

1 Department of Dermatology, Southwest Hospital, Third Military Medical University (Army Medical University), 400038 Chongqing, China
2 Department of Endocrinology, Southwest Hospital, Third Military Medical University (Army Medical University), 400038 Chongqing, China
3 Institute of Wound Repair and Regeneration Medicine, Southern University of Science and Technology School of Medicine, 518071 Shenzhen, Guangdong, China
4 Department of Wound Repair, Southern University of Science and Technology Hospital, 518055 Shenzhen, Guangdong, China
Abstract
Microtubules (MTs) are essential structural elements of cells. MT stability and dynamics play key roles in integrity of cell morphology and various cellular activities. The MT-associated proteins (MAPs) are specialized proteins that interact with MT and induce MT assemble into distinct arrays. Microtubule-associated protein 4 (MAP4), a member of MAPs family, ubiquitously expressed in both neuronal and non-neuronal cells and tissues, plays a key role in regulating MT stability. Over the past 40 years or so, the mechanism of MAP4 regulating MT stability has been well studied. In recent years, more and more studies have found that MAP4 affects the activities of sundry human cells through regulating MT stability with different signaling pathways, plays important roles in the pathogenesis of a number of disorders. The aim of this review is to outline the detailed regulatory mechanisms of MAP4 in MT stability, and to focus on its specific mechanisms in wound healing and various human diseases, thus to highlight the possibility of MAP4 as a future therapeutic target for accelerating wound healing and treating other disorders.
Keywords
- microtubule
- microtubule-associated protein 4
- microtubule stability
- microtubule dynamics
- wound healing
- human diseases
Microtubules (MTs), being found in all characterized
eukaryotic organisms, are hollow tubes composed of
The MAPs are specialized proteins that interact with MTs and induce MTs assemble into distinct arrays [16]. Specifically, MAPs can decorate the MTs lattice and stabilize them by increasing the rate of polymerization and inhibiting the rate of depolymerization, and promote growth and reduce shrinkage speeds, enhancing the stability of MTs [17, 18, 19, 20]. In mammals, MAPs family mainly includes MAP1 (also named MAP1A), MAP2, microtubule-associated protein 4 (MAP4), MAP5 (also named MAP1B), MAP7, MAP8 (also referred to as MAP1S), MAP9 (also known as apoptosis and splicing-associated protein, ASAP), tau protein (also known as MAPT), and so on. Basing on their cellular expression patterns, MAP1, MAP2, MAP5, MAP7, MAP8 and tau are expressed predominantly in neuronal cells, while MAP4, MAP7, MAP9 are mainly in non-neuronal cells [15, 16, 21, 22]. However, it was later found that MAP4 was also expressed in neuronal cells of mammalian brain [23]. All MAPs overexpressed in mammalian cells decorated the MT cytoskeleton, while tau induced microtubule bundling [16]. Some MAPs have a similar structure, such as MAP4, MAP2 and tau, which all have an N-terminal projection domain and a C-terminal MT-binding domain [24].
Since its ubiquitously expression, MAP4 has a strong potential to be implicated in a number of disorders. In the current review, we will comprehensively go through MAP4 and analyze its roles in wound healing and human diseases that have been studied, and discuss the possibility of MAP4 as a precise treatment target for these conditions.
