




 , Assiya Turgambayeva 2, Aigul Almabayeva 3, Marina Zhanaliyeva 3, Lyazat Orakbay 4, Zhanara Shabanbayeva 3, Oryngul Narmanova 2, Marat Kelissovich Syzdykbayev 5
, Assiya Turgambayeva 2, Aigul Almabayeva 3, Marina Zhanaliyeva 3, Lyazat Orakbay 4, Zhanara Shabanbayeva 3, Oryngul Narmanova 2, Marat Kelissovich Syzdykbayev 51 Institute of Biomedical and Genetic Engineering (IBGE), 44000 Islamabad, Pakistan
2 Department Public Health and Management, NJSC, Medical University Astana, 010000 Astana, Republic of Kazakhstan
3 Department of Human Anatomy, NJSC “Astana Medical University”, 010000 Astana, Republic of Kazakhstan
4 Department of Hygiene and Epidemiology, Kazakh Russian Medical University, 050004 Almaty, Republic of Kazakhstan
5 Department of Anesthesiology, Reanimatology and Narcology, Semey Medical University, 071400 Semey, Republic of Kazakhstan
Abstract
Chromosomal rearrangements and recurrent gene fusions were previously presumed to be the primary oncogenic mechanisms of hematological malignancies. However, the discovery of gene fusions in different cancers has opened new horizons to comprehensively investigate how cell type-specific fusion oncoproteins modulate signaling cascades. Prostate cancer (PCa) is a multifaceted and therapeutically challenging disease, and functional genomics have helped us develop a better understanding of the mechanisms underlying prostate carcinogenesis, castration-resistant PCa, and metastasis. Keeping in mind the fact that gene fusions have also been discovered in PCa, there has been rapid expansion in the field of molecular oncology and researchers are uncovering new facets regarding the mechanistic regulation of signaling pathways by fusion oncoproteins.
Keywords
- cancer
- fused oncoproteins
- cell signaling
- metastasis
- therapy
- therapeutics
- prostate cancer
Chromosomal rearrangements and recurrent gene fusions are gradually gaining increasing attention because of their ability to rewire signaling cascades and fuel cancer development and metastasis. Identification of the transmembrane protease, serine 2:ETS-related gene (TMPRSS2:ERG) in prostate cancer (PCa) has paved the way for detailed analyses of its regulatory role in fusion-positive PCa [1, 2, 3]. Our rapidly evolving knowledge related to the mechanisms underlying the generation of genomic rearrangements has helped put together the missing pieces of an incomplete jigsaw puzzle. Importantly, the androgen-activated gene TMPRSS2 frequently fuses with the ERG gene in PCa cells [4, 5, 6, 7]. There are direct pieces of evidence suggesting that the process of genomic rearrangements is not random in PCa cells [8, 9, 10]. In this introductory section, we summarize cutting-edge and groundbreaking discoveries, which have undoubtedly revolutionized our understanding related to mechanisms underlying the genomic rearrangements and formation of fusion genes.
