








 , Ahmed M. El-Khayatt 1,2, Omar K. Al Duaij 3, Ahmed M. Alkaoud 1
, Ahmed M. El-Khayatt 1,2, Omar K. Al Duaij 3, Ahmed M. Alkaoud 11 Department of Physics, College of Science, Imam Mohammad Ibn Saud Islamic University (IMSIU), 11623 Riyadh, Saudi Arabia
2 Reactor Physics Department, Nuclear Research Centre, Atomic Energy Authority, 13759 Cairo, Egypt
3 Department of Chemistry, College of Science, Imam Mohammad Ibn Saud Islamic University (IMSIU), 11623 Riyadh, Saudi Arabia
Abstract
Background: Trans-[Cu (quin)2(EtOH)2], a new copper (II) complex, was characterized using a variety of computational techniques to explore its biological role in pharmacological applications. Methods: The computational methods included density functional theory (DFT), ADMET and molecular docking. Results: The optimized geometrical parameters revealed that the plane containing the Cu ion and the Quinaldinate ligands was confirmed to be nearly planar. DFT findings suggest that the complex has a stable structure with a moderate band gap of 3.88 eV. Highest Occupied Molecular Orbital (HOMO) and the Lowest Unoccupied Molecular Orbital (LUMO) analysis revealed a planar surface intramolecular charge transfer from its donor sites, in the center, to its ends instead of the vertical plane. Two electron-rich regions were observed around the oxygen ions in the molecular electrostatic potential (MEP) map, which were expected to be the sites of molecular bonding and interactions with target proteins. Drug-likeness and pharmacokinetics parameters were determined to provide insight into the safety level of the studied compound. The ADMET (absorption, distribution, metabolism, excretion, and toxicity) results showed favorable pharmacological features, as evidenced by a high oral bioavailability and a low risk of toxicity. A molecular docking study was performed by fitting the copper complex into the active sites of target proteins for Bacillus cereus, Staphylococcus aureus, and Escherichia coli bacteria. The title complex had the strongest antifungal effect within the inhibitory zone of B. cereus with a strong binding affinity of –9.83 kcal/mol. Also, maximum activity was exhibited against S.aureus (–6.65 kcal/mol) compared to the other recently reported Cu complexes within the limits of the screened references. Docking studies implicated modest inhibitory activity against E. coli bacteria. Conclusions: The findings highlighted the compound’s biological activities and identified it as a possible treatment drug for the bacteria B. cereus and S. aureus.
Keywords
- copper complexes
- DFT
- geometrical structure
- HOMO and LUMO
- MEP
- ADMET
- molecular docking
The discovery of novel and innovative drugs is one of the several major issues that the pharmaceutical industry faces today. The inability to successfully develop a large number of drugs is due to the poor pharmacokinetic properties of drugs, which, as a result of absorption and toxicity levels, has negative consequences [1, 2]. Over the years, copper complexes have demonstrated their ability to support biological activity. This is because copper compounds exhibit a broad range of pharmacological effects, such as anti-inflammatory, anti-cancer, and antimicrobial effects. They have vital applications in radiotherapy and nuclear imaging [3, 4, 5, 6]. The ability of copper to coordinate with organic or inorganic biomolecules, to form novel complexes having better oral bioavailability and pharmacological profiles, has recently received a lot of attention [7, 8, 9]. When copper is coordinated with 4, 5, or 6 donors, it forms geometric configurations such as square-planar, distorted square-pyramidal, or distorted octahedral structures due to the Jahn-Teller effect. This feature is just one of the many features that provide copper the benefit of being less toxic than the majority of the 4d and 5d transition metals [10, 11]. Most of the research investigations are currently focusing on the various biochemical functions that could arise from the use of copper complexes in medicine. Copper and its complexes play important roles in in vitro experiments, in vivo research, and clinical studies, and the interest in these metal-based compounds stems from their potential therapeutic applications in a variety of diseases [12]. One of the key issues in this domain is identifying new compounds that are effective against particular cancer cells while simultaneously limiting the side effects [13]. The variety of coordination numbers and different oxidation states of copper, to interact with a wide range of ligand bases, constitute a broad base for the development of anti-cancer drugs [14].
Nowadays, molecular modeling of more complex biomolecular systems has been made easier by improvements in computational capabilities. The design of drugs is a field where molecular modeling is extremely useful [15]. Algorithms and computational techniques are utilized, in modern drug research, to design novel therapeutic drugs or transform drug treatments into effective pharmaceuticals. For novel drug discovery, realistic chemical and biological constraints must be exceeded before further refinement and clinical evaluation [16]. In this regard, computational approaches have been utilized to evaluate the pharmacokinetic characteristics of the new drugs to save the effort and cost of identifying their promising therapeutic properties [17]. The evaluation of new compounds, in computer-aided drug design and development, requires essential criteria like ADMET (absorption, distribution, metabolism, excretion, and toxicity) and molecular docking predictions. ADMET parameters are computed to explore the physicochemical properties of new drugs and determine whether they are suitable for administration in humans [18]. Determining the proper pharmacokinetic behavior for the ADMET profile is essential for increasing the success of drug candidates. Based on the databases and software frequently used to evaluate the relevant properties, molecular modeling provides a systematic classification and definition of biochemical molecules in ADMET prediction categories [19]. Many ADMET software and online web servers could derive the pharmacokinetic and related physicochemical properties based on different molecular descriptors [20, 21, 22]. Over the past ten years, molecular docking methodologies have been more important in the search for new drugs and their development for the treatment of various diseases. This computational technique could support the experimental results by predicting the potential binding interactions between ligand-receptor complexes or protein-protein interactions [23]. Precisely, molecular docking elucidates how the medicine functions within the cell by defining binding sites and determining the binding energies between the drug and protein. There is a significant history of scientific research into molecular docking algorithms and their associated software to identify specific biological activities for a large library of drug candidates [24, 25, 26].
