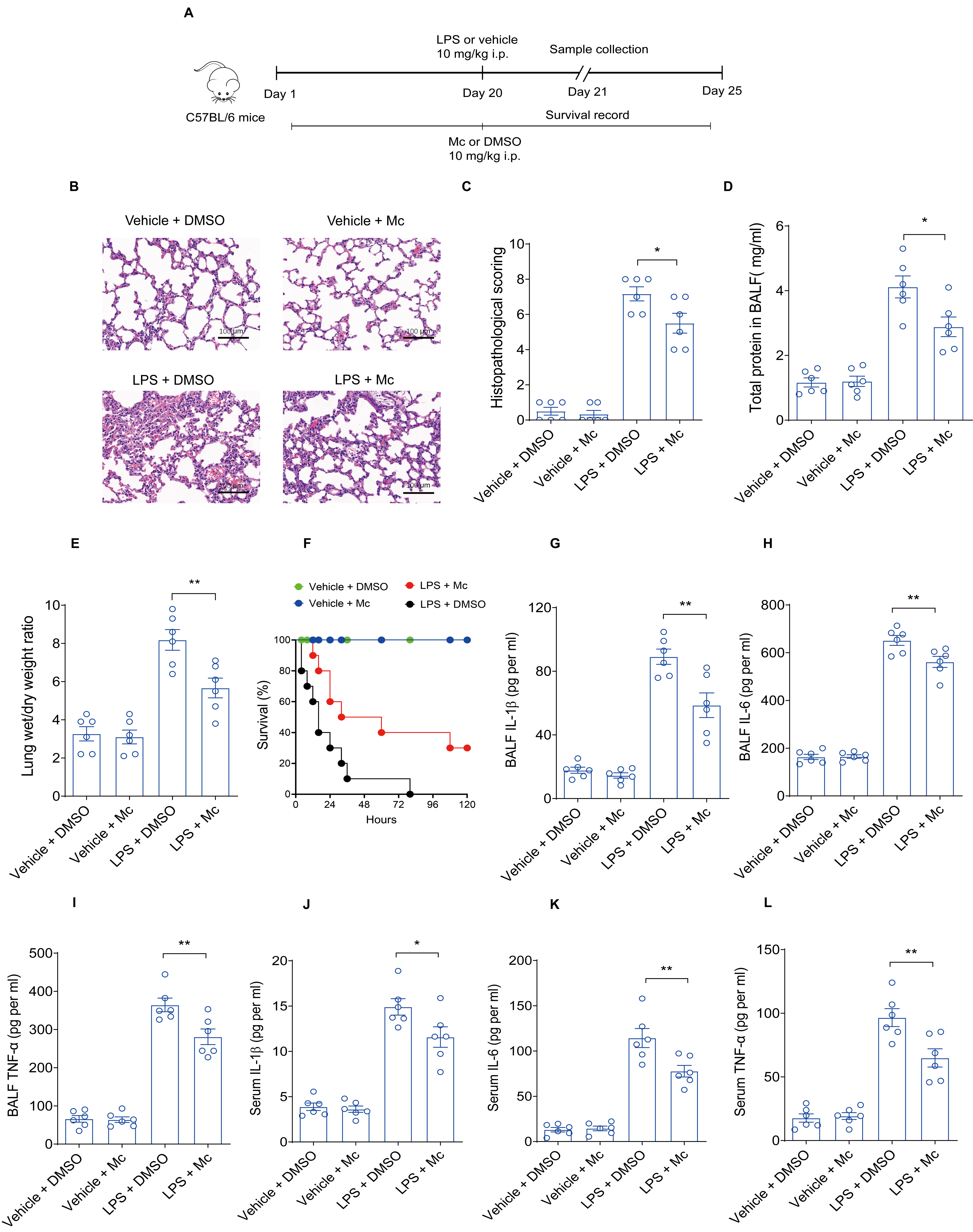 Fig. 2.