Increasingly, it is being recognized through pioneering research that signaling
landscapes are rewired for the growth and progression of TMPRSS2:ERG-positive
PCa. TMPRSS2:ERG and oncogenic KRAS work synergistically to promote
tumorigenesis and metastasis. Intracardiac injection of TMPRSS2:ERG- and
KRAS-expressing cells causes widespread metastatic colonization in mice
[11]. TMPRSS2:ERG and mutant p53 also accelerate the progression of PCa. p53
gain-of-function mutants bind to the promoter region of
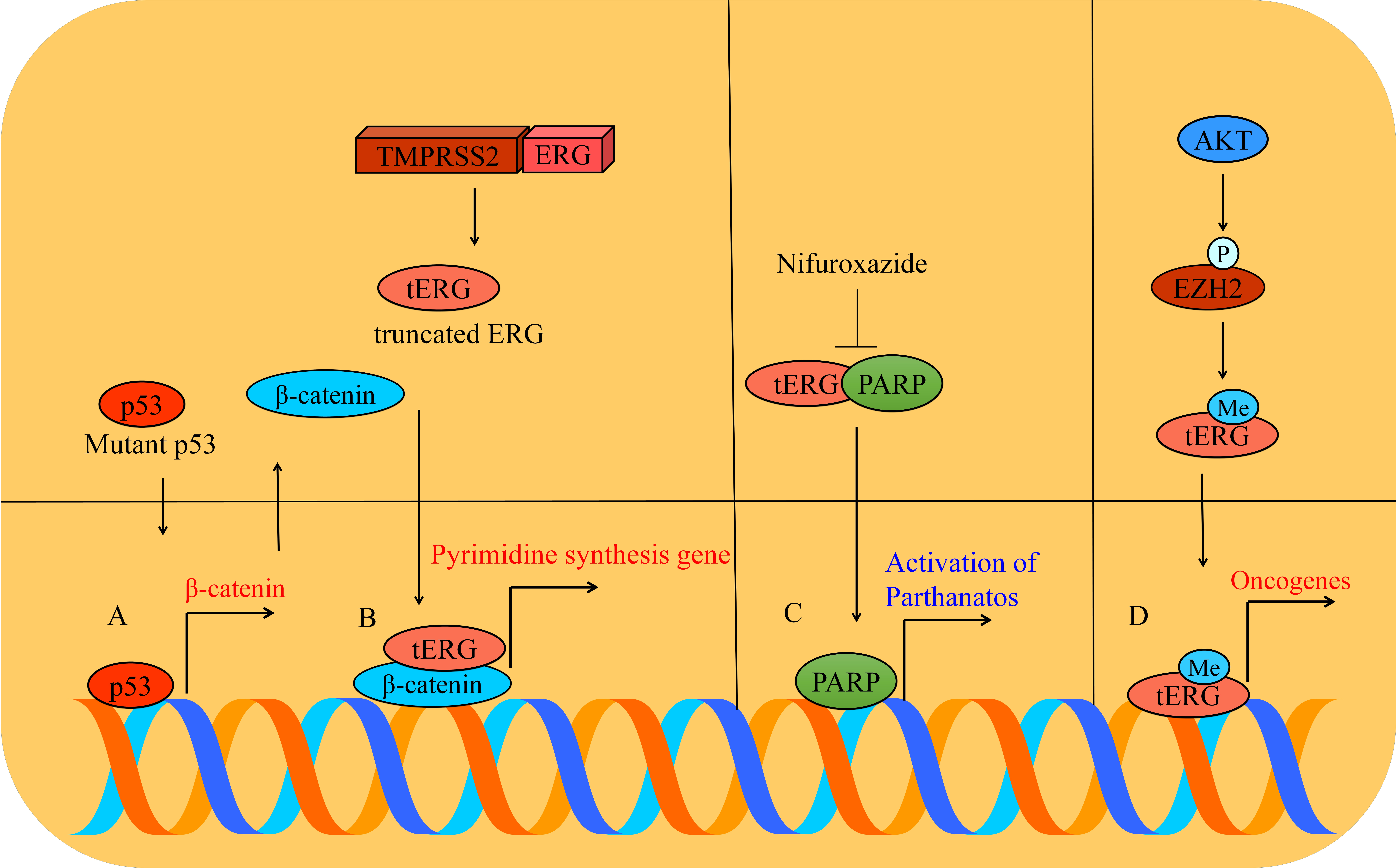 Fig. 1.
Fig. 1.
Crosstalk of tERG with different proteins. (A,B) Mutant p53
stimulates the expression of
Nifuroxazide efficiently targets ERG and inhibits its functions. Nifuroxazide blocks the interactions between ERG and poly (ADP-ribose) polymerase 1 (PARP1) and activates parthanatos (Fig. 1), a type of programmed cell death dependent on hyperactivation of PARP1. Moreover, nifuroxazide also triggers an increase in cleaved PARP1. Intraperitoneally injected nifuroxazide induces tumor regression in mice inoculated with VCaP cells [13].
Frequent overexpression of enhancer of zeste homolog 2 (EZH2) is associated with fusion-positive PCa. EZH2 induces the methylation of ERG at a specific lysine residue (K362) in fusion-positive PCa cells. Methylated K362 potently enhances ERG transcriptional and oncogenic activities. Importantly, preventing methylation by expressing K362A-mutated ERG results in the impairment of ERG-mediated transcriptional activities. Growth rate of tumor xenografts derived from LNCaP cells in mice was enhanced considerably by ERG and not by K362A-mutated ERG. Phosphatase and tensin homolog (PTEN) knockdown not only enhances ERG methylation but also increases the levels and promoter occupancy by ERG, methylated ERG, and EZH2 of different ERG/EZH2 co-regulated target genes. Furthermore, PTEN-depleted VCaP cells demonstrate a significant increase in the ability to invade blood vessels and promote the formation of hepatic metastasis in chorioallantoic membrane assays. However, EZH2 inhibitor causes repression of both functional and transcriptional effects of ERG in PTEN knockdown cancer cells. Importantly, ERG transgenic mice fail to develop invasive lesions; however, mice expressing both ERG and PTEN develop invasive prostate adenocarcinomas. GSK343 (EZH2 inhibitor) causes significant tumor shrinkage in ERG/PTENloss mice. Loss of PTEN and functionally active Protein kinase B (AKT) resulted in the phosphorylation of EZH2 at serine 21 and consequent EZH2-mediated ERG methylation (Fig. 1) [14].