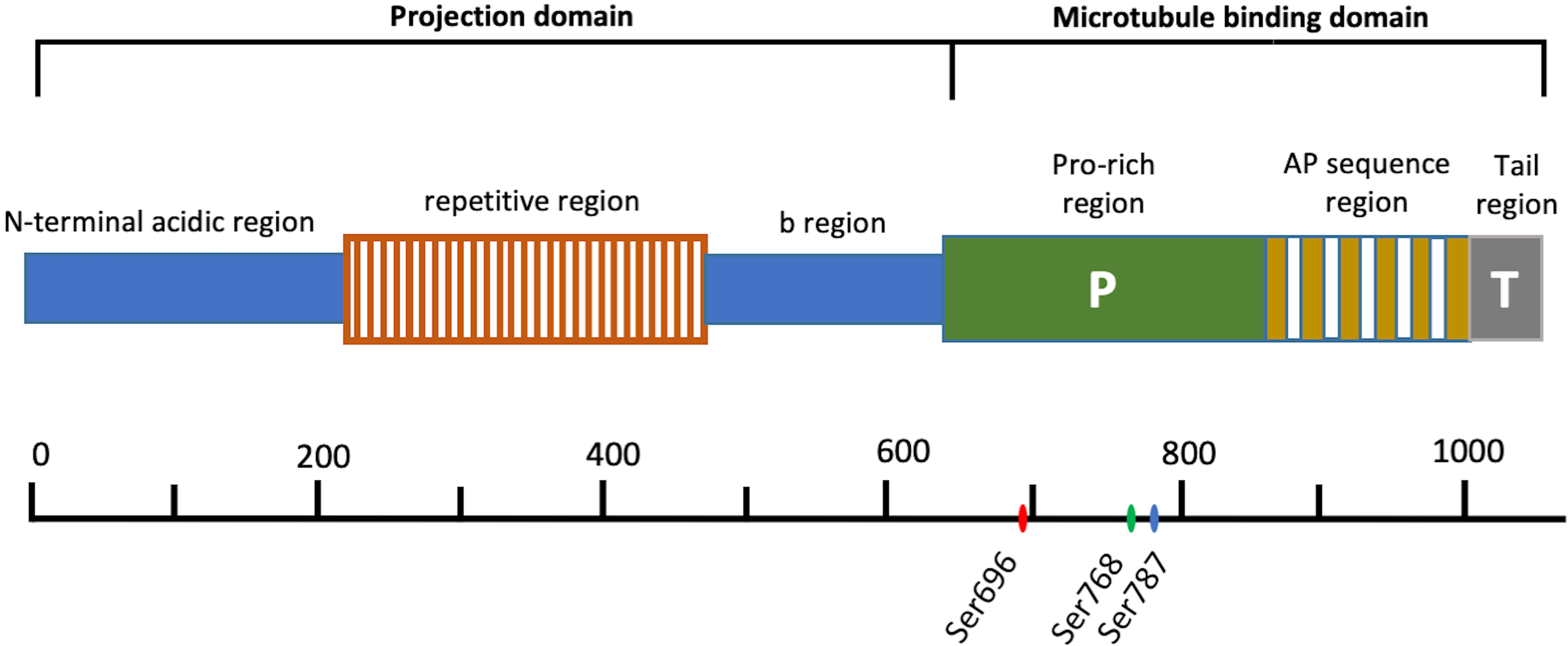
In humans, MAP4 is encoded by a single-copy gene which located on chromosome 3p21. Its open reading frames have three distinct regions consisting of related sequences with different motifs, and alternative RNA splicing leads to expressions of multiple MAP4 isoforms from this gene [25, 26]. MAP4 is a bipolar protein which consists of a carboxyl-terminal MT-binding domain and an amino-terminal projection domain (PJ-domain) (Fig. 1). The MT-binding domain of MAP4 can be divided into three subdomains: a region consisting of 18 amino acid residues, which contans repeats of an assembly-promoting sequences (AP sequence region), a region with rich proline (Pro-rich region) and a hydrophobic tail region (Tail region). Studies have shown that both of the fragments from C domain corresponding to the AP sequence region and the Pro-rich region can promote tubulin polymerization, and the former mainly effects the longitudinal affinity of tubulin dimer in a protofilament, the later mainly effects the lateral protofilament-protofilament interaction, indicating that both of AP sequence region and Pro-rich region are essential for MAP4 to promote MT assembly [27, 28]. Tail region cannot directly interact with MT [28]. MAP4 exists several alternatively spliced variants, with 3–5 different AP sequence repeats [26]. There are four MAP4 cDNA clones with different repeat region organizations have been identified from human, bovine, mouse and rat sources [24]. In rat, different MAP4 isoforms were expressed in different tissues, for example, the 5-repeat isoform was the most abundant isoform expressed in rat lung, liver, kidney, spleen, and testis, the 4-repeat isoform was expressed in rat brain, heart, and skeletal muscle, and the 3-repeat isoform was expressed in heart and, to a lesser extent, in brain, skeletal muscle, and lung [26]. Varied MAP4 isoforms have similar degrees of MT assembly promoting activity and MT binding affinity, but with the number of repeat sequences in the fragments increasing, the MT bundle-forming activity is augmented [29]. On the other side, the PJ-domain of MAP4 also consists of three regions: N-terminal acidic region, a repetitive region, and b-region followed by the MTB domain. It had been found that fragments from N-domain did not bind to MTs, but the PJ-domain could suppress the bundle-forming ability of the MT-binding domain to keep individual MTs separated, and its suppressive activity was correlated with the length of PJ-domain [27, 30].
