



 , Ahmed Bamaga 2, Alaa Alkhotani 3, Thamer Alsharif 4, Ghada A Abdel-Hamid 5, Mohamed E Selim 6, Taghreed Alsinani 7, Ahmed Albeshri 8, Adnan Badahdah 9, Mazen Basheikh 9, Saleh Baeesa 10
, Ahmed Bamaga 2, Alaa Alkhotani 3, Thamer Alsharif 4, Ghada A Abdel-Hamid 5, Mohamed E Selim 6, Taghreed Alsinani 7, Ahmed Albeshri 8, Adnan Badahdah 9, Mazen Basheikh 9, Saleh Baeesa 101 Department of Pathology, Faculty of Medicine, King Abdulaziz University, 21911 Rabigh, Saudi Arabia
2 Department of Pediatrics, Faculty of Medicine, King Abdulaziz University, 21589 Jeddah, Saudi Arabia
3 Department of Pathology, College of Medicine, Umm Al-Qura University, 21955 Makkah, Saudi Arabia
4 Department of Surgery, King Abdulaziz Specialist Hospital, 26522 Taif, Saudi Arabia
5 Department of Clinical Anatomy, Faculty of Medicine, King Abdulaziz University, 21589 Jeddah, Saudi Arabia
6 Department of Microbiology, Faculty of Medicine, King Abdulaziz University, 21911 Rabigh, Saudi Arabia
7 Department of Neurosurgery, King Fahad Hospital, 22332 Jeddah, Saudi Arabia
8 Department of Surgery, King Abdulaziz Medical City, National Guard Hospital, 21452 Jeddah, Saudi Arabia
9 Department of Internal Medicine, Faculty of Medicine, University of Jeddah, 23882 Jeddah, Saudi Arabia
10 Department of Neurosciences, King Faisal Specialist Hospital and Research Center, 21499 Jeddah, Saudi Arabia
Abstract
Metabolic reprogramming within tumor cells involves a shift towards either glycolysis or mitochondrial respiration, depending on the stage of tumor progression. Consequently, irreversible dysfunction of the mitochondria is considered a crucial mechanism driving the progression mechanism. While numerous mutations in mitochondrial DNA (mtDNA) have been identified across various tumor types, including glioblastoma, many studies have been limited in the scope, focusing on small segments of mtDNA or utilizing sequencing methods with restricted sensitivity. As a result, several potentially significant mtDNA mutations may have been underestimated, along with their heteroplasmic states, which play a crucial role in determining the phenotypic impact of mtDNA mutation. Although both somatic and germline mtDNA mutations have been observed in different tumor types, research on the mtDNA mutations linked to glioblastoma remains scarce. The mitochondrial genome encodes thirteen protein-coding genes that are essential for the proper functioning of respiratory complex chains. Alterations in mitochondrial function manifest at various levels, including structural and functional changes, impacting mitogenic, hemodynamic, bioenergetic, and apoptotic signaling pathways. These alterations often signify a reduced efficiency of the oxidative phosphorylation system and energy production in tumor cells. As the crucial role of mitochondrial dysfunction in glioma development grows, mitochondria have emerged as promising targets for therapy aimed at overcoming chemoresistance and eliminating cancer cells. This brief review outlines the association between mtDNA alteration and glioblastoma, as well as the current advancements in therapeutic strategies targeting mtDNA alterations.