It was previously revealed that androgen induces chromosomal proximity between
TMPRSS2 and ERG loci [15]. There is an evident increase in DNA double-strand
breaks (DSBs) and generation of TMPRSS2-ERG gene fusion in gamma
irradiated cells [15]. Tumor necrosis factor alpha (TNF-
It has been established that fusion gene-encoded proteins and PTEN loss work synergistically to promote prostate carcinogenesis. ERG has been shown to transcriptionally downregulate PTEN in PCa cells [17]. Additionally, due to the loss of phosphatases, there is a marked increase in the phosphorylated levels of fibroblast growth factor receptor 1 (FGFR1) and FGFR4 in fusion transcript-expressing PCa cells [18]. Some evidence has emerged suggesting that ERG transgene expression and deletion of forkhead box protein O1 (FOXO1) result in the upregulation of ERG target genes in fusion transcript-expressing PCa cells [19]. Ectopically expressed FOXO1 inhibits ERG recruitment to target genes. The ERG binding region in FOXO1 is central for FOXO1-mediated inhibition of the ERG-directed migratory capacity of PCa cells [19].
High glucose has been shown to increase the number of TMPRSS2:ERG fusion products [20]. Detailed analyses of the underlying mechanism revealed that the glucose-triggered increase in insulin-like growth factor-binding protein-2 stimulates the repair of DSBs by increasing levels of DNA-dependent protein kinase catalytic subunit, which consequently promote gene fusion [20].
Bromodomain-containing protein 4 (BRD4) is a member of the bromodomain and extraterminal (BET) family of chromatin reader proteins, which bind directly to acetylated histones and modulate the transcriptional regulation of target genes [21]. In a previous study, ionizing radiation (IR) treatment induced marked increases in the acetylation of histone H4 in the first intron of TMPRSS2. Furthermore, an increase in acetylated histone H4 efficiently promoted BRD4 recruitment to chromatin upon DNA damage, whereas treatment with JQ1 (BET inhibitor) blocked BRD4 recruitment to the chromatin in response to IR-directed DNA damage [21]. Treatment with a next-generation BET inhibitor severely impaired IR-mediated tethering of 53BP1, BRD4, XRCC4, KU80, and Artemis to the chromatin in LNCaP and 22Rv1 PCa cell lines [21]. These findings suggest that BRD4 plays a central role in activation of the non-homologous end joining DNA repair pathway. BRD4 knockdown using different small interfering RNAs (siRNAs) significantly blocked the formation of TMPRSS2:ERG [21].
