






 , Haiyan Huang 2, Liucheng Wu 3, Mei Ding 1, Xiangyang Zhu 1
, Haiyan Huang 2, Liucheng Wu 3, Mei Ding 1, Xiangyang Zhu 11 Department of Neurology, The Second Affiliated Hospital of Nantong University, 226001 Nantong, Jiangsu, China
2 Department of General surgery, The Second Affiliated Hospital of Nantong University, 226001 Nantong, Jiangsu, China
3 Laboratory Animal Center, Nantong University, 226019 Nantong, Jiangsu, China
Abstract
Vitamin D receptor (VDR) can prevent myocardial ischemia reperfusion injury (MIRI). Hence, we aimed to illuminate the effect of VDR on cerebral ischemia/reperfusion injury (CIRI).
C57BL/6 mice and SK-N-SH cells were utilized to establish CIRI and cellular oxygen deprivation/reoxygenation (OGD/R) models. Mice were injected with 1 μg/kg Calcitriol or 1 μg/kg Paricalcitol (PC) and adenovirus-mediated VDR overexpression or knockdown plasmids. 2,3,5-triphenyl-tetrazolium chloride (TTC) and Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assays were performed to measure the brain infarct volume and the apoptosis of cerebral cells. SK-N-SH cells were treated with 5 mM N-acetyl-L-cysteine (NAC) and transfected with VDR knockdown plasmid. Flow cytometry and Cell Counting Kit-8 (CCK-8) assays were employed to assess the apoptosis and cell viability. Enzyme-Linked Immunosorbent Assay (ELISA), quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR) and Western blot were exploited to quantify the levels of reactive species oxygen (ROS), other oxidative stress-related factors, VDR and apoptosis-related factors.
The level of VDR in mouse cerebral tissue was elevated by CIRI (p < 0.001). CIRI-induced cerebral infarction (p < 0.001) and the apoptosis of cerebral cells (p < 0.001) in mice were mitigated by the activation of VDR. VDR overexpression abrogated while VDR silencing enhanced CIRI-induced infarction, oxidative stress and apoptosis of cerebral cells (p < 0.05). Furthermore, VDR silencing aggravated the oxidative stress and apoptosis in OGD/R-treated SK-N-SH cells (p < 0.05). NAC, a scavenger of oxidative stress, could reverse the effects of VDR silencing on apoptosis and oxidative stress in OGD/R-treated SK-N-SH cells (p < 0.01).
VDR alleviates the oxidative stress to protect against CIRI.
Keywords
- cerebral ischemia/reperfusion injury
- vitamin D receptor
- oxidative stress
- oxygen deprivation/reoxygenation
Despite the great advancement in therapeutic strategies, stroke is one of the major reasons for permanent disability in adults and the second lethal factor around the world [1]. The majority of ischemic stroke has been categorized as observed stroke, which occurs when the blood clot obstructs the cerebral blood flow that transports vital ions, oxygen, and nutrients to the brain [2]. The treatment of ischemic stroke primarily depends on recanalization, which can replenish nutrients and oxygen and remove the toxic metabolites; in addition, the restoration of blood flow is related to the worsening of injury and inflammatory response within the tissue, a condition referred to as cerebral ischemia/reperfusion injury (CIRI) [3]. Regrettably, there is a dearth of effective therapeutic strategies to ameliorate the outcomes of CIRI, and the complexity of the injury cascade has not been fully understood, resulting in the requirement of additional research to develop more effective treatment options [4].
Vitamin D has important effects on the
development and function of the central nervous system [5]. Vitamin D prevents
excitotoxicity injury caused by a sudden increase in cytoplasmic Ca2+,
up-regulates the synthesis of parvalbumin and calbindin, and down-regulates
L-type voltage-gated calcium channels (L-VGCCs) [6]. Glial cell-derived
neurotrophic factor (GDNF) is a potent survival factor for many neurons,
but cannot cross the blood-brain barrier [7]. Vitamin D is a fat-soluble hormone
that can pass the blood-brain barrier and upregulate GDNF [8, 9].
1,25-Dihydroxyvitamin D3 (1,25(OH)2D3), an active metabolite of vitamin
D, exerts its neuroprotective effect by increasing the expression of the GDNF and
then activating downstream intracellular signaling cascades [10]. Calcitriol, an
active form of vitamin D, plays a crucial role in brain development and
neuroprotection, and can be generated by microglia within the central nervous
system to inhibit the synthesis of inducible nitric oxide synthase, leading to
upregulation of glutathione [11]. Calcitriol and vitamin D receptor (VDR) can
synergistically exert effects via genomic (classical) pathways (slow response) or
non-genomic pathways (rapid response) [12]. VDR belongs to the nuclear hormone
receptor superfamily that functions as a transcription factor, expressing in
various tissues and regulating the metabolism- and immune system-related
processes [13, 14]. It has been suggested that the overexpression of VDR in
pancreatic
Oxidative stress is termed the pathological procedure of tissue damage resulting from the exorbitant generation or declined scavenging ability of reactive oxygen species (ROS), which causes the imbalance between antioxidant and oxidative systems [18]. Oxidative stress is a major pathological reason for CIRI, as supported by the recent evidence that excessive oxidative stress results in the nitration of protein, the lipid peroxidation of nerve cell organelles and membranes, the disruption of nucleic acids and severe inflammatory reactions [19]. Meanwhile, it should not be neglected that the supplementation of vitamin D3 ameliorates the CIRI in mice via the protection against CIRI-induced oxidative stress, and the activation of VDR mitigates both microglia polarization and oxidative stress in spontaneously hypertensive rats [20, 21]. These findings have made us wonder whether VDR impacts CIRI via attenuating oxidative stress. Accordingly, to bridge the knowledge gap, the current study constructed the CIRI and oxygen glucose deprivation/reoxygenation (OGD/R) models in both mice and SK-N-SH cells, and probed into the effect of VDR on oxidative stress and CIRI.
The animal experiments were carried out after being approved by the ethics committee of Laboratory Animal Center of Nantong University (NTU) (Ethics Approval No. IACUC20221230-1006) and according to Animal Care and Use Guidelines, and every attempt was made to minimize discomfort and pain to the animals.
The CIRI model was constructed as depicted in a prior study [22] with slight modifications. 8-week-old C57BL/6 mice (male, 22–26 g, n = 160) were ordered from Hangzhou Medical College (Zhejiang, China) and maintained in the specific pathogen-free cages at 26 °C and 50–70% relative humidity. All mice could get standard experimental food and sterile tap water at will and were raised following the circadian rhythm.