Keywords
- mitochondrial DNA mutation
- oxidative phosphorylation glioblastoma
- treatment
Glioblastoma, the most common aggressive malignant central nervous system (CNS) tumor, is associated with high rates of morbidity, recurrence, and mortality [1, 2, 3]. It can develop either de novo or through progression from a low-grade glioma [4, 5]. Prior to 2021, Grade 4 astrocytoma was classified based on Isocitrate Dehydrogenase (IDH) mutation into IDH-mutant or wildtype [6]. In the recent 5th edition of the World Health Organization (WHO) classification of CNS tumors, glioblastoma is now categorized as a separate entity known as IDH-wildtype glioblastoma [7, 8]. This histogenomic change has significantly impacted the biological complexity of high-grade astrocytomas and their response to various treatment approaches. Due to the observed high biological complexity during tumor progression, glioblastoma is highly resistant to chemotherapies and has become less responsive to the commonly used temozolomide (TMZ) chemotherapy [9]. Over the past two decades, researchers have investigated the molecular and chemical pathways of glioblastoma, including its immune microenvironment, molecular alterations, DNA methylation profiling, and immune-related receptors. Despite no significant breakthroughs have been made in treatment and prognosis, one promising scientific discovery has shown to involve the metabolic programming within glioma cells [10]. Metabolic programming is a rising concept in tumor biology, well-known as a key hallmark of cancer, impacting both cancer cells and the surrounding microenvironment. The interplay between metabolic programming and the microenvironment significantly influences disease progression and treatment response [11]. This reprogramming involves alterations in glucose metabolism and uptake within cells, which can be indirectly influenced by changes in mitochondrial function [12]. A recent study has uncovered that specific tumors depend on mitochondrial respiration instead of glycolysis, challenging the conventional understanding known as the Warburg effect [13]. Consequently, irreversible mitochondrial dysfunction is now considered a crucial theory in the malignant progression of cancer cells, particularly in glioblastoma.
Mitochondria are intracytoplasmic organelles that play a crucial role in
metabolic regulation, energy production, and the control of cellular
proliferation and apoptosis [14]. Each mitochondrion possesses its own genetic
material, and its biogenesis is independent of the cell cycle [14]. The
mitochondrial DNA (mtDNA) is located exclusively within the matrix of the
mitochondrion and encodes 13 protein-coding genes responsible for the function of