A marked reduction in androgen receptor (AR) transcripts has been observed in ERG-overexpressing LNCaP and VCaP PCa cells [22]. ERG knockdown induces AR upregulation in VCaP cells. ERG disrupts androgen signaling through several mechanisms including inhibition of AR expression, and binding to and repressing AR target genes. Furthermore, overexpressed ERG remarkably increases cell invasion in an AR-independent manner in LNCaP and VCaP cells [22]. TMPRSS2:ERG induces EZH2-directed epigenetic inactivation of various genes. ERG interferes with androgen-regulated prostatic differentiation and promotes EZH2-induced de-differentiation of cells [22].
The functional role of E3 ubiquitin ligases in different cancers has garnered
significant attention. Importantly, the ubiquitination of proteins can be
reversed by deubiquitinating enzymes [23, 24, 25, 26, 27, 28, 29, 30, 31, 32]. Ubiquitin-specific proteases can
modulate various signaling proteins by the removal of ubiquitin(s) from different
target proteins. DNA damage induces proteasomal degradation of wild-type ERG and
TMPRSS2:ERG. ERG is phosphorylated at threonine 187 and tyrosine 190 by glycogen
synthase kinase 3 beta (GSK3
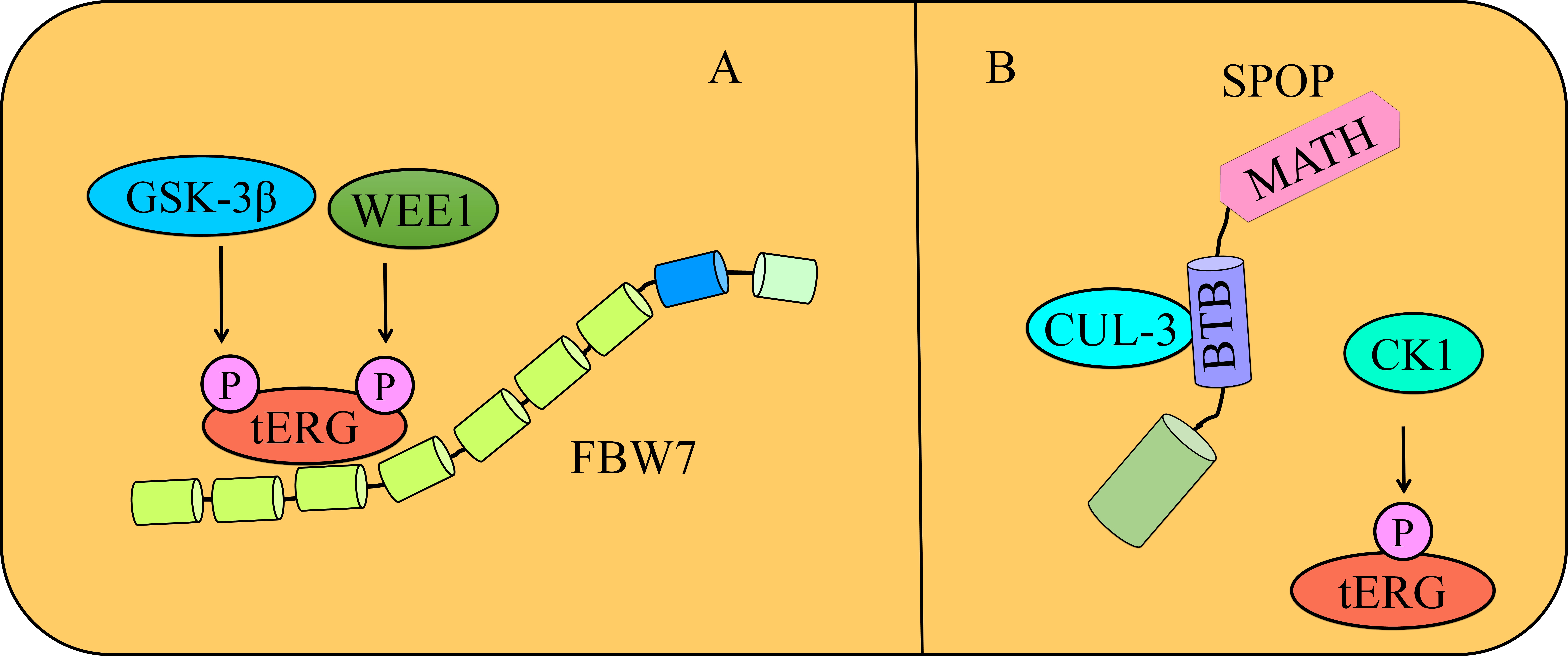 Fig. 2.
Fig. 2.
Degradation of tERG. (A) GSK3
Tripartite motif-containing protein 25 (TRIM25) is another E3 ubiquitin ligase that ubiquitinates target proteins for proteasomal degradation. TRIM25 polyubiquitinates ERG, and inactivation of TRIM25 causes a reduction in the polyubiquitination and stabilization of ERG [34]. These findings are promising; thus future studies should focus on the role of TRIM25 in the inhibition of carcinogenesis in fusion-positive cancers in xenograft models.
Ubiquitin-specific peptidase 9, X-linked (USP9X) stabilizes ERG and promotes prostate carcinogenesis. However, the USP9X inhibitor WP1130 results in the degradation of ERG. WP1130 markedly reduces the levels of ERG in tumors derived from VCaP and 22Rv1 cancer cells. These tumors are poorly vascularized and show a marked reduction in microvessel densities. WP1130 induces shrinkage of the tumor mass in mice xenografted with ERG-overexpressing VCaP cancer cells [35].
Speckle-type POZ protein (SPOP) contains a substrate-binding MATH domain at the amino terminal and a cullin 3-binding BTB domain at the C terminus. Importantly, TMPRSS2:ERG fusion generates amino terminal-truncated ERG proteins. These truncated proteins are resistant to degradation by SPOP. SPOP inactivation-driven increase in the invasive potential of PCa cells is mediated by elevated levels of ERG. ERG contains SPOP-binding consensus (SBC) motifs. The SBC motif 42ASSSS46 is a major degron that effectively controls the SPOP-mediated degradation of ERG and invasion of PCa cells. PCa-associated SPOP mutants are unable to bind, degrade, and impair oncogenic and transcriptional activities of ERG. The majority of TMPRSS2:ERG fusions, such as T1-E4 and T1-E5 have acquired intrinsic mechanisms to counterbalance SPOP-mediated degradation of androgen receptor as well as suppression of androgen receptor-driven transcriptional activation of TMPRSS2:ERG [36].