The process of restoring blood flow and causing reperfusion injury after
cerebral ischemia was called CIRI modeling. For the establishment of animal CIRI
model, after being fasted for 12 h, all mice were inhaled with 2%
isoflurane (I40690, Acmec Biochemical, Shanghai, China) and
anesthetized, with fur and skin pre-disinfected using
povidone-iodine (A606166, Sangon Biotech, Shanghai, China) in a
supine position. The body temperature was retained at 37.0
The specified criteria were listed as follows: grade 0: there was no defect in mice; grade 1: mice were incapable of straightening the contra forelimb contralateral to the embolized artery when their tails were lifted; grade 2: mice circled to the contralateral side of the embolized artery while walking; grade 3: mice titled to the opposite side of the embolized artery while walking; grade 4: there was severe defect without spontaneous motor activity or consciousness.
To determine the change on the endogenous expression level of VDR in mice, mice were stochastically distributed to the groups of Sham, ischemia (30 min), and reperfusion (3, 6, 12 and 24 h) (n = 10/group). For intervention assay to unravel the effect of activating VDR in CIRI-modeled mice, mice were assigned to the groups of Sham, CIRI, Vehicle, Calcitriol and Paricalcitol (PC) (n = 10/group), and those in Vehicle, Calcitriol and PC groups were given the solvent including 95% propylene glycol (P62861, Acmec Biochemical) and 5% ethanol (E99942, Acmec Biochemical), Calcitriol (a natural agonist of VDR, 1 µg/kg, C39241, Acmec Biochemical) and PC (a synthetic agonist of VDR, 1 µg/kg, P44940, Acmec Biochemical) via intraperitoneal injection 15 min before the reperfusion [17]. Subsequently, two schemes were applied for the analyses: mice were either subjected to ischemia for 30 min and reperfusion for 3 h to determine the apoptosis of cerebral cells or subjected to the ischemia for 30 min and reperfusion for 24 h to detect the infarct volume.
Additionally, the mice were distributed to the groups of Sham, CIRI, Vector, short hairpin RNA against VDR (shVDR) and VDR (n = 10/group). The mice in the last three groups received intraventricular injection of adenoviruses of vector (control), shVDR and VDR overexpression plasmid 2 h prior to the construction of CIRI model [24]. Also, to determine the apoptosis of cerebral cells and the infarct volume, mice were subjected to the scheme as mentioned above.
At the end of the experiment, all mice were anesthetized by 1% sodium pentobarbital (45 mg/kg, P3761, Sigma-Aldrich, St. Louis, MO, USA) and euthanized with neck dislocation. Then the brain tissue was removed and snap-frozen in liquid nitrogen until usage.
All procedures here referred to previous studies [17, 24]. The adenoviral shuttle vector pDC316-mCMV-EGFP (FH1691, FengHui Biotechnology, Co., Ltd., Hunan, China) and backbone plasmid pBHGloxdeltaE13Cre (JiRan Biotechnology, Shanghai, China) were prepared in advance. The cDNA of shVDR and VDR was cloned into the shuttle vector to obtain pDC316-mCMV-shVDR-EGFP and pDC316-mCMV-VDR-EGFP. The adenoviruses of VDR and shVDR were created in accordance with the protocol of an adenoviral system (632269, Takara Bio, Osaka, Japan), and the viral titers were assayed by Adeno-X-Rapid Titer kit (632250, Takara Bio). The virus only carrying empty coding sequences was used as the control.
For adenovirus-mediated overexpression or knockdown of VDR, the adenoviruses were dissolved in 0.9% saline (ST341, Beyotime, Shanghai, China) and administrated into mice 2 h prior to CIRI modeling. Specifically, after the mice were anesthetized and placed on 71000-M automatic stereotactic head frame (RWD Lifescience, Guangdong, China), the adenoviruses were slowly infused (0.2 µL/min) into the right lateral ventricle with the help of an microinfusion pump (VOGT Medical, Karlsruhe, Germany). Bone wax was used to seal the burr hole for the prevention of possible leakage. The suture of incision was made and mice were put back to cages for recovery, with free access to water and food.
To determine the infarct volume, the collected tissue was rinsed with saline for 5 min and placed at –20 °C for 20 min, followed by being transferred to a brain slice mold. The remaining tissues were perfused with 0.9% saline, fixed with 4% paraformaldehyde (P39200, Acmec Biochemical), embedded in paraffin and sliced as required [25].
The infarct volume in the brains of mice was determined as follows [26]. The mouse brains in each group were coronally sectioned into 2 mm-thick slices, dyed with 2% 2,3,5-triphenyl-tetrazolium chloride (TTC, T54930, Acmec Biochemical) at 37 °C for 20 min, and finally fixed with 10% formalin (AC11741, Acmec Biochemical) for 24 h. A digital camera (D500, Nikon, Tokyo, Japan) was used to photograph the brain sections and the infarcted area was quantified via ImageJ (v. 1.8, Bio-Rad, Hercules, CA, USA). The infarct volume of mice was determined by the formula:
The apoptosis of cerebral cells was detected as depicted in a previous study
[27]. The TUNEL staining was performed following the producer’s protocols. In
detail, the 5 µm-thick sections of fixed paraffin-embedded brain tissue
were obtained, following which the sections were deparaffinized and rehydrated.
The sections were then immersed in 3% hydrogen peroxide
(H112515, Aladdin, Shanghai, China) and in 0.1 M sodium citrate
(S25420, Acmec Biochemical) in a microwave for 5 min, followed
by treatment with TUNEL assay kit
(11684795910, Roche Diagnostics,
Indianapolis, IN, USA) and dyeing with diaminobenzidine (DAB, AC11043, Acmec
Biochemical) in sequence. The TUNEL-positive cerebral cells were observed in a
fluorescence microscope (
Human neuroblastoma cell line SK-N-SH (AW-CELLS-H0335, AnWei Biotechnology, Shanghai, China) was maintained in minimum essential medium (iCell-0012, iCell, Shanghai, China) blended with 10% fetal bovine serum (iCell-0500, iCell), 1% non-essential amino acid (iCell-01000, iCell), 1% L-GlutaMax (iCell-0900, iCell) and 1% penicillin-streptomycin (iCell-15140-122, iCell). CB-S Solid.Line 170 incubator (BINDER, GmbH, Tuttlingen, Germany) was used to culture the cells with 5% CO2 at 37 °C as needed. Short tandem repeat method was exploited to verify the cell line. The cells used in this study were validated by STR profiling and tested negative for mycoplasma.