Fig. 2.
Sentrin-specific protease 1 (SENP1) inhibition ameliorates
lipopolysaccharide (LPS)-induced acute lung injury (ALI). (A) A schematic
diagram illustrating the process of generating an LPS-induced ALI model. (B)
Representative images of H&E staining (20
Given the significance of macrophages in ALI, we speculated whether the elevated expression of SENP1 is localized in macrophages. Our immunofluorescence staining results showed that overexpressed SENP1 in the lung predominantly overlapped with the macrophage marker F4/80 (Fig. 3A), suggesting that the increased SENP1 was mainly expressed in macrophage. Since pyroptosis is an inflammatory process and caspase-11 serves as an intracellular LPS sensor vital for septic ALI pathogenesis, we evaluated the levels of proteins associated with caspase-11 inflammasome and pyroptosis. Western blot analysis revealed that LPS injection facilitated the activation of caspase-11 in the mouse lungs, as demonstrated by the presence of caspase-11 p26 structure and GSDMD-N release (Fig. 3B–D). Moreover, immunofluorescence staining also demonstrated the co-localization between overexpressed GSDMD and the F4/80 macrophage marker (Fig. 3E).
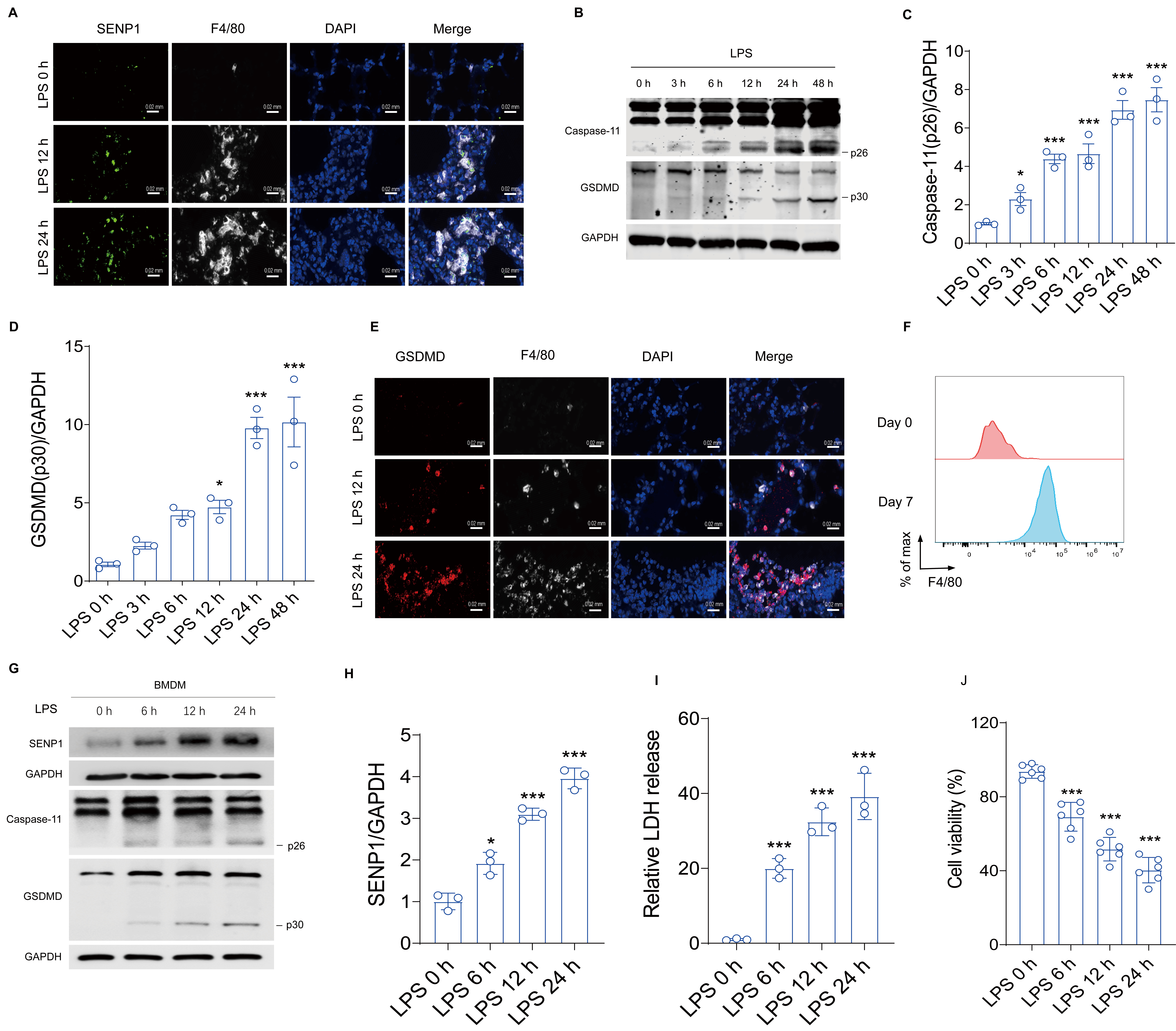 Fig. 3.
Fig. 3.
Sentrin-specific protease 1 (SENP1) expression and caspase-11 inflammasome activation in lung