the respiratory chain complexes, including complex I (Nicotinamide Adenine
Dinucleotide [NADH]), complex II (Succinate Dehydrogenase [SDH]), complex III
(Cytochrome C Oxidoreductase [COR]), complex IV (Cytochrome C Oxidase [COX]), and
complex V (Adenosine Triphosphate [ATPase]) [15] (Fig. 1). These multi-protein
complexes are essential for cellular energy production. Additionally, mtDNA
contains 22 tRNAs, 2 rRNAs, and the D-loop, which serves as the site for
interactions with nuclear-encoded factors that regulate mtDNA transcription and
replication [15]. The proteins encoded by mtDNA are integral to the electron
transport chain (ETC), which generates ATP through oxidative phosphorylation
(OXPHOS) and is vital for enabling cells to carry out specialized functions [16].
The electron transfers within the respiratory complexes are linked to the
regulation of nucleotide pools, carbon metabolism, calcium transport, and
programmed cell death (apoptosis) [17]. Moreover, mitochondria serve as a primary
contributor to the production of reactive oxygen species (ROS), accounting for
nearly 90% of the overall cellular ROS generation. Elevated levels of ROS result
in oxidative stress and associated complications [17]. Therefore, it is not
surprising that mitochondrial dysfunction contributes to cancer progression and
chemoresistance. Various alterations in mitochondrial function are evident across
different levels, encompassing structural and functional modifications that
impact mitogenic, hemodynamic, bioenergetic, and apoptotic signaling [18]. These
changes may signify a decline in OXPHOS function and energy metabolism in tumor
cells [17, 18]. In cancer, tumor cells rely on cytosolic ATP generated through
glycolysis rather than ATP sourced from mitochondria [9]. In instances where
cancer cells, such as those in glioblastoma, exhibit poor responses to
chemotherapy, it suggests that the emergence of therapy-resistant cells is
closely associated with the metabolic reprogramming of the tumor, including
aerobic glycolysis and reduced mitochondrial energy metabolism [19]. This
phenomenon often arises due to tumor hypoxia, regulated by hypoxia-inducible
factor-1
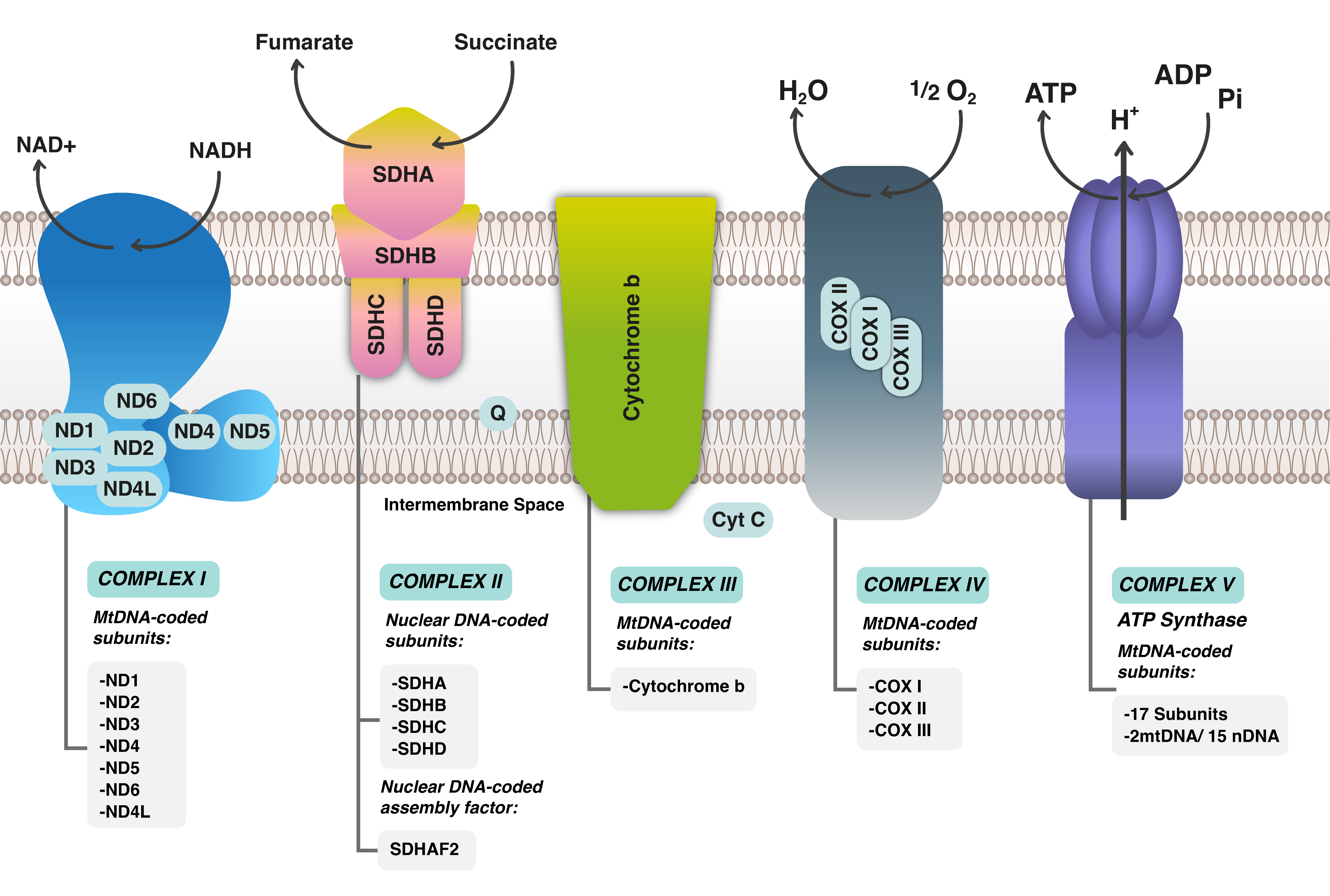 Fig. 1.
Fig. 1.
Mitochondrial Respiratory chain complexes with electron transport chain (ETC) in Mitochondrial DNA. ND stands for NADH Dehydrogenase; NAD, Nicotinamide Adenine Dinucleotide; NADH, Nicotinamide Adenine Dinucleotide H; SDH, Succinate Dehydrogenase (A,B,C,D); SDHAF2, Succinate Dehydrogenase Complex Assembly Factor 2; MtDNA, Mitochondrial DNA; COX, Cyclooxygenase; COMPLEX, Complex (usually refers to the mitochondrial electron transport chain complexes).