To mimic CIRI in vitro, SK-N-SH cells were treated with the OGD/R as described [28]. Briefly, the normal medium for cell culture was replaced by glucose-free Dulbecco’s modified Eagle’s medium (iCell-138-0001, iCell) and cells were incubated in an incubator at 37 °C with 95% N2 and 5% CO2 for 4 h. For reoxygenation process, cells were then maintained in normoxic condition (95% air and 5% CO2) for 24 h. Cells in the control group were cultured normally without OGD/R.
To determine the effect of VDR in SK-N-SH cells, the cells were transfected with the shVDR (C02002) and its control (Vector, C03002) obtainable from GenePharma (Shanghai, China) with the help of lipofectamine RNAiMAX transfection reagent (13778150, Thermo Fisher Scientific, Waltham, MA, USA). The sequences used were available in Table 1.
| Gene | Sequence (5′-3′) |
|---|---|
| sh-VDR forward oligo | CCGGCCTCCTTCCTCTGCCTTTATACTCGAGTATAAAGGCAGAGGAAGGAGGTTTTTG |
| sh-VDR reverse oligo | AATTCAAAAACCTCCTTCCTCTGCCTTTATACTCGAGTATAAAGGCAGAGGAAGGAGG |
| Vector forward oligo | CCGGCTGTCCCTCTTTTATACTGGACGAGTATAAAGGCAGAGGAAGGTTTTCTTCGCC |
| Vector reverse oligo | AATTCAAAACCTCTCTTTATAAAGGCTCGAGTATAAAGGCAGAGGAGACCTCCTTGGC |
Then cells were treated with OGD/R only or additionally treated with N-acetyl-L-cysteine (NAC, N27390, Acmec Biochemical), a scavenger of oxidative stress, at 5 mM for 3 h [29].
2
The apoptosis of SK-N-SH cells with indicated treatment was determined via flow
cytometry using a commercial kit (C0162S, Beyotime). After the centrifugation at
1000
A ROS assay kit (S0033S, Beyotime) was utilized to estimate the level of ROS in
the brain tissue slices (5 µm-thick) and SK-N-SH cells (3
Two schemes were adopted for the analyses on the levels of factors related to oxidative stress [30, 31]. In the first scheme, the tissues harvested from the cerebral peri-infarcted areas of mice were homogenized mechanically and centrifuged for 10 min to collect the supernatant. Following the detection of protein concentration by bicinchoninic acid protein assay kit (AC13858, Acmec Biochemical), the content of malondialdehyde (MDA, KTE71091, Abbkine, Hubei, China) and the level of superoxide dismutase (SOD, CSB-E08556m, Cusabio, Fannin, TX, USA) were determined by a microplate reader according to the protocols.
In the second scheme, SK-N-SH cells (1
The total RNA in the tissue of cerebral peri-infarcted areas and SK-N-SH cells were harvested by Trizol (R0016, Beyotime) and then CM-36dG spectrophotometer (Konica Minolta, Tokyo, Japan) was used to assess the quality and quantity of the RNA. cDNA was synthesized via a QuantiTech Reverse Transcription kit (205311, Qiagen, Hilden, Germany), and the PCR was operated by a multiplex PCR kit (206145, Qiagen) in a PCR system (911001, Qiagen) under the following conditions as per the protocols:
(1) initial heat activation: 95 °C for 15 min
(2) 3-step cycling in a total number of 40 times:
a. 94 °C for 30 seconds (s)
b. 60 °C for 90 s
c. 72 °C for 90 s
(3) 72 °C for 10 min.
Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was the internal control and 2-ΔΔCT method was employed to analyze the relative expressions [32].
Primers were listed below:
(mouse) VDR: Forward: 5′-ATTCGTGCAGACGTAAGTA-3′
Reverse: 5′-GGTGTACAGATCAGAGTTTG-3′
(human) VDR: Forward: 5′-ATGAAGGAGTTCATTCTGAC-3′
Reverse: 5′-CTGAATCCTGGTATCATCTT-3′
(mouse) GAPDH: Forward: 5′-CCTCAACTACATGGTTTACA-3′
Reverse: 5′-TGTTGTCATACTTCTCATGG-3′
(human) GAPDH: Forward: 5′-GATGCTGGTGCTGAGTAT-3′
Reverse: 5′-CACAAAGTTGTCATTGAGAG-3′
All reagents, except for those indicated, were the products of E-Labscience (Hubei, China). Radio immunoprecipitation assay lysis buffer which contained phosphatase inhibitors and protease (E-BC-R327) was utilized for the extraction of proteins in cerebral tissue and SK-N-SH cells. Following the separation on 12% gel reagent (E-IR-R305), the samples were transferred to the polyvinylidene fluoride membranes (E-BC-R266). Subsequently, the membranes were soaked in 5% defatted milk for 60 min and, under the condition of 4 °C overnight, were incubated with the primary antibodies (1:1000; Cell Signaling Technology, Danvers, MA, USA) against VDR (#12550, 54 and 48 kDa), Bcl-2-associated X (Bax, #2772, 20 kDa), B-cell lymphoma-2 (Bcl-2, #3498, 26 kDa), Cleaved Caspase-3 (#9661, 19 and 17 kDa), and housekeeping gene GAPDH (#5174, 37 kDa). After that, the membranes were rinsed using TBST (E-BC-R347) and continuously incubated with horseradish peroxidase conjugated goat anti-rabbit IgG (E-AB-1003, 1:5000) at room temperature for 60 min. Enhanced chemiluminescence reagent (E-BC-R347) was applied to detect the protein bands, and the chemiDoc imaging system (Bio-Rad) with Quantity One software 4.6 (Bio-Rad) was employed to analyzed the results [33].
GraphPad Prism 8 (GraphPad, Inc., San Diego, CA, USA) was utilized to analyze
the data of three independent tests to reduce errors and obtain the mean value.
All data were displayed as mean
Former findings made us wonder if VDR could impact mice with CIRI, and
accordingly, we measured its mRNA and protein expressions in cerebral
peri-infarcted areas of mice by qRT-PCR and Western blot, and found similar
results that the level of VDR was evidently increased in the cerebral tissue of
CIRI mice compared to mice with 30 min ischemia (Fig. 1A,B, p
 Fig. 1.