tissue and bone marrow-derived macrophage (BMDM) after lipopolysaccharide (LPS)
stimulation. (A) Representative immunofluorescence staining of pulmonary tissue
for SENP1 (green) and F4/80 (grey) in the ALI mouse model (100
To further validate the potential correlation between SENP1 and caspase-11 activation in macrophages, we conducted in vitro experiments. Flow cytometry analysis indicated that a high level of purity was observed in BMDMs on day 7. (Fig. 3F). We observed the significant enhancement of SENP1 expression in BMDMs following intracellular LPS stimulation (Fig. 3G,H). Additionally, we measured the levels of caspase-11 and GSDMD in BMDMs and found that cleaved caspase-11 and GSDMD-N were upregulated in response to LPS infection. Furthermore, LPS-treated BMDMs exhibited increased LDH release (Fig. 3I) and decreased cell viability (Fig. 3J). These findings support the correlation between upregulated SENP1 and caspase-11 activation, both of which are primarily localized in macrophages.
To evaluate the role of SENP1 in macrophages during ALI, we employed the
flox/flox Lyz2-cre cKO strategy to specifically KO SENP1 in macrophages,
generating SENP1cKO and SENP1flox/flox mice. Subsequently, we established an
LPS-induced ALI model in both SENP1cKO and SENP1flox/flox mice. As shown in
Fig. 4A,B, SENP1 deficiency obviously alleviated pulmonary damage.