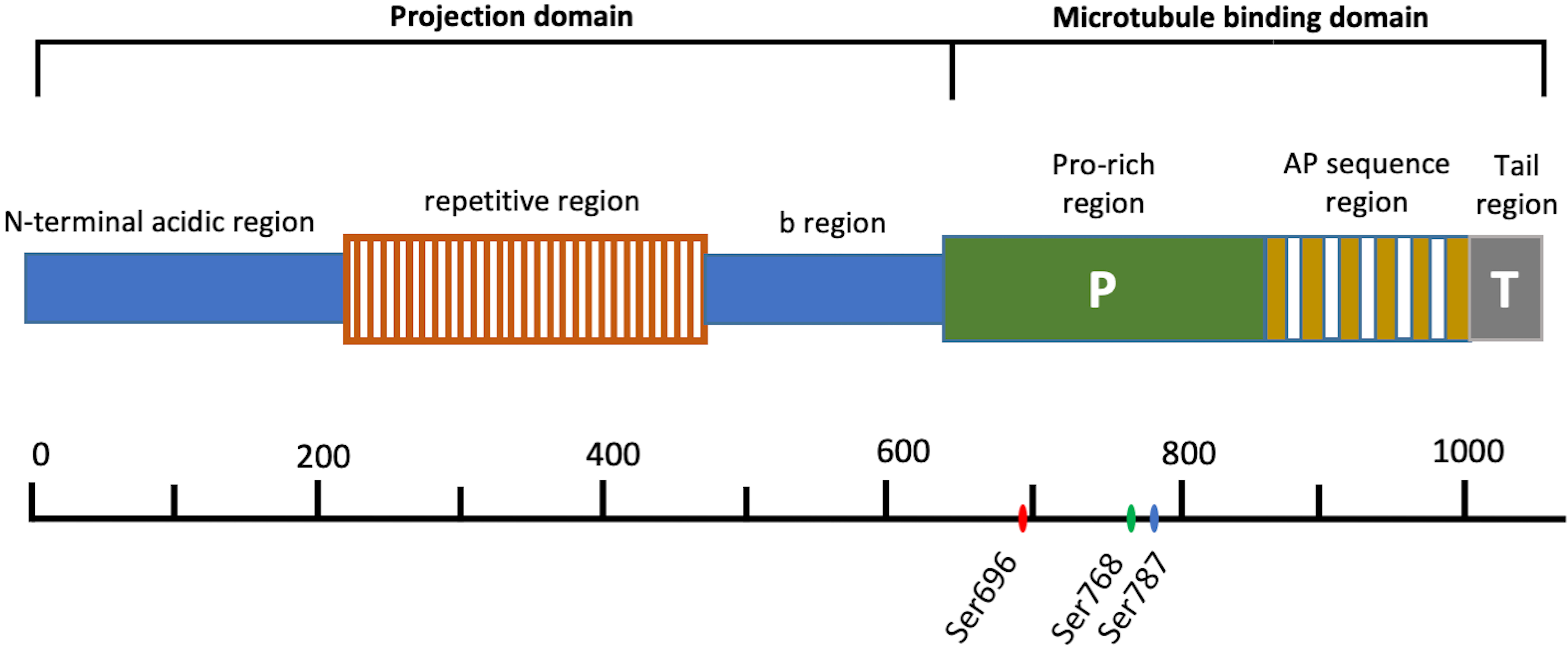 Fig. 1.