Fig. 1.
VDR was expressed in mouse cerebral tissue and its expression
was elevated by CIRI. (A,B) The protein and mRNA expression levels of VDR in the
cerebral tissue of mice with CIRI were determined via Western blot (A) and
qRT-PCR (B). GAPDH was used as the housekeeping gene. All data of three
independent tests were expressed as mean
Subsequently, in order to determine whether the activation of VDR could protect
against CIRI-induced cerebral injury in mice, we treated CIRI mice with vehicle
(the solvent only), Calcitriol or PC, and
then calculated the cerebral infarct volume based on TTC staining. A sharp
increased infarct volume was seen in CIRI mice compared with Sham mice (Fig. 2A,B, p
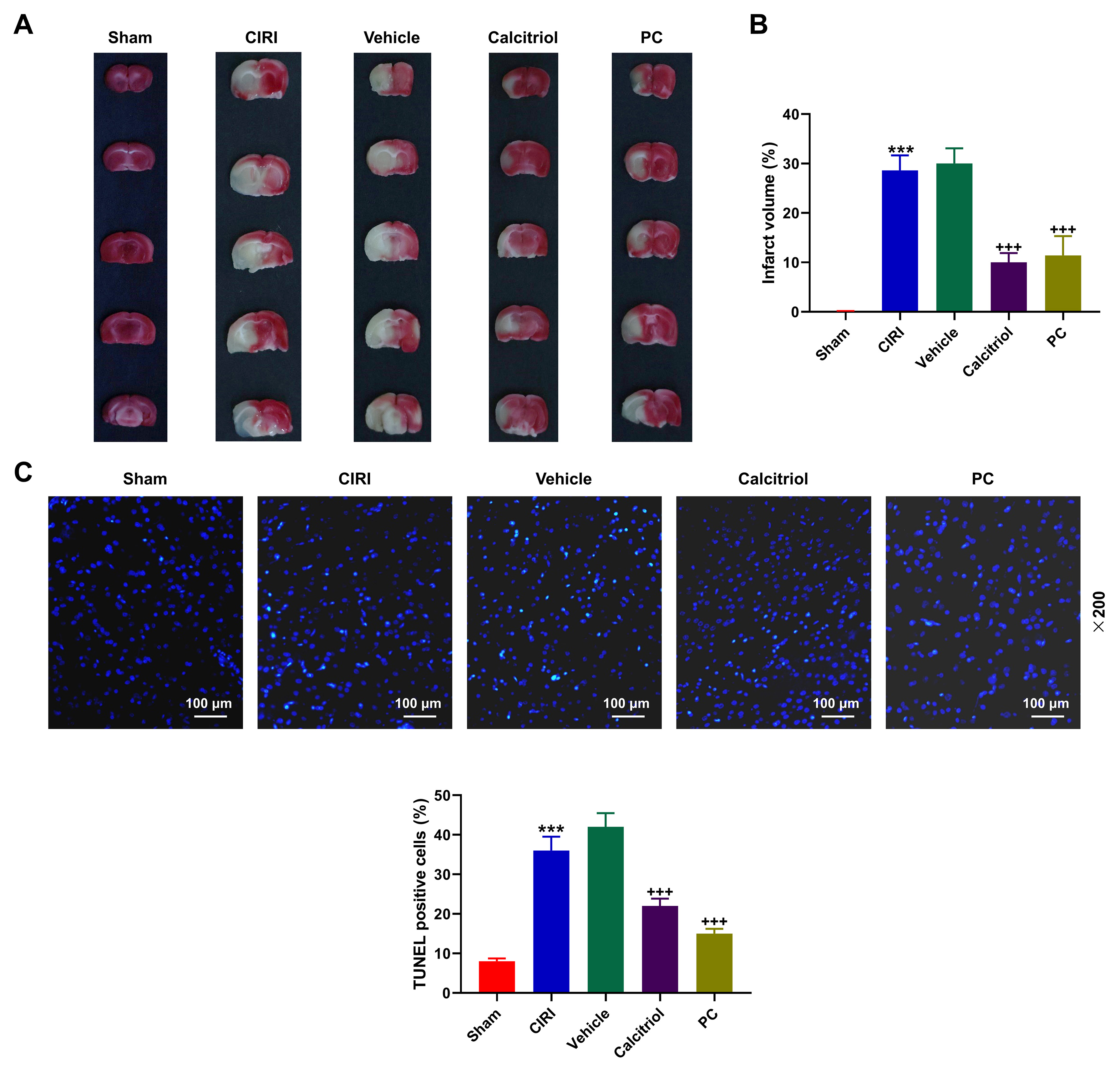 Fig. 2.
Fig. 2.