While some studies have examined the impact of mtDNA alterations on cancer initiation and maintenance, the precise underlying mechanism remains elusive [21, 22]. Mutations in mtDNA are present in over 50% of solid tumors [23], with these mutations primarily affecting common respiratory coding chains, leading to disruptions in ATP production and heightened oxidative stress [14]. These mtDNA mutations often result in changes in the expression of proteins coded by the respiratory chain, impacting the functionality of respiratory chain complexes when pathways associated with mitochondrial function are altered. Understanding the role of mtDNA in the development, progression, and drug resistance of high-grade astrocytomas, particularly glioblastoma, is crucial as mitochondrial dysfunction is now recognized as a potential marker for glioblastoma etiology [24]. However, the mitochondrial alterations or dysfunctions observed in glioblastoma may stem from mtDNA mutations or be a consequence of dysregulations in intracellular pathways. The majority of mtDNA mutations in glioblastoma are somatic point mutations that are distributed throughout the genome [23, 25]. These frequent alterations have been discussed in a previous study and their existence showed no potential role for specific therapeutic targets [23]. Despite detecting a high number of mutations, glioblastoma carries a limited burden of pathogenic mutations that do not seem to lead to a general impairment of the respiratory chain [9]. In a comprehensive study conducted by Yeung et al. [26], next-generation sequencing (NGS) was utilized to identify mtDNA mutations in germ cell lines. Among the glioblastoma cell lines analyzed, 30% of the detected variants, previously unreported, were located in the non-coding region, with 63% found in the coding region [26]. Significantly, no mutations were detected in the NADH 1 (ND1) gene or the 22 tRNAs. Among the coding region, ND6 and ND4 were identified as the most susceptible genes, whereas the origin of light strand replication was found to be the most vulnerable non-coding gene.
According to the study by Yeung et al. [26], protein-coding genes related to NADH are highly impacted in cancers, particularly in glioblastoma [27]. NADH plays a crucial role in cellular responses to oxygen deficiency and the development of hypoxia mechanisms. Genetic alterations leading to NADH dysfunction can result in resistance to chemotherapeutic agents that rely on the activation of the redox cycle [28]. Approximately 65% of truncating somatic missense mutations in mtDNA in glioblastoma are found in the NAD5 [29]. Additionally, the A10398G polymorphism in the NAD3 gene can lead to complex dysfunction, increased production of ROS, oxidative stress, and potentially promote carcinogenesis [25]. Mutations in genes encoding complexes III and IV are also common in glioblastoma which may cause structural and functional changes in proteins that are considered independent predictors of poor prognosis [30, 31]. For example, the T14798C mutation, resulting in an amino acid alteration in the central subunit of cytochrome b, can impact the activity and sensitivity to drugs targeting complex III, affecting ROS production, cell behavior, and patient response to treatment, ultimately leading to a poor prognosis [21]. Furthermore, mutations in the cytochrome b complex of complex III have been associated with increased drug sensitivity to atovaquone and clomipramine [32].
COX, an enzyme involved in the ETC of cellular respiration, plays a vital role in ATP energy generation. The impact of COX or NADH levels on mitochondrial function in glioblastoma, whether they increase or decrease, remains uncertain and may contribute to mitochondrial dysfunction or enhancement [23]. Decreased expression of the respiratory chain is typically linked to impaired mitochondrial function, potentially linked to the metabolic reprogramming seen in cancer cells. Consequently, the activity of COX and NADH in glioblastoma may be contingent on the specific context and could vary between cases. Further research is necessary to comprehensively grasp the precise alterations in COX and NADH activities and their functional consequences in glioblastoma. Previous research has shown that COX plays a crucial role in the resistance to apoptosis in cervical cancer cells and gliomas [33, 34]. Increased levels of COX activity have also been linked to the development of chemoresistance to TMZ in high-grade gliomas [35]. As COX serves as the terminal enzyme in the mitochondrial respiratory chain, facilitating the transfer of electrons from cytochrome c to oxygen, heightened COX activity enhances the electron flux capacity of the ETC, resulting in decreased ROS production [36]. These changes promote adaptive chemoresistance by suppressing apoptotic signaling pathways [34]. Another possible explanation is that elevated COX activity may provide a competitive advantage during tumor progression under conditions of oxidative stress, nutrient deprivation, and/or hypoxia [33, 34]. In fact, inhibiting COX has been shown to reverse tumor chemoresistance to chemotherapy [31]. Griguer et al. [31], demonstrated a correlation between COX activity and both overall survival and progression-free survival in patients newly diagnosed with glioblastoma. They also noted that tumor COX activity alone does not serve as a confirmed prognostic marker in glioblastoma. The combination of low COX activity and a methylated O6-methylguanine-DNA methyl-transferase (MGMT) promoter may be associated with improved long-term survival outcomes [31]. Additionally, a higher respiratory capacity in glioma cells could impede cancer cell apoptosis by preventing the premature release of COX into the cytosol [37].