Fig. 1.Schematic structures of MAP4. The amino acid residue numbers are presented on the bottom. Ser696, Ser768 and Ser787 are phosphorylation sites of MAP4 in human.
MAP4 has been implicated as a MT stabilizer, which stabilizes MTs to an extent similar to very low concentrations of MTs stabilizing agents (like Taxol) [9]. It has been confirmed that both of AP sequence region and Pro-rich region in the MT binding domain of MAP4 take part in the promotion of tubulin polymerization [28]. What’s more, a study of Xiao et al. [31] strongly suggested that similar to the Taxol, MAP4 induces MT stability via transforming the MT conformation from curved to straight, and they speculated the driving force for this straightening could be the direct result of simultaneous binding of MT binding domain of MAP4 to both the outside and the luminal portion of the MT, which needs to be further verified. The effect of MAP4 on MT stability may be influenced by interacting with other proteins. Septins are a class of GTP-binding proteins that can form heterooligomers in mammalian cells. It has been found that suppression of septin expression in HeLa cells caused a pronounced increase in microtubule stability. Further study revealed that MAP4 is a septin binding partner and that the trimer formed by septins can directly bind to the proline-rich region in the C-terminal region of MAP4 which blocked the ability of this MAP4 fragment to bind and bundle MTs [32]. In addition, PTMs of tubulin are crucial regulators of the MT network, and they are emerging mechanisms that can directly and most likely selectively control the interactions of MTs and MAPs [33]. For example, it has been showed that MAP4 interactions occurs preferentially with detyrosinated MTs that may increase MT stability [9]. Since the MT stabilization effect of MAP4, the expression level of MAP4 can directly effect the stability of MTs. The phenomenon that cells with increased MAP4 expression display an increase in polymerized MTs has been well established, and MAP4 in cancer cells being repressed by DNA-damaging agent (like doxorubicin) can decreased MT polymerization [34, 35, 36].
The function of MAP4 in stabilizing of MTs is directly affected by the PTMs
of itself. Previous studies have shown that
the phosphorylation of MAP4 causes a remarkable decrease in
its ability to stimulate MT assembly, induces the detachment
of them from MT, ultimately increase MT instability [37, 38]. The
phosphorylation sites of
MAP4 have been reported at Ser696 [39], Ser768, and Ser787
[40] in the proline-rich region of human (Fig. 1) (corresponding to Ser667,
Ser737, and Ser760 in mouse), and the phosphorylation kinase including protein
kinase C (PKC), cyclin-dependent kinase family (CDKs), microtubule affinity
regulating kinases 4 (MARK4), P38 mitogen-activated protein kinase (MAPK),
protein kinase A (PKA), monopolar spindle 1 (Mps1) kinase, and
Ca
Wound healing is an important physiological process to maintain the integrity of skin after trauma, which is a topic of great concern to burn, dermatology and endocrinology. It is a complex and precisely regulated process which includes hemostasis, inflammation, proliferation, and remodeling. Proliferation stage, including angiogenesis, fibroplasia, and reepithelialization, aims to diminish the lesioned tissue area by establishing a viable epithelial barrier to activate keratinocytes, contraction and fibroplasia [51]. Reepithelialization, including keratinocyte proliferation and migration, is a crucial step of wound healing [52]. An acute hypoxic microenvironment is commonly formed in acute wound as the oxygen supply is reduced and excessive oxygen consumption after acute injury. It has been revealed that hypoxic stress post wounding stimulates gene expression and growth factor synthesis that contribute to wound repair [53]. Such is the case, a large number of studies have reported that hypoxia in the early stage can promote the migration and re-epithelialization of epidermal cells, our study also confirms this phenomenon [54, 55]. But the underlying mechanism is a great mystery.