Quantitatively, we observed a robust increase in the lung injury score in
SENP1flox/flox mice, which was markedly reduced in SENP1cKO mice. A similar
pattern of changes was also observed in the pulmonary W/D weight ratio, total
protein levels in BALF (Fig. 4C,D), and the production of pro-inflammatory
cytokines such as IL-1
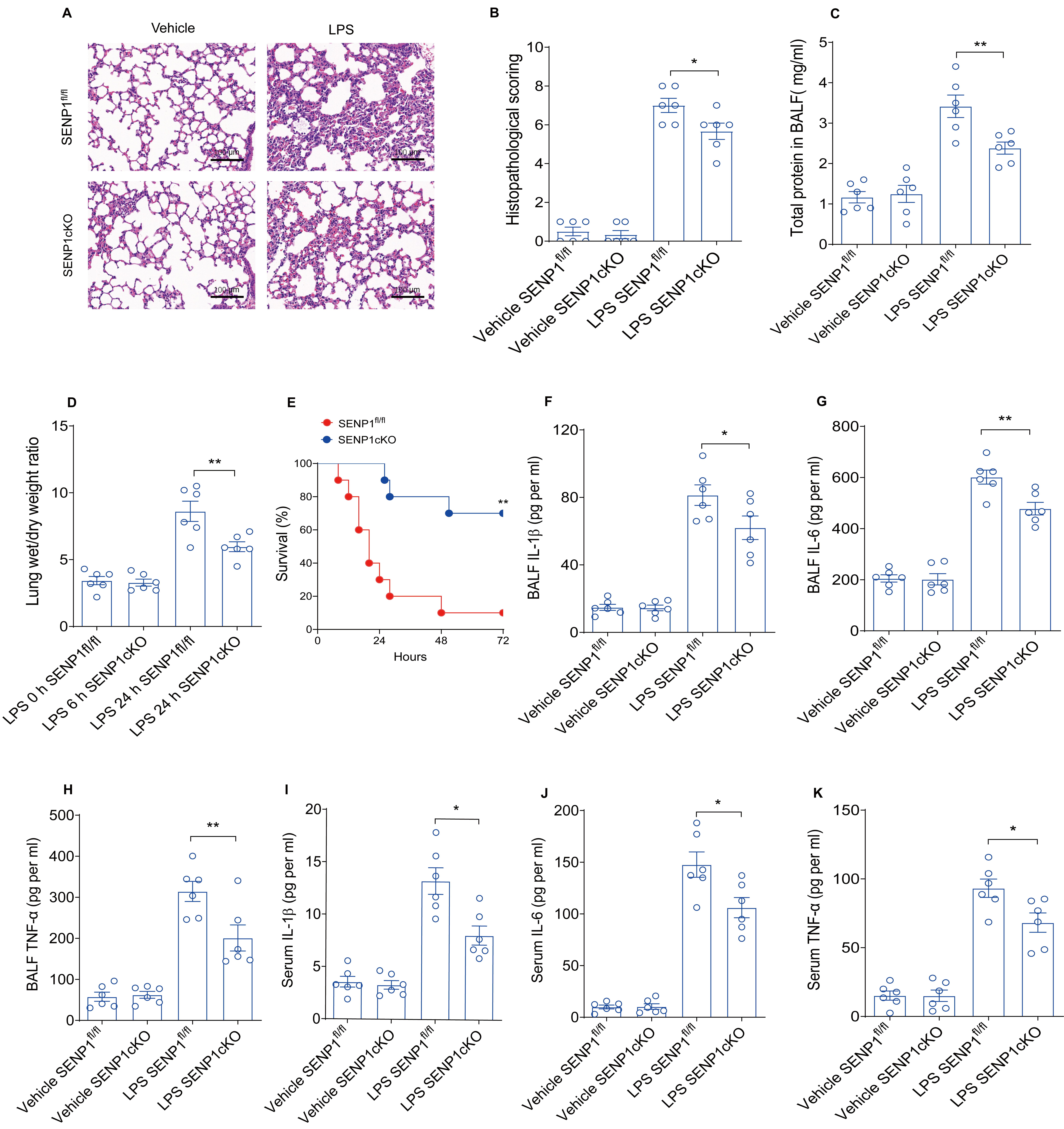 Fig. 4.
Fig. 4.