The association between IDH and NADH primarily revolves around their
critical roles in cellular metabolism and energy production. IDH1 and IDH2,
enzymes encoded by the IDH1 and IDH2 genes, catalyze the
oxidative decarboxylation of isocitrate to
Mitochondria have been identified as important targets for therapy in overcoming chemoresistance and eliminating cancer cells. As a result, various compounds have been developed to target essential pathways that control mitochondrial biogenesis, autophagy, and antioxidant mechanisms. Reprogramming tumor metabolism through treatment is another significant mechanism of resistance to therapy, and targeting these abnormalities could offer effective novel therapeutic strategies [42]. It is important to emphasize that pharmacological inhibitors have the capability to inhibit all primary mitochondrial fuel sources. Currently, no active clinical trials are registered to try different medications or compounds on altered mtDNA in glioblastoma. Various natural compounds have shown significant anticancer properties by targeting mitochondrial dysfunction, thereby influencing OXPHOS and apoptotic signaling either directly or indirectly by regulating metabolic abnormalities. Zhang et al. [43], illustrated that curcumin induces apoptotic cell death in RN5 murine cells by inhibiting the PI3K-Akt-mTOR signaling pathway. Additionally, the combination of curcumin with gallic acid has been shown to significantly inhibit cell growth and induce apoptosis in cancer cells. However, this combination has not yet been tested in glioblastoma cells [44]. Sulforaphane, an isothiocyanate compound, hinders the growth of human cancer cells by the engagement into intricate cellular processes and triggering the mitochondrial apoptotic pathway, resulting in a reduction in mitochondrial membrane potential, COX nuclear translocation, and the production of ROS [45].
Number of chemical drugs exhibit anti-cancer activity by targeting various mitochondria-related pathways. The combination of diclofenac and lumiracoxib can enhance the sensitivity of cancer cells to the Rapidly Accelerated Fibrosarcoma (RAF)-inhibitor vemurafenib. Together, they amplify the antiglycolytic effects of vemurafenib and prevent the RAF-inhibitor-induced metabolic shift towards OXPHOS [46]. Metformin is an oral inhibitor of mitochondrial complex I and is a widely used drug in diabetic and non-diabetic patients, safe and well tolerated in association with radiotherapy and chemotherapy. Metformin’s in cancer cells has been shown to halt cancer cell growth in glioblastoma by promoting glycolysis and subsequent extracellular acidification [47] (Fig. 2). Mohammad et al. [47], discovered a survival benefit associated with metformin use in glioblastoma patients with methylation of the MGMT promoter. An intriguing recent discovery highlights metformin’s ability to reduce Programmed Cell Death Ligand-1 (PDL-1) levels by interfering with energy metabolism, thereby linking the respiratory chain with the immune system and the tumor microenvironment [48]. Heat shock Protein 90 (HSP90) chaperones also have long been considered promising targets for anticancer therapy due to their cytoprotective properties and overexpression in cancer cells [49]. Geldanamycin, developed to inhibit HSP90 ATPase activity, demonstrated an unfavorable toxicity profile and anticancer efficacy [50] (Fig. 2). To enhance their effectiveness, HSP90 inhibitors have been engineered to penetrate mitochondria without inducing the upregulation of HSP70. Notably, gamitrinib has been shown to impact SDH, highlighting it as a critical target [51]. Gamitrinibs, a second generation of mitochondria-targeted HSP90 inhibitors, represent a class of fully synthetic small molecules modified with diverse mitochondria-targeting sequences [52].