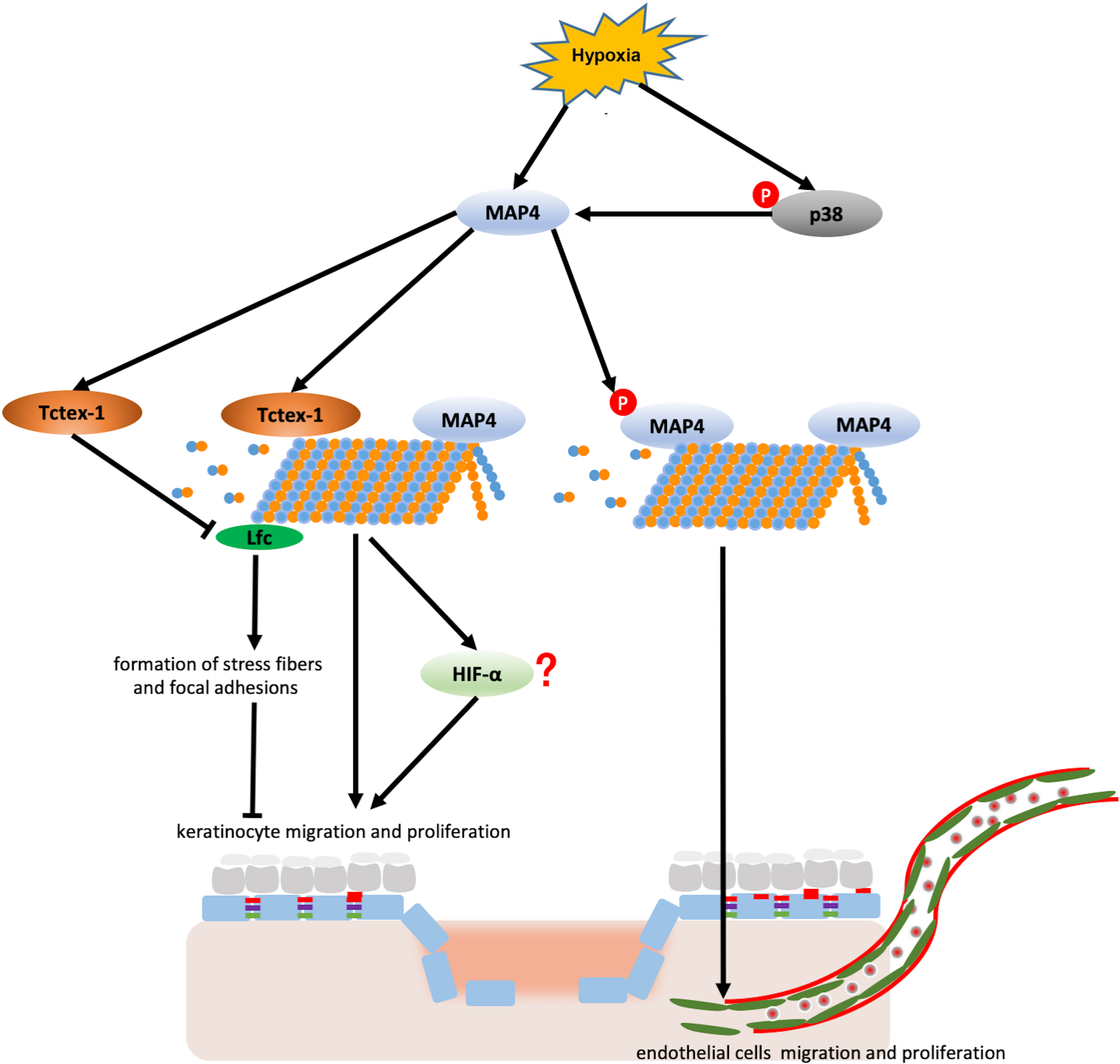
Chen et al. [54] first reported that MAP4 may play an important role in epidermal cell migration in an acute hypoxic microenvironment. They found early hypoxia induces HaCaT cells migration, and MT instability in HaCaT cells increases under hypoxia, hypoxic stimulation increased the expression of MAP4, however, shRNA-mediated knockdown of MAP4 inhibits HaCaT cell migration under hypoxia. Further research underlying regulatory molecular mechanism of this effect, they found keratinocyte migration ability was improved significantly but no any changes in the MT dynamics after the overexpression of MAP4, and keratinocyte migration ability was also significantly enhanced but MT depolymerization showed significant increase after the overexpression of light chain protein DYNLT1 (Tctex-1), further study showed that after interference of MAP4 expression, the expression of Tctex-1 showed a downward trend, but after interference of Tctex-1expression, the expression of MAP4 did not change. Therefore, MAP4 does not directly affect MT dynamics, but promotes the expression of Tctex-1, which induces MT depolymerization and thus promotes the migration of epidermal cells under hypoxia [56]. Tctex-1, a member of the Tctex family, was originally characterized as a dynein motor light chain [57], and was later found to have dynein-independent functions that regulating branched actin polymerization and endocytosis and being involved in neuronal growth [58, 59]. It is well known that actin and MTs are the two major components of the cellular cytoskeleton. MT disassembly promotes actin stress fiber formation and enhances cell contraction [60]. The interplay between the MT and actin cytoskeletons is partly determined by Rho signaling. It has been revealed that MTs repress Rho signaling is attributed to the MT sequestration of Lfc in an inactive state [61]. Meiri et al. [62] have identified that Tctex-1 is the link responsible for anchoring Lfc to polymerized MTs. The the overexpression of Tctex-1 impaires the activity of Lfc by enhancing MT sequestration of it. Overall, Tctex-1 