ERG phosphorylation may facilitate the interaction between ERG and SPOP. Casein
kinase 1 phosphorylates ERG to trigger SPOP/ERG interactions and the degradation
of ERG. Etoposide promotes the association of TMPRSS2:ERG with endogenous SPOP,
which subsequently results in a marked increase in the ubiquitination of
TMPRSS2:ERG. Etoposide suppresses the migration of cancer cells mainly through
degradation of both ERG (wild-type) as well as ERG fusion oncoproteins in SPOP-
and casein kinase I
Traf2 and Nck-interacting kinase (TNIK) is upregulated and phosphorylated on multiple sites in ERG-overexpressing PCa cells [38]. Gene silencing or chemical inhibition of TNIK reduces the viability, colony formation, and anchorage-independent growth of PCa cells [38]. Soluble guanylyl cyclase (sGC) is involved in the synthesis of cyclic guanosine monophosphate (cGMP). cGMP levels are markedly elevated in VCaP cells treated with a high dose of riociguat (sGC activator) [39]. cGMP levels are significantly higher in VCaP-derived tumor xenografts compared to tumors derived from fusion-negative PCa cells. Essentially, sGC-mediated cGMP signaling potentiates the proliferation potential of TMPRSS2:ERG-positive PCa cells. Intraperitoneal injection of an sGC inhibitor (NS2028) considerably reduces tumor growth in mice xenografted with VCaP cancer cells [39]. Mechanistically, it has been shown that TMPRSS2:ERG interacts with Brahma-related gene 1/Brahma (BRG1/BRM) associated factor chromatin remodeling complexes and transcriptionally modulates a wide range of target genes [40]. The presence of a catalytically active BRG1 ATPase is necessary for the regulation of ERG target genes. Changes in the expression levels of ERG target genes have not been observed in BRG1 mutant-expressing PCa cells [40].
There is a marked increase in the number and infiltration of regulatory T cells in PCa tissues in mice subcutaneously injected with TMPRSS2:ERG-expressing PC3 PCa cells [41]. Excitingly, fusion-positive PCa responds to enzalutamide [42]. Overexpression of TMPRSS2:ERG in PCa cells increases the osteoblastic phenotype of bone lesions and inhibits osteoclastic destruction [43]. The ET-1 gene encodes a 21-amino acid secreted peptide that plays a role in the formation of osteoblastic lesions in PCa bone metastases. ET-1 also stimulates osteoblast differentiation and prevents osteoclast-mediated bone resorption. ET-1 expression is notably higher in TMPRSS2:ERG PCa cells, whereas, there is a 40% reduction in the expression of ET-1 in ERG-silenced PCa cells [43].
A positive correlation in the expression of ERG and estrogen-related receptor
alpha (ERR
CREB binding protein (CBP)/p300-interacting transactivator with E/D-rich carboxy-terminal domain-2 (CITED2) is transcriptionally upregulated by ERG in PCa cells [45]. CITED2 interacts with a multimeric complex consisting of nucleolin (NCL), protein arginine N-methyltransferase-5 (PRMT5), and p300. NCL is arginine dimethylated by PRMT5 and lysine acetylated by p300. More importantly, CITED2-mediated NCL modifications are severely impaired by knockdown of PRMT5 and p300, which clearly suggest that post-translational changes in NCL are regulated synchronously by CITED2, PRMT5, and p300 (Fig. 3) [45]. The CITED2-NCL axis promotes the epithelial-to-mesenchymal transition and cell migration. Metastasis is significantly enhanced by CITED2 overexpression but these effects can be reversed by knockdown of NCL [45].