In the current study, the Trans-[Cu (quin)2(EtOH)2] molecule (shown in Fig. 1) has been screened as a new copper complex in an attempt to identify a potential drug that could be effective in treating microbial infections. density functional theory (DFT) analysis has been reported to describe molecular structure and chemical characterization. The ADMET properties have been evaluated to determine whether the target compound has a safe and optimal pharmacokinetic profile with regard to human bioavailability. The molecular docking technique has been used to clarify the mode of molecular interaction with related proteins from the B.cereus, S. aureus, and E.coli bacteria. The antibacterial activities of the compound were revealed by identifying its inhibition capabilities in light of the calculated binding affinities.
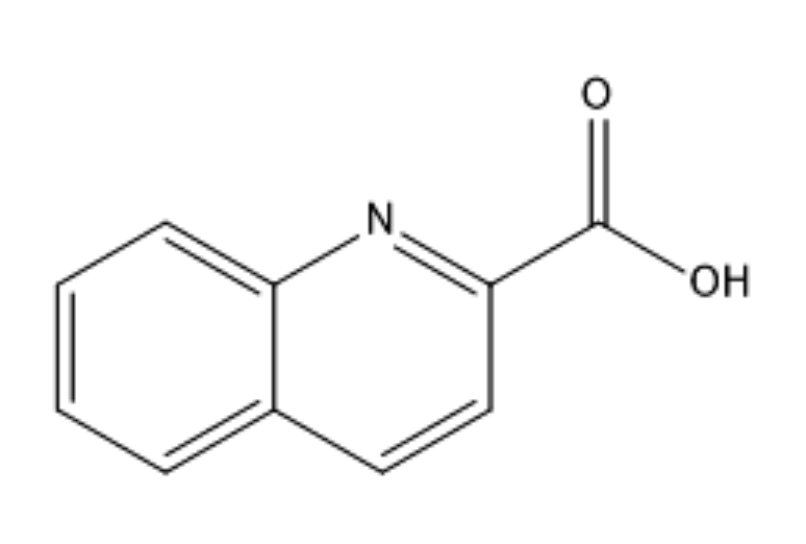 Fig. 1.
Fig. 1.Molecular structure of the quinaldinic acid.
Trans-[Cu (quin)2(EtOH)2] compound has been synthesized by Nina Podjed et al. [27] during a study on copper(II) Quinaldinate complexes with several O- and N-donor ligands. Using stirring and a hot water bath, to maintain the reaction mixture in hot water, copper (II) acetate hydrate (100 mg, 0.50 mmol) was dissolved in ethanol (15 mL). Then, 86 mg (0.50 mmol) of quinaldinic acid was added to the mixture. A light turquoise solid formed was filtered out and dried. The turquoise filtrate was kept at ambient conditions in a closed container. Trans-[Cu (quin)2(EtOH)2] was identified in a few needle-shaped crystals that formed within two days.
At the DFT level of theory, the optimal molecular structure of the studied Cu-complex was computed with a mix basis set of 6-311G (d, p) and LANL2DZ. The well-known polarized basis set 6-311G (d, p) has a triply divided valence basis set and a core region of six contracted Gaussian primitives. For the first-row elements, the polarization functions are composed of a single set of five pure d-functions, whereas for hydrogen, a set of three p-type orbitals are added, i.e., feature d functions for heavy atoms plus p functions for hydrogen atoms. The LANL2DZ, commonly referred to as effective core potential, has been successfully used in calculations on transition metal complexes [28, 29]. The Gaussian 09 program was used to accomplish all the calculations [30]. GEN keyword was used to specify the basis set 6-311G (d, p) for C, H, O, N atoms and LANL2DZ effective core potential for Cu atom [31]. The geometrical structural parameters, as well as HOMO& LUMO and MESP graphs, were visualized using the Gauss View 6.0.16 graphical interface (Semichem Inc., Shawnee Mission, KS, 2016) [32].
The ADMET properties, associated with the bioavailability, pharmacokinetics, and physicochemical properties of the compound under study, were predicted and analyzed using the OSIRIS property explorer code (https://www.organic-chemistry.org/prog/peo/) and the online web server Molinspiration (https://www.molinspiration.com/). A set of pharmacokinetic properties were also evaluated using the integrated online service for ADMET properties prediction (ADMET Prediction Service). In this web-based prediction system, artificial neural networks and fragmental descriptions were used to construct a predictive model of the relationship between the structure of organic molecules and their related pharmacokinetic parameters [33, 34].
The molecular docking study was carried out using AutoDock 4.2.6 Software (version 4.2.6, https://autodock.scripps.edu/). [35]. A Protein Data Bank webserver (https://www.rcsb.org/) was used to download the 3D structures of the target proteins Bacillus cereus, Staphylococcus aureus, and Escherichia coli bacteria, with protein data bank (PDB) identifiers 5NCD, 2DHN, and 1WXH, respectively. The DFT-optimized structure was provided as the ligand molecule in the docking simulation. Using the Autodock, the proteins were first prepared by removing water ions, and, then, polar hydrogens and Gasteiger partial charges were added to it. The dimension and x, y, and z coordinates of the grid box were determined from the PDB site records using the Discovery Studio software [36]. The Lamarckian genetic algorithm was used with Genetic Algorithm (GA) runs that were set to 100 to find the ligand’s optimum binding pose. The protein-ligand complex and the hydrogen bond interactions, between the protein and ligand molecule, were visualized using the Molegro Molecular Viewer software [37].