Sentrin-specific protease 1 (SENP1) depletion in macrophages ameliorated lipopolysaccharide (LPS)-induced acute lung injury (ALI). (A)
Representative images of H&E staining (20
To determine whether SENP1 contributes to ALI progression via modulation of the
caspase-11 inflammasome, we analyzed the expression of caspase-11 and GSDMD,
along with their cleaved forms, in BMDMs isolated from SENP1flox/flox and
SENP1cKO mice. Following cytosolic LPS stimulation, the expression of cleaved
caspase-11 and GSDMD-N was enhanced in all groups (Fig. 5A). However, BMDMs from
SENP1cKO mice exhibited lower levels of cleaved caspase-11 and GSDMD-N compared
to SENP1flox/flox mice (Fig. 5B,C). Subsequently, we evaluated the levels of
various inflammatory factors in the BMDMs after cytosolic LPS stimulation. Our
results showed that LPS stimulation dramatically increased the levels of
IL-1
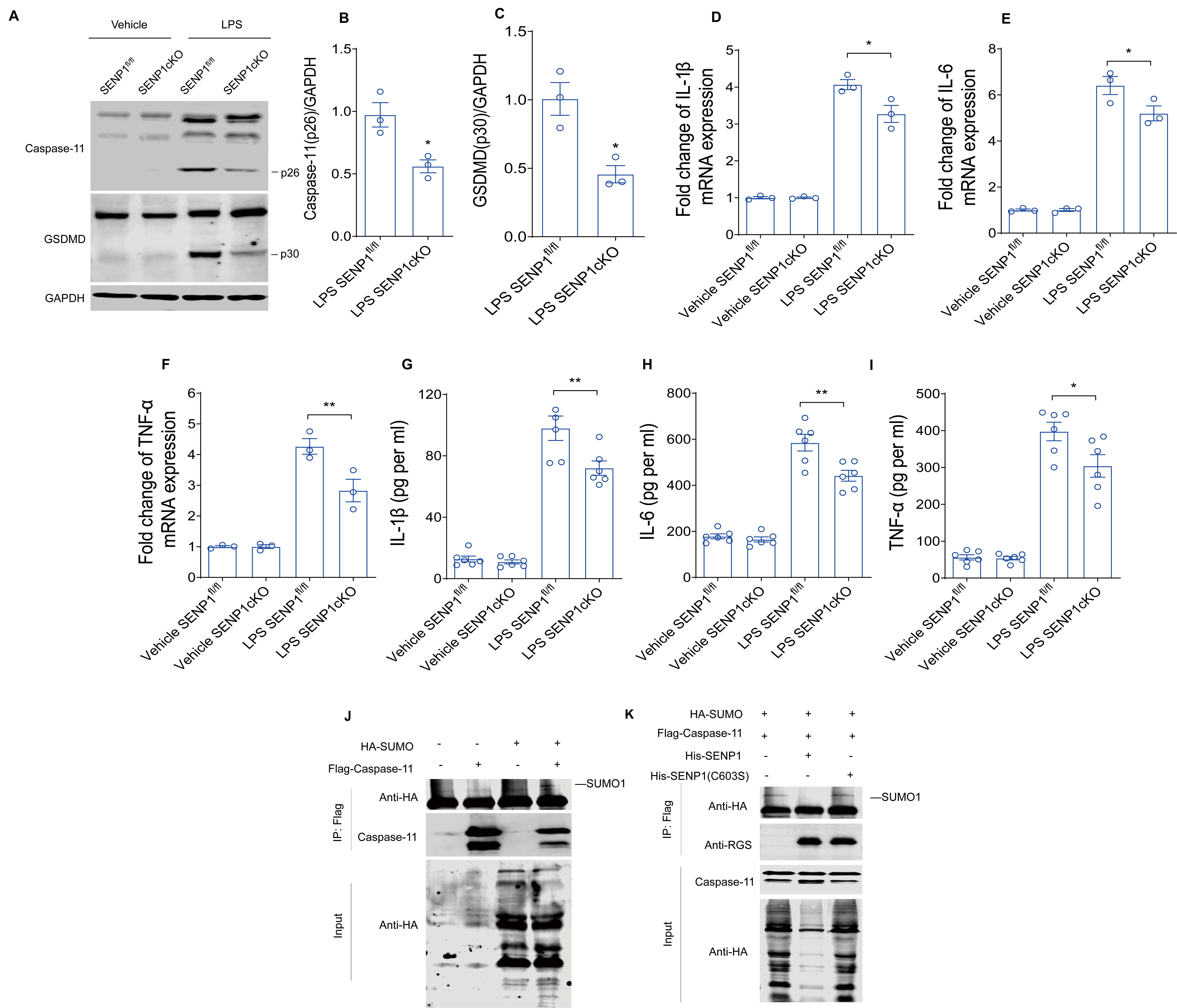 Fig. 5.