Activation of VDR attenuated CIRI-induced cerebral infarction
and apoptosis of cerebral cells in mice. (A,B) Cerebral infarct size in mice,
which underwent the ischemia for 30 min and reperfusion for 24 h, with CIRI and
the administration of VDR agonist or the solvent only was measured using TTC
staining. (C) The apoptosis of cerebral cells in mice with CIRI and the
administration of VDR agonist or the solvent only was detected according to TUNEL
staining (magnification:
To unravel the effect of VDR in CIRI mice, we subsequently constructed the
adenoviruses of both VDR overexpression plasmid and shVDR to manipulate VDR
expression. In light of Western blot and qRT-PCR data, both protein and mRNA
expressions of VDR were increased in CIRI mice relative to Sham mice (Fig. 3A,B,
p
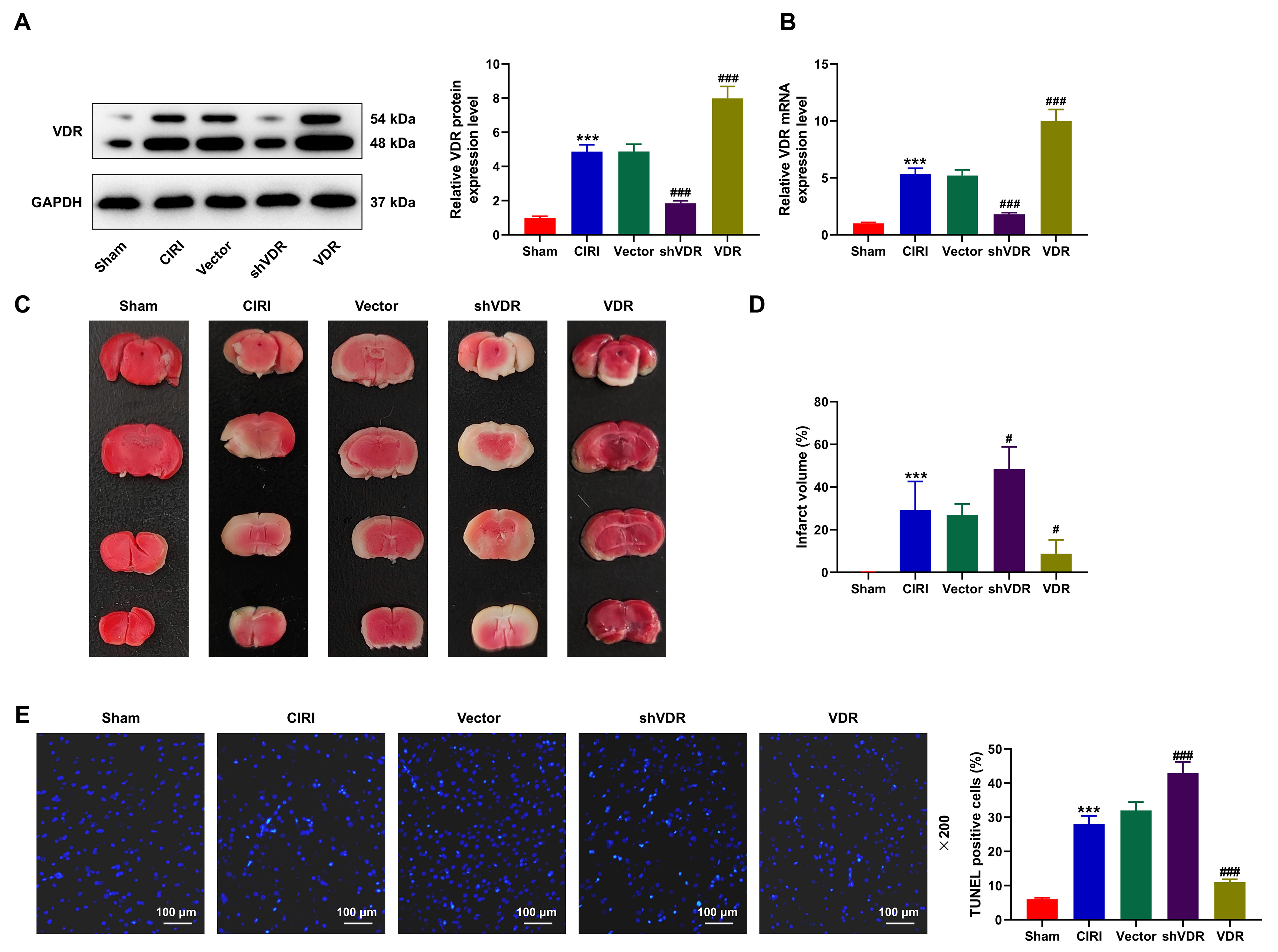 Fig. 3.
Fig. 3.
The overexpression of VDR attenuated the effects of CIRI on
aggravating the cerebral infarction and apoptosis of cerebral cells. (A,B) The
protein and mRNA expressions of VDR in CIRI mice with the overexpression or
depletion of VDR were measured via Western blot (A) and qRT-PCR (B). GAPDH was
used as the housekeeping gene. (C,D) The infarct volume of CIRI mice with the
overexpression or depletion of VDR was calculated based on the results of TTC
staining. (E) The apoptosis of cerebral cells in CIRI mice with the
overexpression or depletion of VDR was detected based on the results of TUNEL
assay (magnification:
Additionally, as for the levels of oxidative stress-related factors, in CIRI
mice, an upward trend was determined in the levels of ROS and MDA, whilst a
downward trend in the level of SOD was found in comparison to Sham mice (Fig. 4A–C, p
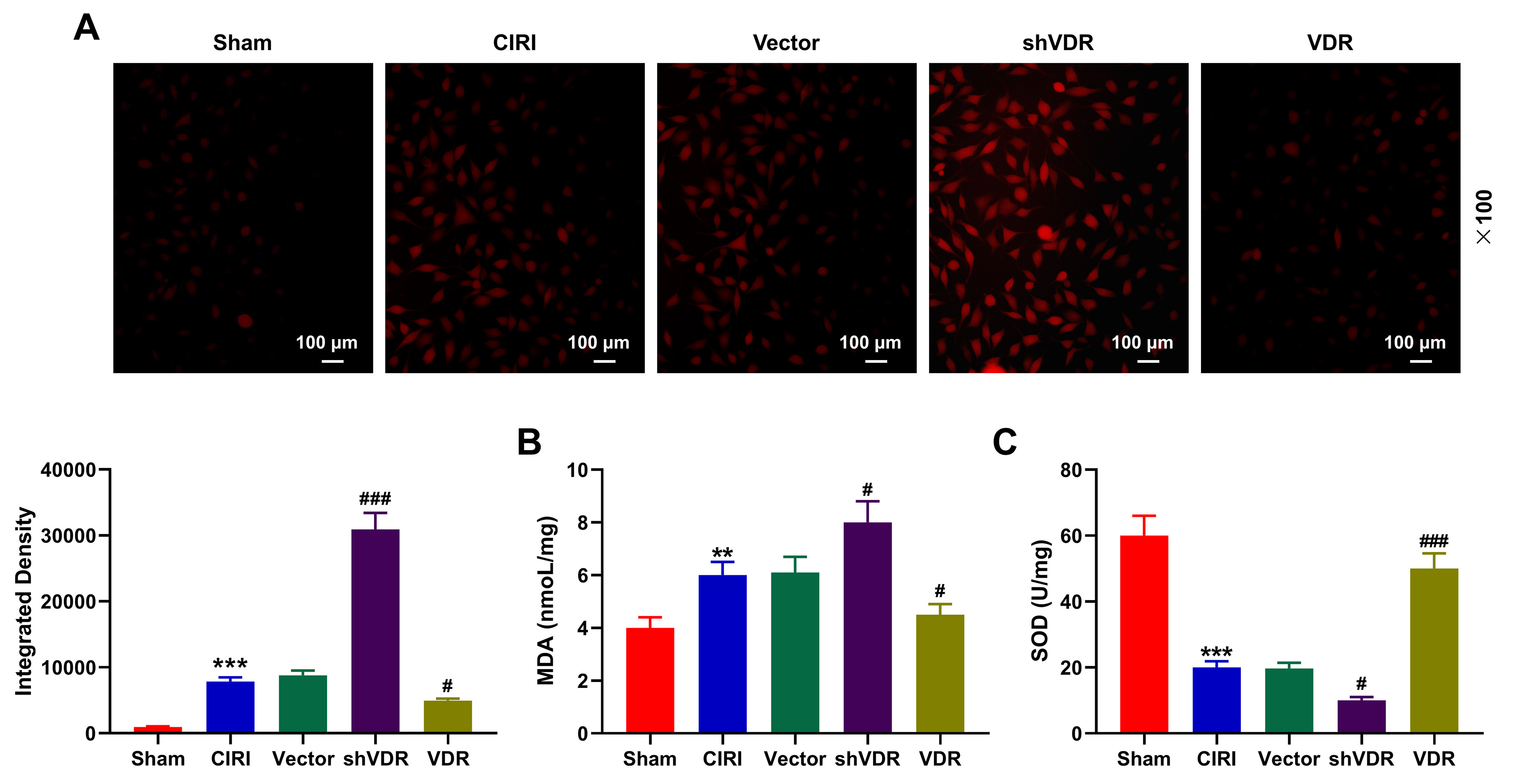 Fig. 4.