Copper (II) can be coordinated in complexes, by four, five, or six donors, due
to its d9 electronic configuration. Quinaldinic acid (known as
quinoline-2-carboxylic acid and referred to as Hquin), on the other hand, can
coordinate the transition of metal ions because it contains N and O-donor ligands
with their functional groups OH and NH
Fig. 2 shows how Quinaldinate and ethanol ligands are arranged in the [Cu
(quin)2(EtOH)2] composition. The ligands are arranged in a trans geometry within
the coordination environment of the complex. The Jahn-Teller effect induced the
deformation of an octahedron defined by the arrangement of the N
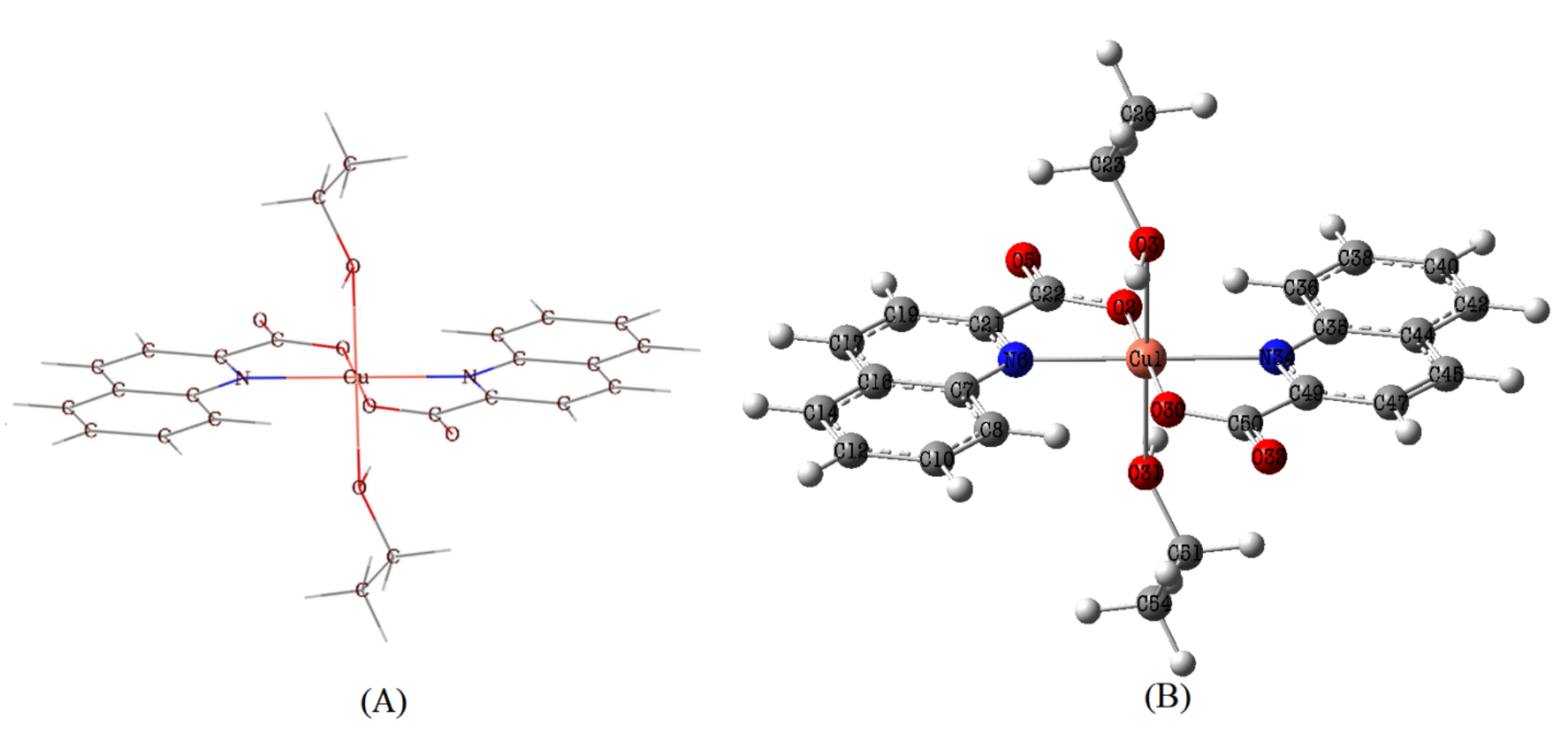 Fig. 2.
Fig. 2.Molecular structure (A) and optimized structure (B) of Trans-[Cu (quin)2(EtOH)2] molecule.
A selection of some basic geometrical parameters is shown in Table 1. The
optimized molecular parameters and those included in the reported experimental
work have been compared (Supplementary Table 1).
Long bond lengths of Cu
| Geometrical Parameters | DFT | |
|---|---|---|
| Bond length (Å) | Cu |
2.395 |
| Cu |
2.395 | |
| Cu |
2.149 | |
| Cu |
2.149 | |
| Cu |
1.958 | |
| Cu |
1.959 | |
| Bond Angle (°) | N |
179.9860 |
| O |
179.99 | |
| Dihedral angle (°) | C |
179.346 |
| C |
–178.727 | |
| C |
178.833 | |
| C |
–179.601 | |
| C |
–178.034 | |
| C |
178.727 | |
| C |
–178.836 | |
| C |
179.620 | |
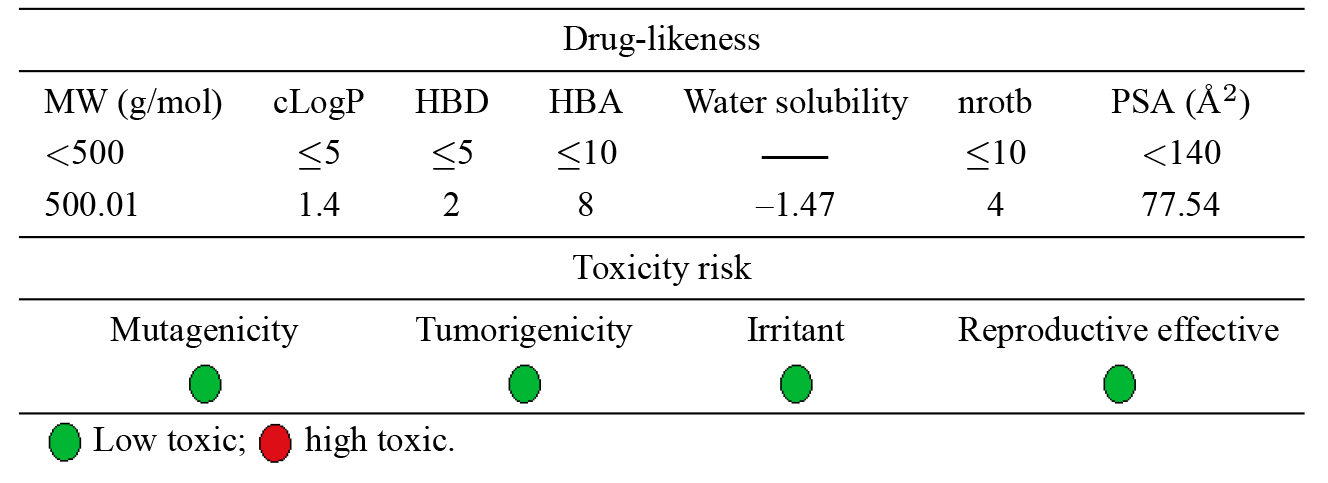
The oral bioavailability of a drug is often predicted using drug-likeness assessments. Lipinski’s rule of five was established as a straightforward criterion to exclude molecules that lack the optimum physicochemical characteristics. The parameters addressed by this rule are molecular weight, octanol/water coefficient (logP), hydrogen bond donors (HBD), and hydrogen bond acceptors (HBA) [40]. Later, additional relevant parameters such as polar surface area (PSA) and the number of rotatable bonds (nRotb) were incorporated [41]. The computed molecular properties of the title complex are shown in Table 2 along with the minimum constraints of the rule of five.