Fig. 5.
Effects of Sentrin-specific protease 1 (SENP1) on the caspase-11 inflammasome activation and
cytokine production in macrophages after lipopolysaccharide (LPS) insult. (A–C) Western blot
analysis of caspase-11 and gasdermin D (GSDMD) expression in bone marrow-derived macrophages
(BMDMs) from SENP1flox/flox or SENP1cKO mice. (D–F) mRNA levels of
interleukin (IL)-1
These findings suggest that SENP1 promotes caspase-11 activation and enhances inflammatory cytokine release in macrophages during cytosolic LPS insult.
As SENP1 is responsible for removing the SUMO modification from substrate proteins, thereby influencing their activity and function, we hypothesized that caspase-11 may be a substrate protein of SENP1. To validate this, we conducted co- IP assays utilizing antibodies targeting SUMO1, tagged caspase-11, SENP1, and endogenous caspase-11. We co-transfected plasmids encoding HA-SUMO1 and FLAG-tagged caspase-11 into RAW264.7 cells and detected the SUMO1-caspase-11 conjugation from IP’d caspase-11 protein (Fig. 5J). Moreover, we observed that WT SENP1 effectively removed SUMO1 from caspase-11, whereas the SENP1 mutant (lacking enzymatic activity) was incapable of performing this function (Fig. 5K). These findings revealed that SENP1 interacts with caspase-11 and subsequently deSUMOylates caspase-11 under LPS insult, which further enhances the progression of ALI.
During intracellular LPS stimulation, the transcription of SENP1 is enhanced, leading to the upregulation of SENP1 protein expression. Then, SENP1 de-SUMOylates and activates caspase-11 to promote the pyroptosis of macrophage.
SENPs have recently emerged as regulators of specific macrophage activities; however, their involvement in macrophage pyroptosis during ALI remains uncharted territory. In the current study, we uncovered the significant role of SENP1 in the development of ALI triggered by LPS. Our research reveals that ALI promotes the upregulation of SENP1 expression in macrophages. Notably, SENP1 deSUMOylates and activates caspase-11, intensifying inflammatory reactions during ALI (Fig. 6). Translationally, our preclinical data reveled the defensive impact of Mc, a small molecule designed to target SENP1, in mitigating ALI. These findings provide novel insights into the molecular mechanisms of ALI and may pave the way for future application of an SENP1 inhibitor in the treatment of ALI.