Fig. 4.
Overexpressed VDR diminished the effects of CIRI on the levels
of oxidative stress-related factors. (A–C) The levels of oxidative
stress-related factors including ROS (magnification:
To further confirm the interplay between VDR and oxidative stress, we employed
SK-N-SH cells to construct an OGD/R-treated cell model, and OGD/R-treated cells
were transfected with shVDR together with/without treatment of NAC, a scavenger
of oxidative stress. It could be seen that the level of VDR was sharply increased
in OGD/R-treated SK-N-SH cells in comparison to Control SK-N-SH cells (Fig. 5A,
p
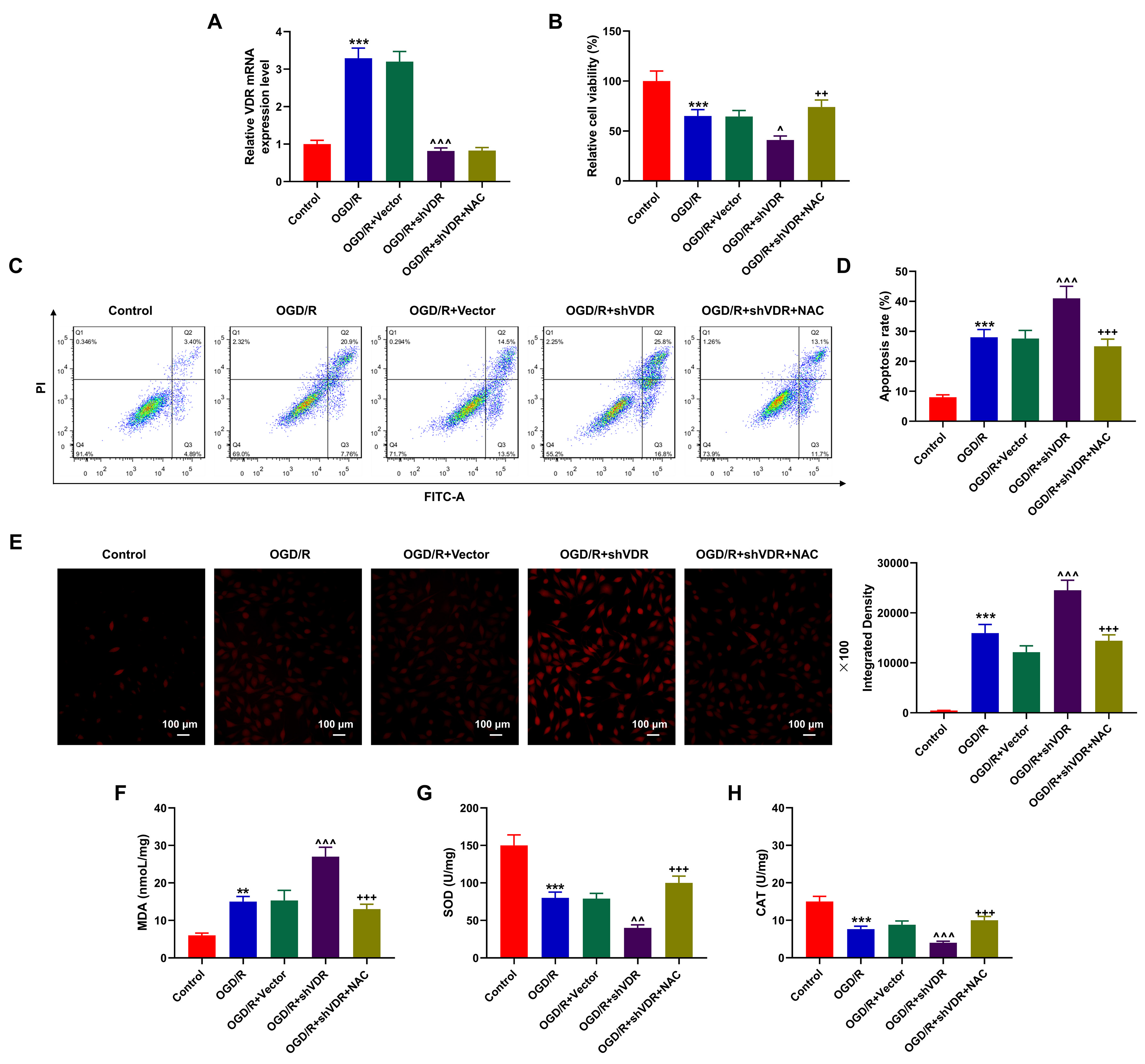 Fig. 5.
Fig. 5.