|
The results revealed that Lipinski’s rule of five has been violated except for
the molecular weight, which barely exceeded the maximum range (by 0.01). The
lipophilic nature of a molecule is directly correlated with the c Log p value,
and good lipophilicity is indicated as the c Log P number that further decreases
below 5. The calculated c Log p value (1.42), is in the range of values
1 to 4, where oral drugs are expected to have the optimum physicochemical
features [42]. The reduction in polar surface area is a sign of the high oral
bioavailability of drugs. In Table 1, the small PSA (77.54 Å
It is necessary to estimate the potential toxicity risk for each fragment in the drug candidate molecules. This toxicity prediction determines if the suggested structure will be toxic with regard to the identified risk levels. The estimation of mutagenicity, tumorigenicity, irritant, and reproductive effects was indicated by color coding. Each category is distinguished by a red or green color, with green denoting a low potential for toxicity and red denoting a high toxic risk. The results in Table 1 reveal that the compound under test would be safe and is predicted to exhibit minimal to no toxicity.
Further, pharmacokinetic processes that affect the corresponding bioavailability
of the drug were predicted by ADMET Prediction Service [43]. The relation between
the structure of organic compounds and their pharmacokinetic properties is
established by a color-coded distribution graph that displays the predicted value
and its position within a known, tested library of compounds. The optimal
pharmacological profile, determined in this regard, is depicted in Fig. 3. The
calculated permeability through the blood-brain barrier (BBB) was –0.51,
surpassing 79% of the database compounds built for the predictive model, this
value is in the range of 0.3 to –1, and it indicates a moderate level of access
to the central nervous system [44]. A high oral bioavailability was anticipated,
based on the high human intestinal absorbance (HIA) value (
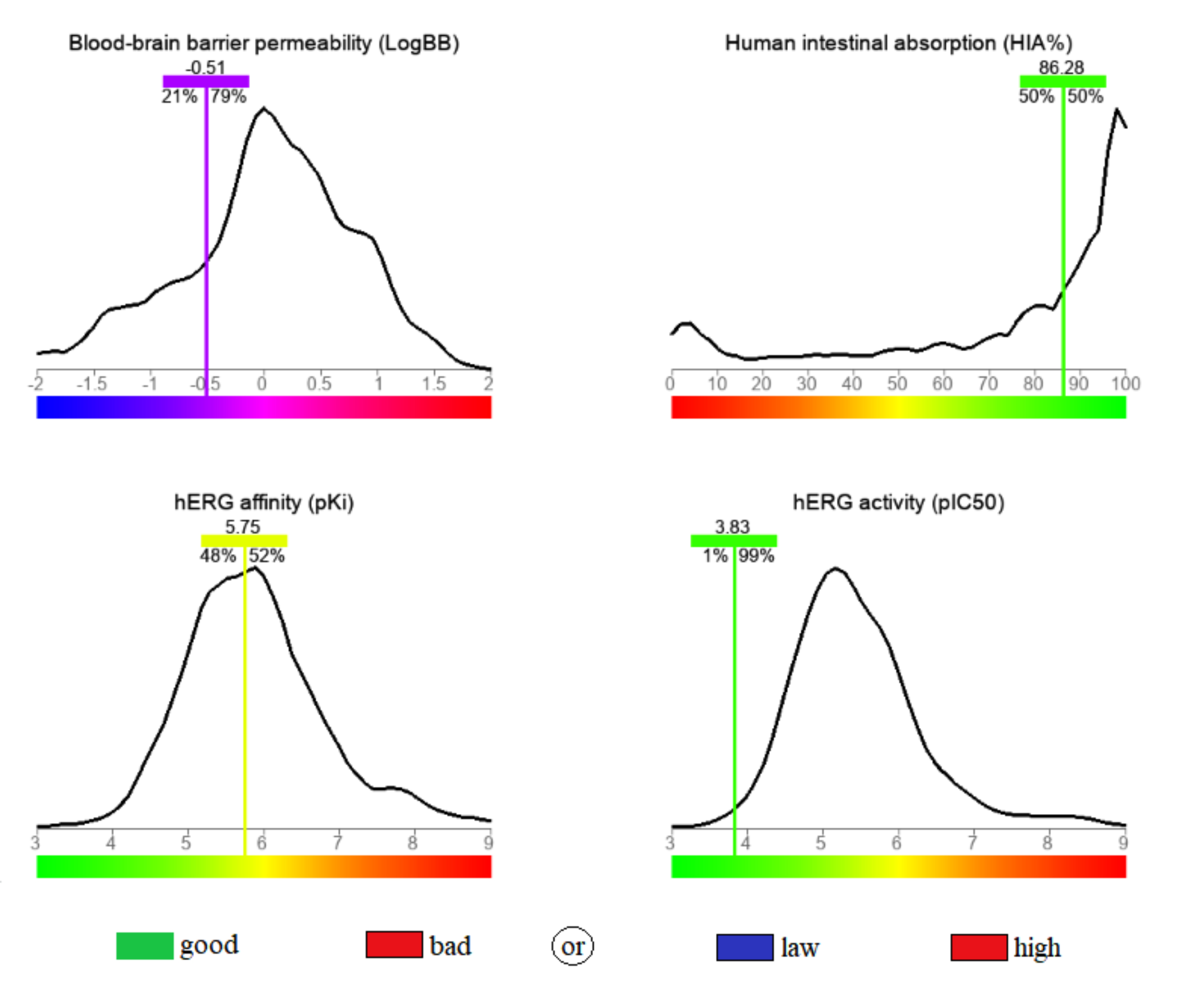 Fig. 3.