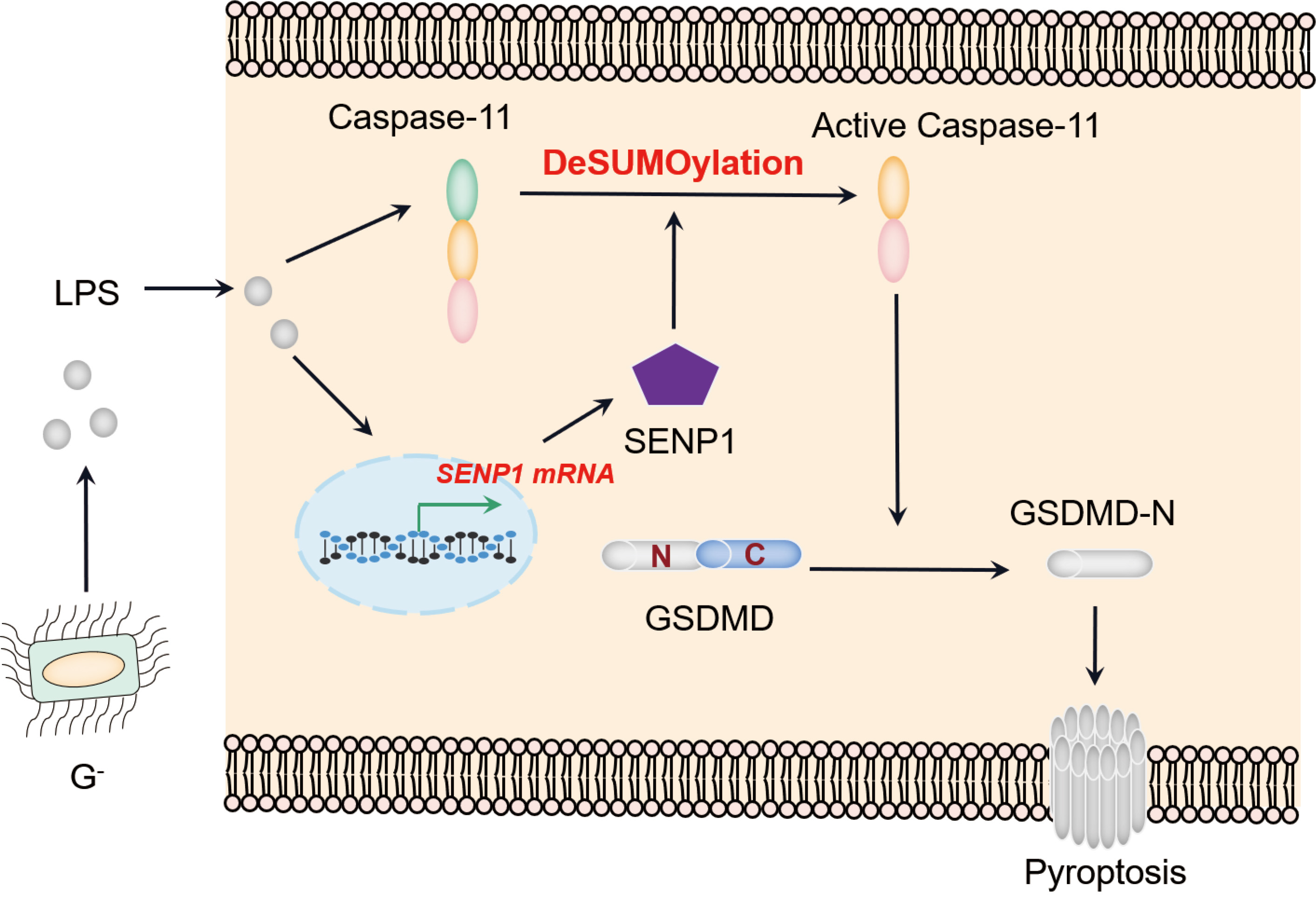 Fig. 6.
Fig. 6.
A working model for Sentrin-specific protease 1 (SENP1) promoting caspase-11 inflammasome activation of macrophages in acute lung injury (ALI). GSDMD, gasdermin D; LPS, lipopolysaccharide.