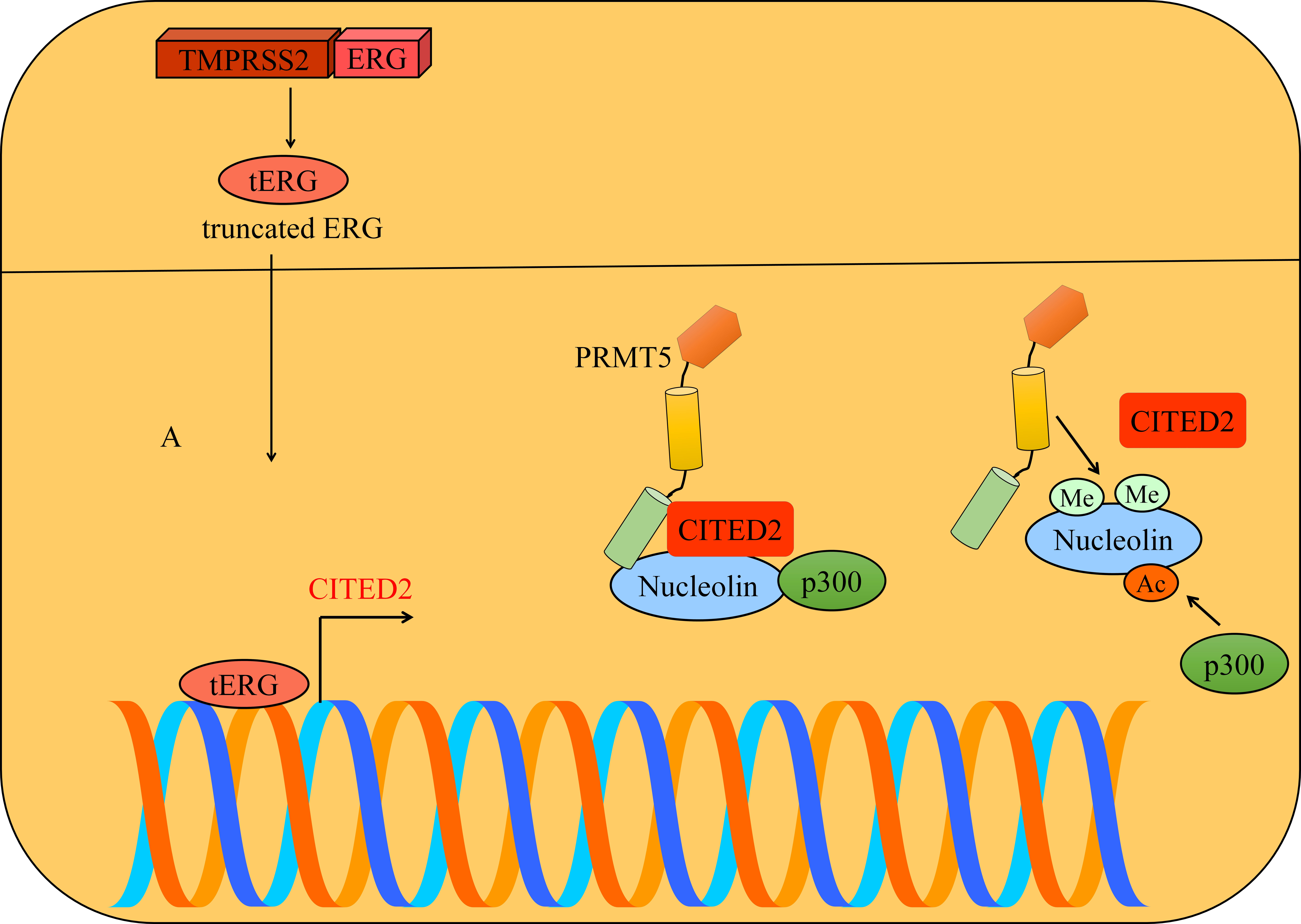 Fig. 3.
Fig. 3.
tERG-mediated upregulation of oncogenic CITED2. Moreover, CITED2 enhanced the interaction of PRMT5 and p300 histone acetyltransferase with nucleolin. PRMT5 mediated methylation and p300-mediated acetylation of nucleolin promoted cancer progression and metastasis. CITED2, CREB binding protein (CBP)/p300-interacting transactivator with E/D-rich carboxy-terminal domain-2; PRMT5, protein arginine N-methyltransferase-5; Me, methyl; Ac, Acetyl.
Extracellular signal-regulated kinase (ERK)-driven phosphorylation of ERG at a serine residue induces conformational changes that further promote ERK phosphorylation at serine 96 [46]. Polycomb repressive complex 2 (PRC2) members (EZH2 and SUZ12) are involved in the transcriptional repression of target genes. Serine 96 phosphorylation results in the dissociation of PRC2 members (SUZ12 and EZH2) and consequent derepression of ERG target genes and enhanced migration of PCa cells. By contrast, loss of ERG phosphorylation at serine 96 facilitates EZH2 binding across the ERG cistrome. Findings suggest that EZH2 recruitment results in a genome-wide loss of the ERG-driven transcriptional regulatory network of target genes and markedly reduces the migratory potential of cancer cells [46].
Secreted frizzled-related protein (SFRP1) induces an increase in the expression of ERG in VCaP cells [47], and promotes the nuclear translocation of AR in VCaP cells. In one study, VCaP cells were injected subcutaneously into mice and when the tumor size reached about 300 mm3, the testicles were removed. Then SFRP1 was administered subcutaneously surrounding the tumor once per week for 10 weeks. The results showed that SFRP1 enhanced the growth of xenografts up to 1000 mm3. Higher levels of ERG were also observed in the tumors of mice that received SFRP1 [47].
Destabilization of runt-related transcription factor 1 (RUNX1)/ETO high molecular weight complexes abrogate the oncogenic activity of RUNX1/ETO [48]. Celastrol, a compound extracted from Tripterygium wilfordii Hook, is effective against fusion oncoproteins. Celastrol markedly reduces the levels of acute myeloid leukemia 1 (AML1)-ETO in the AML cell lines SKNO-1 and Kasumi-1 [49]. siRNA-mediated targeting of the fusion sites of fused oncogenes is also an effective strategy for studying the response of cancer cells. In one study, Kasumi-1 cells were transfected with siRNAs targeting the RUNX1/RUNX1T1 fusion sites [50]. Pretreatment with RUNX1/RUNX1T1 siRNA resulted in an extended median survival of 73 days in xenografted mice [50]. Proton pump inhibitors selectively suppress mixed lineage leukemia (MLL)-rearranged leukemia cells by pharmacological disruption of MLL1-WD repeat-containing protein 5 protein–protein interactions (PPIs) [51].
Derazantinib, a multi-kinase inhibitor, has shown notable pharmacological activity against fibroblast growth factor receptor 2 (FGFR2) fusion-positive intrahepatic cholangiocarcinoma [52]. Results obtained from a phase 1 study of derazantinib in patients with advanced solid tumors have also been encouraging [53]. Therefore, targeted therapy using pan-FGFR inhibitors is advantageous.