NAC reversed the effects of VDR silencing on the apoptosis and
oxidative stress in OGD/R-treated SK-N-SH cells. (A) The mRNA expression of VDR
in OGD/R-treated SK-N-SH cells after the silencing of VDR and the treatment of
NAC was quantified via qRT-PCR. GAPDH was the housekeeping gene. (B) CCK-8 assay
was performed to measure the viability of OGD/R-treated SK-N-SH cells after the
silencing of VDR and the treatment of NAC. (C,D) Flow cytometry was applied to
determine the apoptosis of OGD/R-treated SK-N-SH cells after the silencing of VDR
and the treatment of NAC. (E–H) The levels of oxidative stress-related factors
ROS (magnification:
Also, in flow cytometry and CCK-8 assays, the cell viability was suppressed yet
the apoptosis was enhanced in OGD/R-treated SK-N-SH cells compared with Control
SK-N-SH cells (Fig. 5B–D, p
Furthermore, the results indicated that the levels of ROS and MDA were
upregulated yet those of SOD and CAT were downregulated in OGD/R-treated SK-N-SH
cells (Fig. 5E–H, p
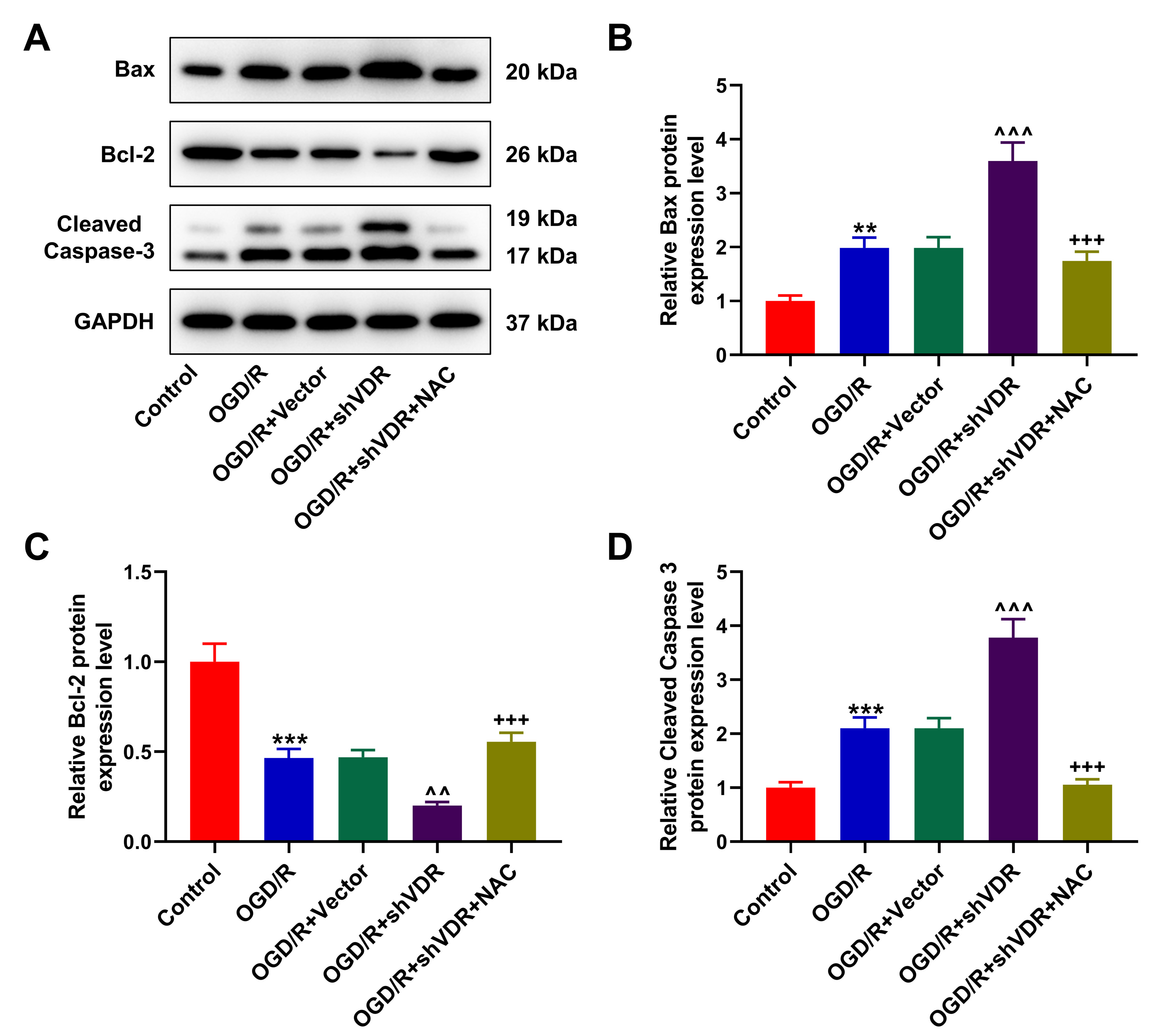 Fig. 6.
Fig. 6.
NAC reversed the effect of VDR silencing on the expressions of
apoptosis-related factors in OGD/R-treated SK-N-SH cells. (A–D) Western blot
was used to quantify the expressions of apoptosis-related factors Bax (B), Bcl-2
(C), and Cleaved Caspase-3 (D) in OGD/R-treated SK-N-SH cells after the silencing
of VDR and the treatment of NAC. GAPDH was the housekeeping gene. All data of
three independent tests were expressed as mean
The VDR is recognized as a chromatin modifier that facilitates expressions of vitamin D-dependent genes, which is highly expressed in the tissues of colon, kidney, small intestine, bone, skin and even brain [34, 35]. A previous study showed that VDR exhibits a predominant upregulation in peri-infarct microglia/macrophages following cerebral ischemia [36]. VDR attenuates ischemia-reperfusion-induced acute kidney disease by suppressing endoplasmic reticulum stress partly via transcriptional regulation of activating transcription factor 4 [37]. Additionally, a prior publication has indicated that VDR also expresses in the cardiovascular system, and can protect against MIRI in mice [17]. However, to our knowledge, the effect of VDR on CIRI has not been expounded yet. Here, we revealed that the endogenous expression of VDR was evidently enhanced in mice after CIRI, which was possibly associated with the protective effect of VDR during the process of CIRI-induced injury. The pharmacological agonists of VDR, including Calcitriol and PC, or overexpressed VDR both diminished the infarct size and the apoptosis of cerebral cells in CIRI-modeled mice. Furthermore, in OGD/R-treated SK-N-SH cells, we confirmed that the silencing of VDR, on the contrary, aggravated the effects of OGD/R on suppressing the viability, and promoting the apoptosis and the oxidative stress. Therefore, it could be concluded that VDR could protect against CIRI in mice and OGD/R in SK-N-SH cells.