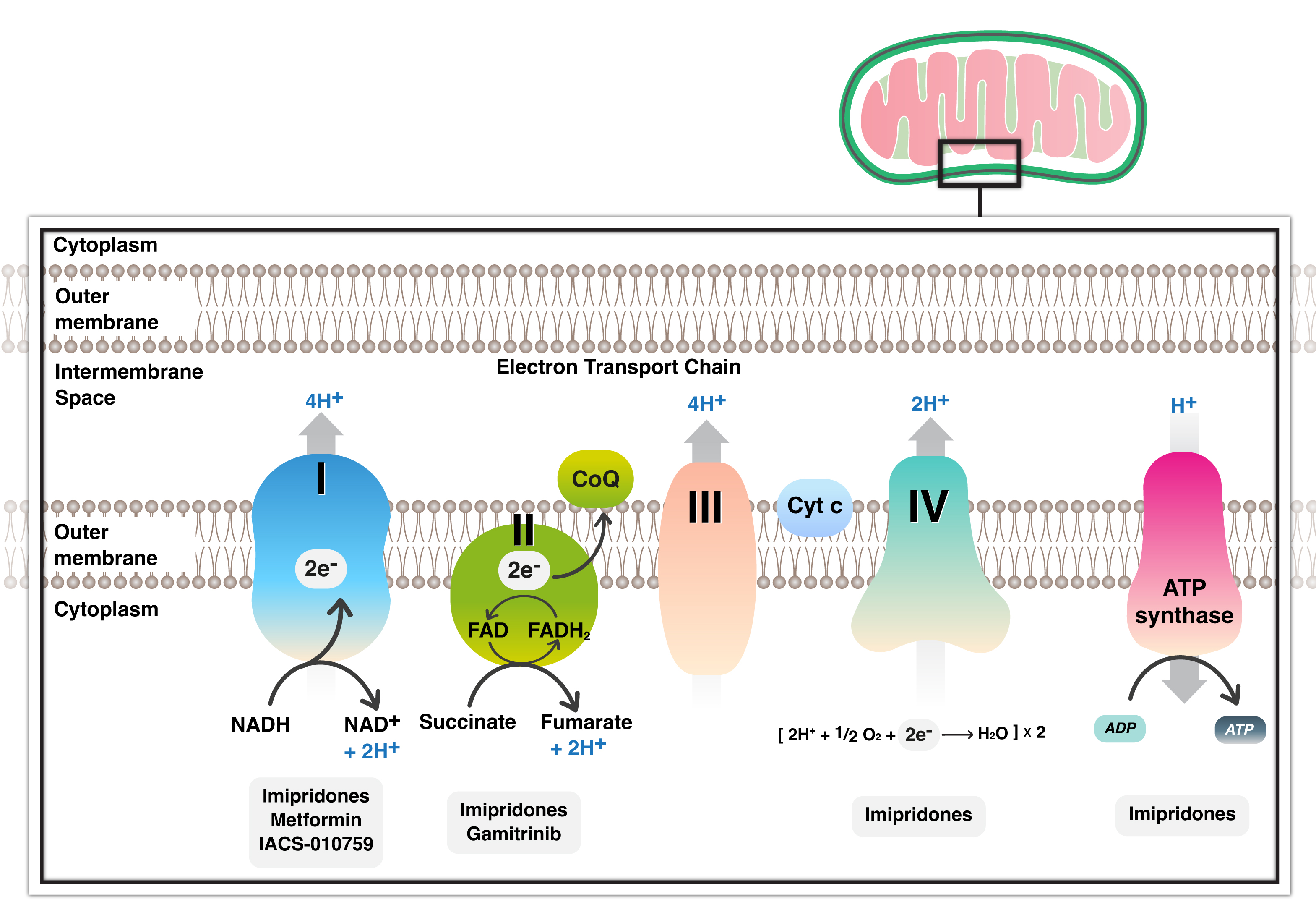 Fig. 2.