The clinical efficacy of tarloxotinib in patients with non-small cell lung cancer is being carefully analyzed. These patients harbor either a human epidermal growth factor receptor (EGFR) exon 20 insertion or EGFR2-activating mutations. Furthermore, tarloxotinib is also being tested in patients with advanced solid tumors with neuregulin 1/ERBB fusion genes (NCT03805841). The safety, tolerability, and efficacy of BOS172738 is being tested in patients with rearranged during transfection (RET) gene-altered tumors (NCT03780517).
Initial promising results demonstrated by nanoparticle-based experimental therapeutics have galvanized their transition into early phases of clinical trials. CALAA-01, a targeted, polymer-based nanoparticle containing siRNA has been reported to be well-tolerated during the initial dose escalation duration of the phase Ia study [54].
Different treatment combinations previously tested to be effective against fusion positive prostate cancers in xenografted mice did not generate promising results in clinical trials. PARP1 interacts with TMPRSS2-ERG gene fusion as well as different other ETS fusion rearrangements. Olaparib (PARP inhibitor) efficiently induces regression of the tumors in mice xenografted with VCaP (ERG positive) cancer cells [55]. Thus, pharmacological targeting of PARP1 provides a conceptual framework for evaluation of PARP inhibitors in ETS-rearranged prostate cancers. Essentially, some clinical trials have reconnoitered the clinical benefits of PARP inhibitors. For example, the multi-institutional randomized phase II trial NCT01576172 assessed the efficacy of prednisone and abiraterone acetate with or without veliparib (a PARP inhibitor) according to the status of “ETS fusion” in metastatic castration-resistant prostate cancers [56]. However, veliparib treatment and ETS status did not influence response in this trial.
ERG-overexpressing prostate cancers have twice the chances of the development of resistance against docetaxel as compared to ERG-negative cancers [57]. However, optimization of the treatment is clinically challenging because of many factors in castration-resistant prostate cancer (CRPC) patients. Targeted inhibition of AR and ERG improves the efficacy of taxane-based drugs. Combination of enzalutamide and taxanes exerted synergistic cytotoxic effects. However, prior treatment with enzalutamide impaired docetaxel activity [58].
“Clickable” siRNA-Polyisoprenoid-conjugated nanoparticles have emerged as novel formulations for the delivery of oligonucleotides. These formulations effectively targeted TMPRSS2-ERG and inhibited tumor progression in mice xenografted with VCaP cells [59]. ERG inhibitory peptides and peptidomimetics interact with ERG with higher specificity and affinity that consequently result in the proteolytic degradation of ERG [60]. ERG inhibitory peptides interfere with ERG-mediated transcriptional regulation, recruitment to chromatin, PPIs, cell invasive ability, and tumor growth [60]. Intraperitoneal injection of 1-[2-Thiazolylazo]-2-naphthol (ERGi-USU) efficiently reduces tumor growth in mice xenografted with TMPRSS2:ERG-expressing VCaP cells [61].
Antisense morpholino oligonucleotides (splice-switching oligonucleotides) reportedly target the 5′ and 3′ splice sites of the 4th exon of ERG [62]. Splice-switching oligonucleotides effectively induce skipping of the exon 4, which consequently reduce the levels of ERG protein. Splice-switching oligonucleotides induce the regression of tumors in mice inoculated with ERG-positive MG63 cells [62].
siRNA TMPRSS2-ERG-squalene nanoparticles have been reported to work efficiently with flutamide and inhibited cancer progression in mice inoculated with VCaP cells [63].
Intravenous injections of nanoparticles of siRNA TMPRSS2-ERG-squalene in SCID mice remarkably suppressed the growth of VCaP xenografted tumors [64].
Targeting of TMPRSS2-ERG fusion mRNA using liposomal nanovectors potently enhanced the efficacy of docetaxel in prostate cancer [65].
TMPRSS2:ERG, a cell type-specific fusion, has been identified in PCa tissues and can also be detected in the urine of PCa patients [66, 67]. Circumstantial evidence has provided proof-of-concept about cell type-specific expression. Findings indicated 94% positive predictive values and 93% specificity in urine samples in a cohort of 108 men [67]. However, because of intra- and interpopulation genetic variability, the TMPRSS2:ERG fusion transcript has not yet gained approval as a diagnostic biomarker for PCa. Furthermore, molecular pathologists have queries related to its predictive value for aggressive disease.