Fig. 3.The calculated blood-brain barrier permeability (BBB), human intestinal absorbance (HIA), human Ether-à-go-go-Related Gene (hERG) activity and hERG affinity for the stated compound.
Frontier orbitals, which are defined by the Highest Occupied Molecular Orbital
(HOMO) and Lowest Unoccupied Molecular Orbital (LUMO), are the major elements in
quantum chemistry. HOMO designates the molecular locations that have the highest
propensity for electron donation, while the molecular locations with the highest
propensity for electron acceptance are indicated as LUMO. The HOMO and LUMO
frontier orbitals are important to understand the molecular properties of a
molecule, which include its chemical stability and reactivity. Fig. 4 illustrates
the graphical representation of HOMO and LUMO, their associated energy values,
and the energy gap (Eg), which describes the total reactivity of the molecule.
The calculated energy gap of the studied ligand is 3.88 eV, which makes it more
stable than the previously reported metal complexes [48, 49, 50]. The HOMO electron
density of the stable ligand is mainly observed as anti-bonding orbitals on the
Cu ion and the nitrogen and oxygen atoms of the Quinaldinate fragment; whereas
the LUMO electron density has a bonding nature that is localized on the
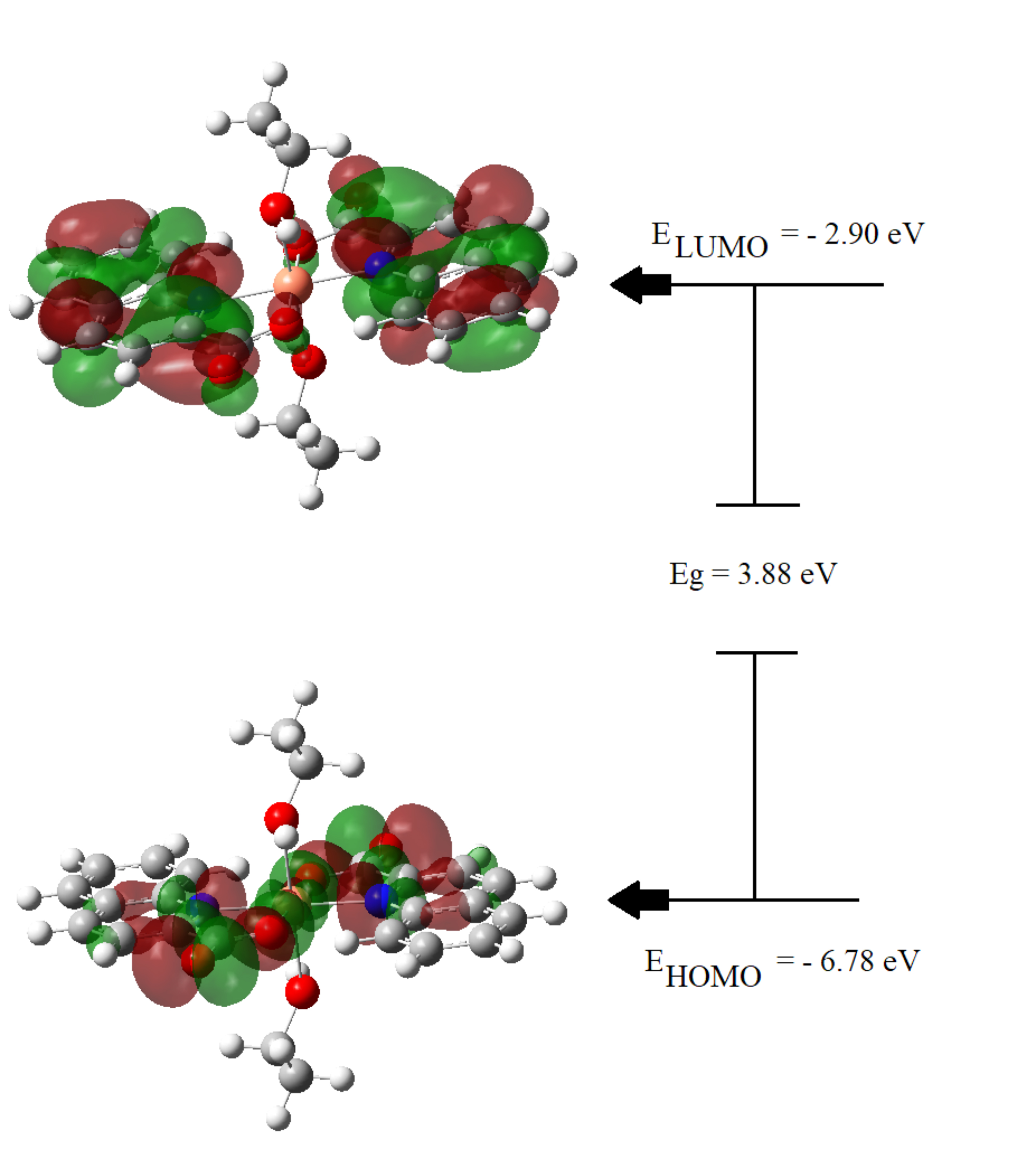 Fig. 4.
Fig. 4.Electron density distributions in HOMO and LUMO frontier orbitals diagram.
The term “molecular electrostatic potential” (or “MEP”) is a crucial reactivity descriptor that is frequently used to characterize the overall electron density surface of a molecule that displays its positive, negative, and neutral electrostatic potential regions. On the MEP surface, negative, neutral, and positive locations are indicated by red, green, and blue colors, respectively. The blue region contains electron-poor sites that are subject to nucleophilic attack, whereas the green areas correspond to neutral sites or zero potential, and the red spot represents electron-rich sites that are subject to electrophilic attack. Electronic charge density, for a drug surface, is used in computer-aided drug design to examine the relationship between that drug’s structure and the associated biological activity. Therefore, by using this information on charge distribution in the compounds, MEP could be used to interpret the hydrogen bonding interactions and biological properties.