The thrombus in blood that leads to insufficient blood supply to the brain is the major reason for ischemic stroke, and the thrombolysis often causes the reperfusion of the infarcted cerebral tissue and thus increases the risks of reperfusion injury [38, 39]. The cerebral ischemia can result in the necrosis of the cerebral tissue within the nucleus of cerebral area and the tardive neuronal death, which finally triggers the damage within the brain [40]. In other words, a necrotic area within the infarct focus and a surrounding ischemic penumbra constitute the area of the ischemic event [41]. Necrotic cells mostly distribute within the center of the infarction, whilst apoptotic cells are discovered in the ischemic penumbra [42]. It’s worth noting that vitamin D hormone has the potential to reduce the infarction in adult rats following post-stroke systemic inflammation [43]. Cellular apoptosis is a physiological process which can be both internally and externally activated and regulated by several genes, and apoptosis is one of the cell death programs activated by CIRI [42, 44]. The Bcl-2 family, for instance, control the intrinsic apoptosis pathway, and specifically, the pro-apoptotic Bcl-2 family protein Bax can commit cells to the programmed death, whereas Bcl-2 itself has been documented to function as an anti-apoptotic molecule that prevents against normal apoptosis [45, 46]. In rats with cerebral ischemia, following the CIRI modeling, Bax not only represses the expression of Bcl-2 indirectly to form the Bax-Bcl-2 heterodimers and to initiate cell apoptosis, but also directly activates caspase-3, which contributes to cell death substantially both in the penumbra and the ischemic core [47, 48]. Also, in a cellular OGD/R model of SK-N-SH cells, the shRNA-mediated VDR knockdown aggravates the effects of OGD/R on inhibiting the viability yet enhancing the apoptosis through upregulating the expressions of cleaved caspase-3 and Bax and downregulating that of Bcl-2, but such effects of VDR knockdown were neutralized via NAC, a scavenger of oxidative stress. A previous study showed that vitamin D-vitamin D receptor alleviates oxidative stress in ischemic acute kidney injury via elevating glutathione peroxidase 3 [49]. Therefore, we had reason to believe that the effects of VDR on OGD/R in vitro and CIRI in vivo were associated with the attenuation of oxidative stress.
Meanwhile, it has been
indicated that CIRI is also attributed to the disturbances in energy homeostasis,
as indicated by the fact that CIRI enhances the mitochondrial production of ROS,
which causes ROS interference in the regulation of gene expression and the
interaction of ROS with the brain-enriched lipids, promoting the generation of
hydrogen peroxide free radicals, the lipid peroxidation within the membrane and
the damage of cells [50, 51]. As a biomarker of lipid peroxidation, MDA causes a
secondary cerebral injury by interacting with biomacromolecules [52]. Also,
CIRI-induced excessive production of ROS is further worsened via the damaged
cellular antioxidant defense system under the ischemic conditions [53]. SOD and
CAT have been previously discussed and evaluated for their protective roles in
ROS-mediated cerebral injuries, those related to ischemia/reperfusion in
particular [54]. SOD can remove the
superoxide anions via the transformation of these anions into
hydrogen peroxide to ameliorate the CIRI, and CAT, an
antioxidant enzyme, can decompose hydrogen peroxide, the byproduct of aerobic
metabolism, into water and oxygen [55, 56, 57]. Considering the effects of VDR on
these processes, including the apoptosis and oxidative stress, existing research
has revealed that VDR activation elicits an anti-apoptotic effect on
ischemia/reperfusion-treated myocardium via downregulating markers of both
endoplasmic reticulum stress (the activation of caspase-12 protein) and
mitochondrial stress (the activation of caspase-9 and the release of cytochrome
c), and the activator of VDR Calcitriol generates an anti-inflammatory effect on
spontaneously hypertensive rats [17, 21]. Reciprocal activation between SMAD
family member 3 and VDR transcription factors defines vitamin D-mediated
oxidative stress to prevent CIRI [58]. In our current research, the
adenovirus-mediated VDR overexpression alleviated the oxidative stress via
upregulating the level of antioxidant SOD yet downregulating those of pro-oxidant
ROS and MDA. Meanwhile, the shRNA-mediated VDR knockdown potentiated the effect
of OGD/R on the oxidative stress via promoting ROS and MDA levels yet suppressing
those of SOD and CAT in OGD/R SK-N-SH cell models. Additionally, the NAC
administration also diminished such effects of VDR knockdown, leading us to
suspect that the effects of VDR on both OGD/R in vitro and
CIRI in vivo were indeed achieved via attenuating
oxidative stress. Moreover, a previous study showed that vitamin D prevents
hypoxia/reoxygenation-induced blood-brain barrier disruption
via vitamin D receptor-mediated nuclear factor kappa-B (NF-
Besides, although the model used in this study has the advantages of simple operation, good reproducibility, easy control of reperfusion and physiological changes, and easy control of ischemia and reperfusion time, the modeling requires complex operations and the infarct volume is difficult to control, which in turn affects the results. Therefore, future studies need to improve the model or further validate the results with different models. Further, dynamic changes of selected parameters after different durations of ischemia and reperfusion should also be tested.
Collectively, we additionally confirm that VDR, a known protective receptor against MIRI, could also be utilized as a protector against CIRI in mice in vivo and OGD/R in SK-N-SH cells in vitro as well, which is achieved via diminishing the CIRI-induced cerebral infarction in vivo, attenuating the oxidative stress, and inhibiting the apoptosis of CIRI-induced cerebral cells and OGD/R-treated neuroblastoma cells both in vivo and in vitro. These results thus additionally provide evidence on VDR as a possibly viable molecular target for both prevention and treatment of ischemic diseases. Further, considering the neuroprotective effects of vitamin D and the role of vitamin D in CIRI, interventional prospective studies on vitamin D supplementation in CIRI patients should be conducted in the future.
The analyzed data sets generated during the study are available from the corresponding author on reasonable request.
Substantial contributions to conception and design: JD. Data acquisition, data analysis and interpretation: HH, LW, MD, XZ. Drafting the article or critically revising it for important intellectual content: JD. All authors have participated sufficiently in the work to take public responsibility for appropriate portions of the content and agreed to be accountable for all aspects of the work in ensuring that questions related to its accuracy or integrity. All authors read and approved the final manuscript. All authors contributed to editorial changes in the manuscript.
The animal experiments were carried out after being approved by the ethic committee of Laboratory Animal Center of NTU (Ethics Approval No. IACUC20221230-1006) in compliance with the specifications of China Council on Animal Care and Use, and every attempt was made to cause minimized discomfort and pain to the animals.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/j.fbl2911389.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.