In 2007, a research team reported a high frequency of TMPRSS2:ERG in either poorly or moderately differentiated cancers compared to well-differentiated PCa [68]. Previous study has shown a positive correlation between TMPRSS2:ERG in urine and higher prostate-specific antigen (PSA) levels, Gleason score, and pathological stage [69]. However, these findings have not been verified and validated by contemporary research groups. Large number (1180) of men were evaluated. The results were intriguing because TMPRSS2:ERG overexpression was identified in 49% of patients, and there was no evidence of statistically significant association with tumor grade or Gleason score [70]. Combinatorial analysis of oncogenic fusion transcripts with other markers has also been evaluated. Keeping in mind the heterogeneous nature of PCa, use of a multiplex panel of markers will be helpful in getting a step closer to individualized medicine. It has been shown that the combinatorial evaluation of TMPRSS2:ERG and PCA3 in urine provide additional prognostic and diagnostic significance in PCa [71]. Notably, the validated Michigan Prostate Score (MiPS) offers the combined measurement of TMPRSS2:ERG and PCa antigen 3 (PCA3) in post-digital rectal exam urine samples along with the evaluation of serum levels of PSA.
PCA3 score, TMPRSS2:ERG score, and PSA density are linked independently to prostate biopsy outcomes in multivariable analyses with an area under the curve (AUC) of 0.734. Importantly, in multilogistic regression models, PSA density and PCA3 score are significantly associated with Gleason grade 4 and there is a positive correlation with TMPRSS2:ERG score [72]. The combination of serum PSA, TMPRSS2:ERG, and PCA3 in a multivariable algorithm has also been evaluated for the accurate prediction of PCa with an AUC of 0.88 [73]. The MiPS test has improved the prediction of high-grade PCa on a biopsy. There are several risk calculators designed for clinical use. The Prostate Cancer Prevention Trial (PCPT) risk calculator has been validated in many published studies. Decision curve analyses have indicated the additional benefits of the MiPS test together with the multivariate PCPT risk calculator. Therefore, the MiPS test may prove to be useful for the stratification of risk for high-grade PCa and better exclusion of unnecessary biopsies [74, 75, 76].
The possibility of the occurrence of False-negative results cannot be ruled out, mainly with RNA sequencing performed on archival tissues and specifically when conducted using RNA of poor-quality. Therefore, detection of certain fusions is challenging at RNA [77, 78]. Conversely, RNA sequencing methodologies have enabled the identification of chimeric RNAs that result from cis-splicing and cannot be detected at DNA levels [79].
Results from bigger, prospective trials are necessary to realistically and rationally analyze the irrefutable evidence about therapeutic, diagnostic and prognostic significance of TMPRSS2-ERG and other fusion genes in prostate cancer patients. Intensive bioinformatics and biostatistics research should be expanded for the refinement of fusion detection tools (for example, FusionSeq [80], TopHat-Fusion [81] and defuse [82], candidate fusion prioritization algorithms and dedicated fusion databases, to reduce the likelihoods of false-negative and false-positive results.
Molecular biologists have started to disentangle the convoluted interwoven regulatory pathways in TMPRSS2:ERG-positive PCa cells. Overexpression of ERG promotes the development of widespread changes in chromatin landscapes and gene expression, resulting in the redistribution of key transcription factors in TMPRSS2:ERG-positive PCa cells.
Dynamic nature of prostate cancer needs to be clinically assessed repeatedly along with longitudinal evaluation of the genomic characteristics of a patient’s tumor during treatment. Therefore, non-invasive methods (coupled with tissue-based assessments) are being used to capture a broader analysis of a patient’s tumor heterogeneities in response to treatment in real-time by examination of circulating tumor cells, serum and urine biomarkers, cell-free DNA and molecular imaging.
Phenomenal breakthroughs in genome technologies and the ensuing outpouring of genomic information related to carcinogenesis and metastasis have galvanized the convergence of discovery science and translational medicine. Accordingly, successful examples of translating cancer genomics into diagnostics and therapeutics reinforce its potential to enhance the possibility of personalized cancer medicine. Nonetheless, debacles and bottlenecks along the path of transformation of a genome discoveries into tangible clinical endpoints are challenging and high in number.
Large-scale integrative genomics have revolutionized in-depth characterization of the genetic and epigenetic landscapes of prostate cancer. Still, the extent to which genomics can elucidate racial disparities in clinical outcome of prostate cancer is vague. Therefore, multi-omics data should be cautiously interpreted, owing to variability in specimen types and assay platforms, as well as geographical and ethnic classifications.
AAF conceptualized the study and selected high-quality research articles for preparation of review article. AT, AA, LO and MZ searched the literature, prepared the initial draft and made the revisions. ZS, ON and MKS analyzed and double-checked the data, prepared the diagrams and proofread the article. AAF supervised the revisions of the draft, technically edited the manuscript. All authors have participated sufficiently in the work to take public responsibility for appropriate portions of the content and agreed to be accountable for all aspects of the work in ensuring that questions related to its accuracy or integrity. All authors read and approved the final manuscript.
Not applicable.
Not applicable.
This research received no external funding.
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.