Fig. 2.
Inhibitors of mitochondrial respiratory chain with all complexes currently present in glioblastoma treatment. Several inhibitors target complex I and II and several complexes with metformin, gamitrinib and imipridone respectively. NADH, Nicotinamide adenine dinucleotide; FADH, Flavin adenine dinucleotide; CoQ, Coenzyme Q; Cyt, Cytochrome; ATP, Adenosine triphosphate.
The imipridone ONC201 is being explored as a potential anti-cancer therapy [53] (Fig. 2). Imipridone has demonstrated the ability to extend overall survival in the U87 glioblastoma model system in vivo [53]. Nguyen et al. [42], also discovered that FDA-approved Histone Deacetylase (HDAC) inhibitors may significantly impact energy metabolism in glioblastoma. While HDAC inhibitors partially suppressed glycolysis by interfering with enhancers, they activated cellular respiration fueled by fatty acid oxidation [42]. In an orthotopic glioblastoma xenograft model, the combination treatment of imipridone and HDAC inhibitors led to increased survival, suggesting potential translational relevance [42].
Temozolomide (TMZ) is the primary chemotherapy treatment for glioblastoma;
however, its resistance poses a significant challenge in the clinical practice
and patients’ clinical outcomes. Understanding the specific mechanisms behind TMZ
resistance is crucial for overcoming this obstacle. One of the rare mechanisms
associated with TMZ resistance is mtDNA mutation. Limited research has indicated
a potential link between mtDNA mutations and TMZ resistance in glioblastoma.
Leão Barros et al. [23] have highlighted that mitochondrial gene
alterations may contribute to TMZ resistance in glioblastoma. mtDNA mutations can
affect cellular energy metabolism and play a role in drug resistance mechanisms.
Its substantial role in the development of TMZ resistance is evident through
various pathways, including changes in ROS production, metabolic reprogramming,
regulation of apoptosis, biogenesis, dynamics, stress response, and mtDNA
mutations [23, 54]. Variations in mitochondrial DNA copy numbers have been also
observed in TMZ-resistant glioblastoma cells in certain patient samples [55].
Reduced mtDNA copy number has been noted with more aggressive glioblastoma
phenotype and treatment resistance. Specific nuclear DNA mutations in genes
encoding proteins related to IDH mutations have been shown to contribute
to TMZ resistance in IDH-mutant WHO-grade 4 astrocytoma by influencing
the production of oncometabolites, a phenomenon not typically found in
IDH-wildtype glioblastoma [55]. In certain cases, mtDNA mutations have
been linked to changes in ETC activity, potentially impacting ROS production,
apoptosis regulation, and energy metabolism [56]. Additionally, resistance to TMZ
has been associated with mitochondrial biogenesis. Glioblastoma cells resistant
to TMZ demonstrated enhanced mitochondrial function, possibly due to the
upregulation of mitochondrial biogenesis regulator such as peroxisome
proliferator-activated receptor-gamma coactivator 1-alpha (PGC-1
Furthermore, mitochondrial dynamics, such as fission and fusion, play crucial roles in preserving mitochondrial integrity [58]. Wang et al. [58] discovered that TMZ-resistant glioblastoma cells exhibit amplified mitochondrial fusion, resulting in mitochondrial elongation and the survival of TMZ-resistant cells. This process is commonly facilitated by the upregulation of fusion-related proteins. Conversely, the downregulation of the fission-related proteins supports the survival of glioblastoma cells following treatment [59]. In 2020, Mulcahy Levy and Thorburn [60] found that autophagy is another mechanistic process affecting the response of glioblastoma to TMZ treatment Autophagy is a process concerned with destruction and rebuilding of damaged protein under specific stress conditions while mitophagy is a specific type of autophagy that destruct or damage mitochondria. Mitophagy is activated by OXPHOS activity to stimulate the renewal of mitochondria and save their efficiency [61]. The affected cells stimulate autophagy as a survival mechanism to generate energy and removed damaged substances or stress nutrients. However, the role of autophagy and mitophagy are controversial as it may promote tumor suppression or progression based on the tumor invasion or stability. Autophagy and mitophagy promotes tumor progression, metastasis and drug resistance in advanced tumors. Treatment with TMZ induces autophagy and mitophagy through the accumulation of ROS and promotion of ER stress [61]. TMZ treatment combined with the inhibition of autophagy, may enhance TMZ cytotoxicity.