Fig. 5 displays the contour map for the electrostatic potential of the title compound. There are only two symmetrically negative regions (red), which basically cover C=O groups and extend slightly to the two oxygen atoms of the ethanol fragment. These groups exhibit high electronegativity, which represents the most reactive component or the optimum sites for an electrophilic attack on the ligand. Therefore, it is expected that the oxygen atoms will be the key sites of interaction between the molecule and the target protein which could lead to biological activity.
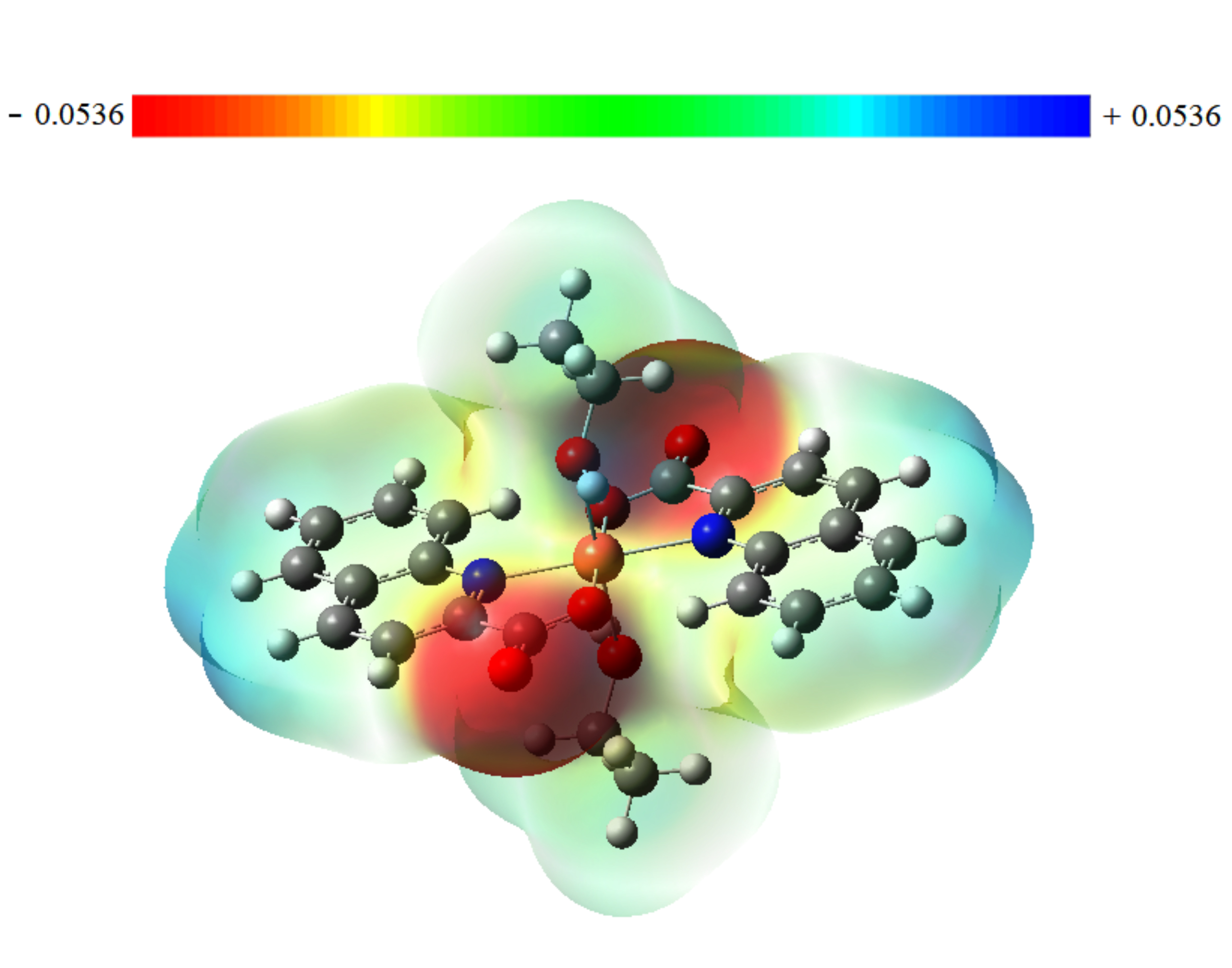 Fig. 5.
Fig. 5.Molecular electrostatic potential map for Trans-[Cu (quin)2(EtOH)2] compound.
To validate the docking protocol, the ligands Zinc Metalloenzyme, 6-hydroxymethyl-7, 8-dihydropterin, and natural substrate were taken from the X-ray structures of B. cereus protein (5NCD with 2.45 Å resolution), S. aureus protein (2DHN with 2.2 Å resolution), and E. coli protein (1WXH with 1.90 Å resolution) respectively, and they have been re-docked into its protein co-crystallized structure. The calculated root-mean-square deviation (RMSD) between docked and experimental poses for the same order of the three proteins was 1.472, 1.759, and 2.04 Å. These reasonably low values verified that the docking protocol was adequate.