ALI is primarily induced by Gram-negative bacterial infection. The inflammatory reaction plays a crucial role in the development of ALI [31]. The innate immune system detects Gram-negative bacteria partly by recognizing LPS, a molecule consisting of a lipid and polysaccharide found in the outer membrane of Gram-negative bacteria [32]. Macrophages, due to their crucial role in initiating host defense and preserving tissue homeostasis, constitute the predominant immune cell population within lung tissues. During LPS stimulation, macrophages possess the ability to release inflammatory cytokines and interact with other immune cells, thus initiating a cycle of auto-amplification that results in escalation of the inflammatory response [13]. Therefore, timely repression of heightened inflammatory responses associated with macrophages is crucial for maintaining lung homeostasis [33]. The significance of SENPs in different immune and inflammatory disorders has been increasingly emphasized by a growing body of evidence [34, 35, 36]. However, the understanding of the roles and functionalities of SENPs in ALI remains limited. Our study showed that exposure to LPS elicited a notable upregulation of SENP1 in damaged lung. Then we found that overexpressed SENP1 was largely located in the macrophages. Given the relationship among macrophages, immunity, and inflammation, we postulate that SENP1 may be involved in the intense inflammatory response in ALI. To assess the function of SENP1 in a simulated clinical scenario, we employed the SENP1 inhibitor Mc to investigate its impact on the pathological process of ALI induced by LPS. The results demonstrated that Mc treatment relieved pulmonary damage, as indicated by the reversal of hydrostatic alveolar edema, suppression of increased inflammatory cell infiltration, and attenuated pathological alterations. In addition, inhibition of SENP1 was associated with increased survival rates and decreased secretion of proinflammatory cytokines in the ALI mouse model. These findings suggest that the increase in SENP1 plays an important role in the development of pulmonary damage and inflammatory reaction in macrophages, offering profound insights into the therapeutic potential of targeting SENP1 in the treatment of ALI.
Caspase-11 is an intracellular LPS sensor of innate immune system.
Caspase-11-dependent pyroptosis is inflammation-related and mainly triggered by
the formation of transmembrane pores through the active N-terminal fragments of
GSDMD, leading to rupture of the plasma membrane and the release of inflammatory
cytokines [37, 38, 39]. During ALI, intracellular LPS and other inflammatory
substances can trigger macrophage pyroptosis through the caspase-11-dependent
inflammasome. The release of intracellular contents by pyroptotic macrophages
further amplifies the inflammatory response, ultimately leading to a cytokine
storm and promoting the progression of ALI [40]. It is challenging that the
efficacy of inflammation-targeted therapy in clinical applications remains
unclear [41]. Recent clinical studies have demonstrated the failure of
TNF
Our current study had some limitations. Notably, the mechanism responsible for the upregulation of SENP1 following LPS exposure, and the precise site where SENP1 mediates the deSUMOylation of caspase-11, were not thoroughly examined in this study. In addition, since the Lyz2Cre system affects not only macrophages but also other myeloid cells (such as neutrophils), the role of SENP1 in the progression of ALI in this study cannot exclude the influence of other myeloid subsets. This is a scientific question worth exploring in the future. Moreover, human samples and data were not included in this study, necessitating further in-depth investigation.
In summary, our data demonstrate that upregulated SENP1 exacerbates the progression of LPS-induced ALI by promoting activation of the caspase-11 inflammasome and facilitating the progression of inflammatory responses in macrophages. These observations provide compelling evidence for targeting the SENP1-caspase-11 axis as a potential therapeutic strategy for the treatment of infectious ALI.
All analyzed data supporting the study are included in this published article and its supplementary information files. And the raw data will be made available from the corresponding author on request.
Conceptualization: HZ, CBZ and MJD; methodology: MJD, WHW, SYZ; data curation: MJD, WHW, JMG; formal analysis: WHW, SYZ, JMG; investigation: MJD, CBZ, HZ, WHW; resources: HZ, CBZ, MJD; software: JMG, SYZ, WHW; supervision: HZ, CBZ; writing—original draft: MJD, HZ, CBZ; writing—review and editing: HZ, CBZ; All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
The research protocol was approved by the Ethics Committee of The First Affiliated Hospital of Nanjing Medical University (Ethic Approval Number: 2023-SRFA-126).
We sincerely appreciate the time and effort of all the researchers involved.
This study was supported by Natural Science Foundation of Jiangsu Province (BK20210956), the High-level Innovation and Entrepreneurship Talent Introduction Plan of Jiangsu Province (JSSCBS20211459) and the Interdisciplinary Program of Shanghai Jiao Tong University (YG2019QNA51).
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/j.fbl2911397.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.