Altered expression of Bcl-2 family proteins were also observed in TMZ-resistant glioblastoma cells [62]. Increased expression of Bcl-2 can promote cell survival and TMZ resistance by inhibiting the release of pro-apoptotic factors [62]. In contrast, the upregulation of pro-apoptotic Bcl-2 family proteins can sensitize glioblastoma in response to TMZ-induced apoptosis. Additionally, the tumor microenvironment, including hypoxia and nutrient availability, can affect mitochondrial function and may contribute to the development of TMZ resistance [62].
To overcome TMZ resistance in the treatment of glioblastoma, targeting the metabolic reprogramming observed in resistant glioblastoma cells is central. Combining TMZ with drugs that target mitochondria or modulators of related signaling pathways may enhance the effectiveness of TMZ in drug-resistant glioblastoma cells. This approach has been explored in various trials, including the use of metformin that has been discussed earlier. Additionally, targeting mitochondrial ROS levels shows promising results in overcoming TMZ resistance. Pro-oxidants like elesclomol were shown to stimulate excess ROS production, induce oxidative stress, and potentially sensitize drug-resistant glioblastoma cells to TMZ-induced apoptosis [63]. Modulating the Bcl-2 family proteins can also impact apoptosis in TMZ-resistant glioblastoma cells [64]. Navitoclax, for example, selectively inhibits anti-apoptotic Bcl-2 family proteins, promoting apoptosis and potentially increasing sensitivity of resistant cells to TMZ [65]. Inhibitors of mitochondrial biogenesis (resveratrol) suppress the expression of key regulators impairing mitochondrial function and sensitizing resistant cells to TMZ [61]. Drugs like chloroquine and hydroxychloroquine are also known to inhibit autophagy [64]. Clinical trials such as NCT00486603 and NCT02378532 are currently assessing the safety and tolerability of hydroxychloroquine in combination with TMZ and radiotherapy in newly diagnosed glioblastoma patients.
Some clinical trials have been assessed the efficacy of using IDH inhibitors (Ivosidenib) in treating patients with IDH-mutant gliomas [66]. The IDH inhibitor may enhance sensitization of glioma cells to TMZ therapy. Moreover, everolimus and temsirolimus, in combination with TMZ or other therapies were also used currently in some clinical trials (NCT00704080) to assess their efficacy in TMZ treatment resistance.
The role of mitochondria in TMZ resistance in glioblastoma is a significant area of study, with various mechanisms identified, including changes in mitochondrial ROS production, metabolic reprogramming, apoptosis regulation, biogenesis, dynamics, stress responses, mtDNA mutations, and interactions with other cellular pathways. Further research is needed to fully understand the specific mechanisms underlying this relationship.
Mitochondria play a crucial role in various cellular processes such as energy production, metabolism, and cell division. The transcriptional regulation of mitochondrial function is primarily governed by mtDNA, which codes for essential proteins in the ETC necessary for Oxidative Phosphorylation. Dysregulated mtDNA expression in cancer cells leads to metabolic reprogramming and dysfunction of mitochondria, contributing to tumor progression. Understanding the impact of mitochondrial transcription in cancer has significant implications for the diagnosis, prognosis, and treatment of cancer. Targeting mitochondrial transcription or associated pathways could offer promising therapeutic approaches for treating glioblastoma.
MK, AlA and SB have made substantial contributions to the concept and the design of the work. AhB, TaA, AhA and ThA performed the literature review. GAAH and MES designed the diagram and the illustrations. AdB and MB performed additional literature. All the manuscripts drafted and revised it critically by all authors for important intellectual content and approved the final version. All authors have participated sufficiently in the work to take public responsibility for appropriate portions of the content and agreed to be accountable for all aspects of the work in ensuring that questions related to its accuracy or integrity. All authors contributed to editorial changes in the manuscript.
Not applicable.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.