The docked binding energy, inhibition constant, and hydrogen bond for the
molecular poses (lowest energy), associated with the interaction of the proteins
5NCD, 2DHN, and 1WXH with the stated copper complex, are shown in Table 3. Also,
the binding interactions within the active domain of the enzymes, along with the
labeled adjacent amino acid residues, are shown in Figs. 6,7,8. Out of the
obtained results, B. cereus (5NCD) bacterium had the best docking pose,
with the lowest binding energy and lowest inhibition constant of –9.83 kcal/mol
and 0.06261
| Antimicrobial activity | Target protein | Binding energy (kcal/mol) | Inhibition constant (µM) | Hydrogen bonds | Interacting residues | Bond distance (Å) |
|---|---|---|---|---|---|---|
| B. cereus | 5NCD | −9.83 | 0.06261 | 2 | MET170 | 2.75 |
| TRP191 | 3.22 | |||||
| S. aureus | 2DHN | −6.65 | 13.38 | 1 | HIS75 | 3.75 |
| E. coli | 1WXH | −6.62 | 13.96 | 4 | LYS261 | 2.60 |
| LYS261 | 2.81 | |||||
| ASN136 | 2.95 | |||||
| ASN136 | 3.08 |
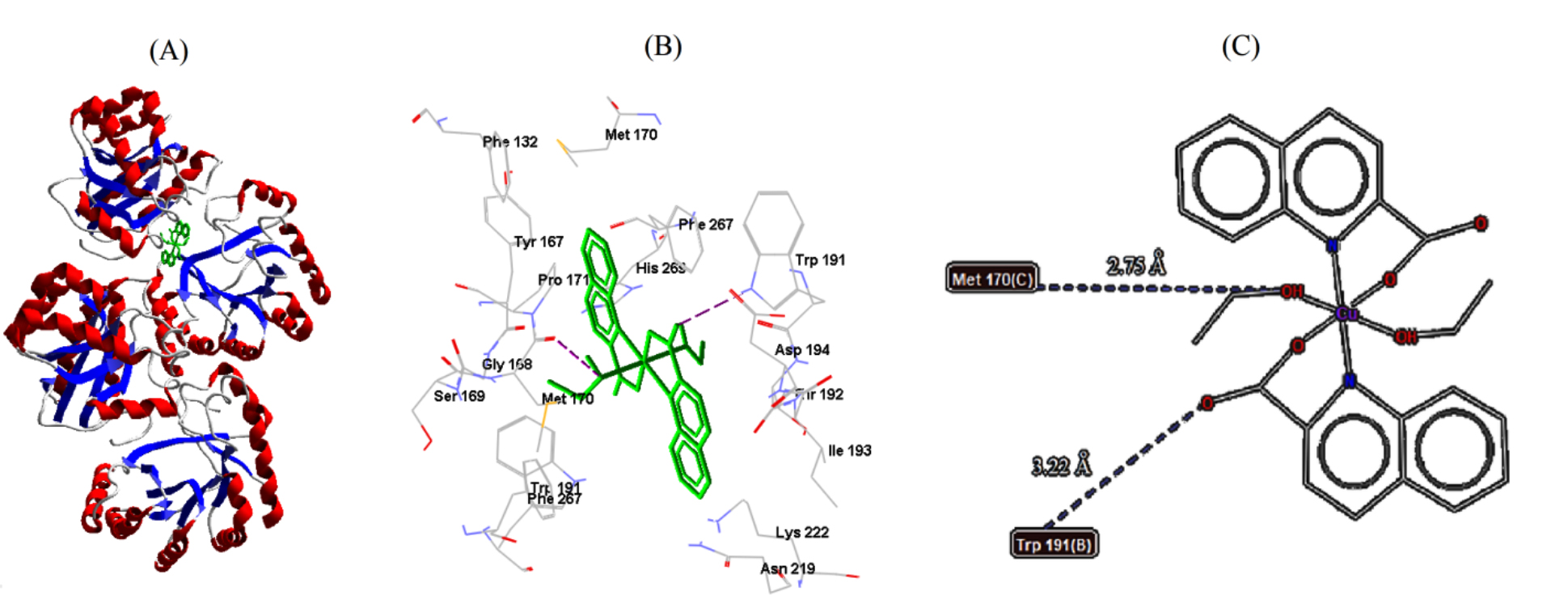 Fig. 6.
Fig. 6.Cu-complex with B. cereus. (A) 3D representation of Cu-complex in the protein cavity. (B) 3D representation of the interactions within the binding pocket. (C) 2D representation of the interactions with amino acid residues.
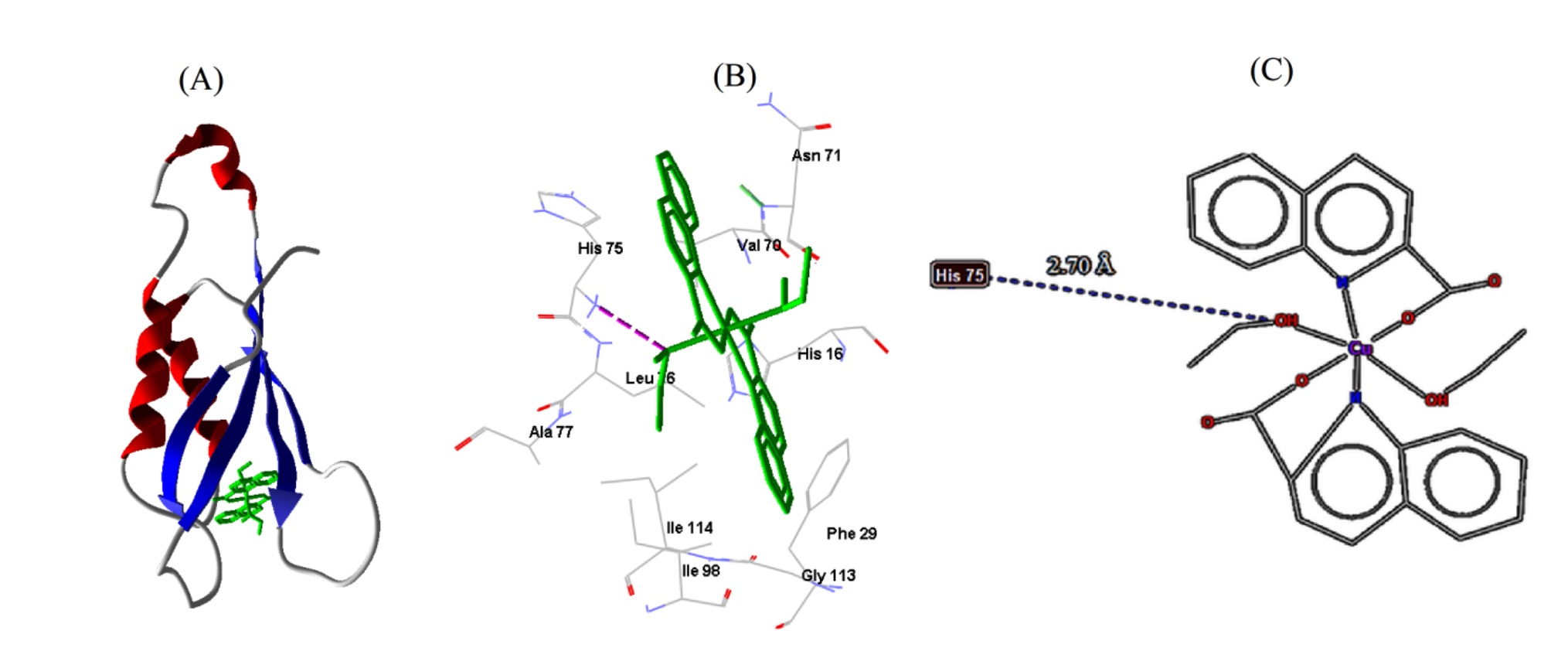 Fig. 7.
Fig. 7.Cu-complex with S. aureus. (A) 3D representation of Cu-complex in the protein cavity. (B) 3D representation of the interactions within the binding pocket. (C) 2D representation of the interactions with amino acid residues.
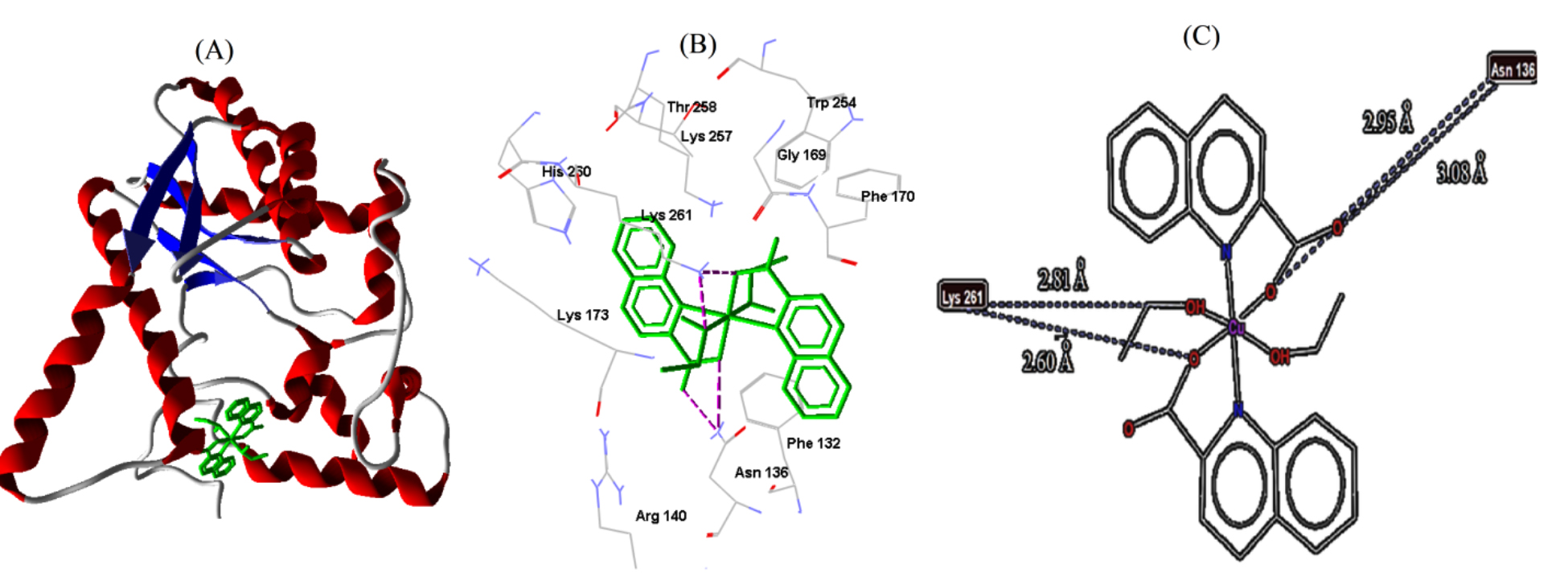 Fig. 8.
Fig. 8.Cu-complex with E. coli. (A) 3D representation of Cu-complex in the protein cavity. (B) 3D representation of the interactions within the binding pocket. (C) 2D representation of the interactions with amino acid residues.
Throughout in silico investigation, the role of a selected copper complex was examined in relation to biological activities. DFT calculations at B3LYP, with mixed basis sets of 6-311G (d, p) and LANL2DZ, were utilized to provide the structural and chemical properties. The octahedral configuration of the compound was analyzed using the calculated geometrical parameters. Frontier molecular orbital energies (HOMO-LUMO) provided information on the path of intramolecular charge transfer as well as an assessment of the energy gap. The MEP map revealed the electronegative potential regions located around C=O groups and the two other oxygen atoms, which are expected to be the binding sites of the title molecule to the target protein. The ADMET prediction satisfied the requirements for a successful drug and demonstrated a high level of oral bioavailability. To evaluate the antimicrobial activities, three different biological targets were selected for molecular docking studies. The Cu-complex formed the most stable complexes with significant binding affinities in the cavity sites of B. cereus and S. aureus protein. The stated compound was able to potentially inhibit the two bacterial strains in comparison to other recently reported studies. This study is considered a positive step for further experimental investigations on the development of biologically active copper complex drugs.
The data used to support the results of this work are available upon request.
RKH—methodology; RKH and AME—software; RKH—validation; RKH, AME and OKAD—formal analysis; RKH and OKAD—investigation; RKH—resources; RKH, AME and OKAD—data curation; AME, AMA and OKAD—writing - original draft preparation; RKH, AME and OKAD—writing - review and editing; AME, AMA and OKAD—visualization; RKH, AME and OKAD—supervision; RKH, AME and OKAD—project administration. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript.
Not applicable.
The authors extend their appreciation to the Deanship of Scientific Research, Imam Mohammad Ibn Saud Islamic University (IMSIU), Saudi Arabia, for funding this research work through Grant No. (221412025).
This research was funded by Deanship of Scientific Research, Imam Mohammad Ibn Saud Islamic University (IMSIU), Saudi Arabia, for funding this research work through Grant No. (221412025).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.