has a pivotal role in negative regulating actin filament organization through its control of Lfc in the crosstalk between microtubule and actin cytoskeletons [62]. As the formation of stress fibers and focal adhesions which limit the cell migration are mediated by actin filament organization, the ability of cell migrate is significantly enhanced when Tctex-1 is overexpressed [56]. Therefore, MAP4 promoted the expression of Tctex-1, which could not only induce MT depolymerization, but also inhibit the formation of stress fibers and focal adhesions by inhibiting the function of Lfc. Both pathways promote epidermal cell migration.
Our team mainly focused on phosphorylation of MAP4 in the regulation of epidermal keratinocyte migration and proliferation under hypoxia. Initially we found that MAP4 phosphorylation was markedly upregulated at the wound margins, and hyperphosphorylation of MAP4 (S737 and S760) induced MT depolymerization and rearrangement, subsequently promoted keratinocyte migration and proliferation, thereby accelerated skin wound healing. We futher confirmed that MAP4 phosphorylation was induced by MKK6 (Glu) activating p38/MAPK pathway under hypoxia. Not surprisingly, the effects that promoting keratinocyte migration and proliferation of MAP4 phosphorylation, which relied on MT depolymerization and rearrangement, was abolished by MAP4 dephosphorylation [55]. In addition, we found hypoxia also promoted the migration and proliferation of endothelial cells (ECs), which are critical processes for angiogenesis during wound repair, and the underlying mechanism being revealed is similar to that in keratinocyte [63].
Besides, hypoxia inducible factor-1
Overall, MAP4 plays importat roles in wound healing under hypoxia through different pathways (Fig. 2). However, all the results reported so far are about the roles of MAP4 in keratinocytes and endothelial cells under hypoxia duiring wound healing, the roles of MAP4 in other wound-healing associated cells, such as fibroblasts, immune cells, under other oxygen concentrations or at other stages of the wound healing process need further study.
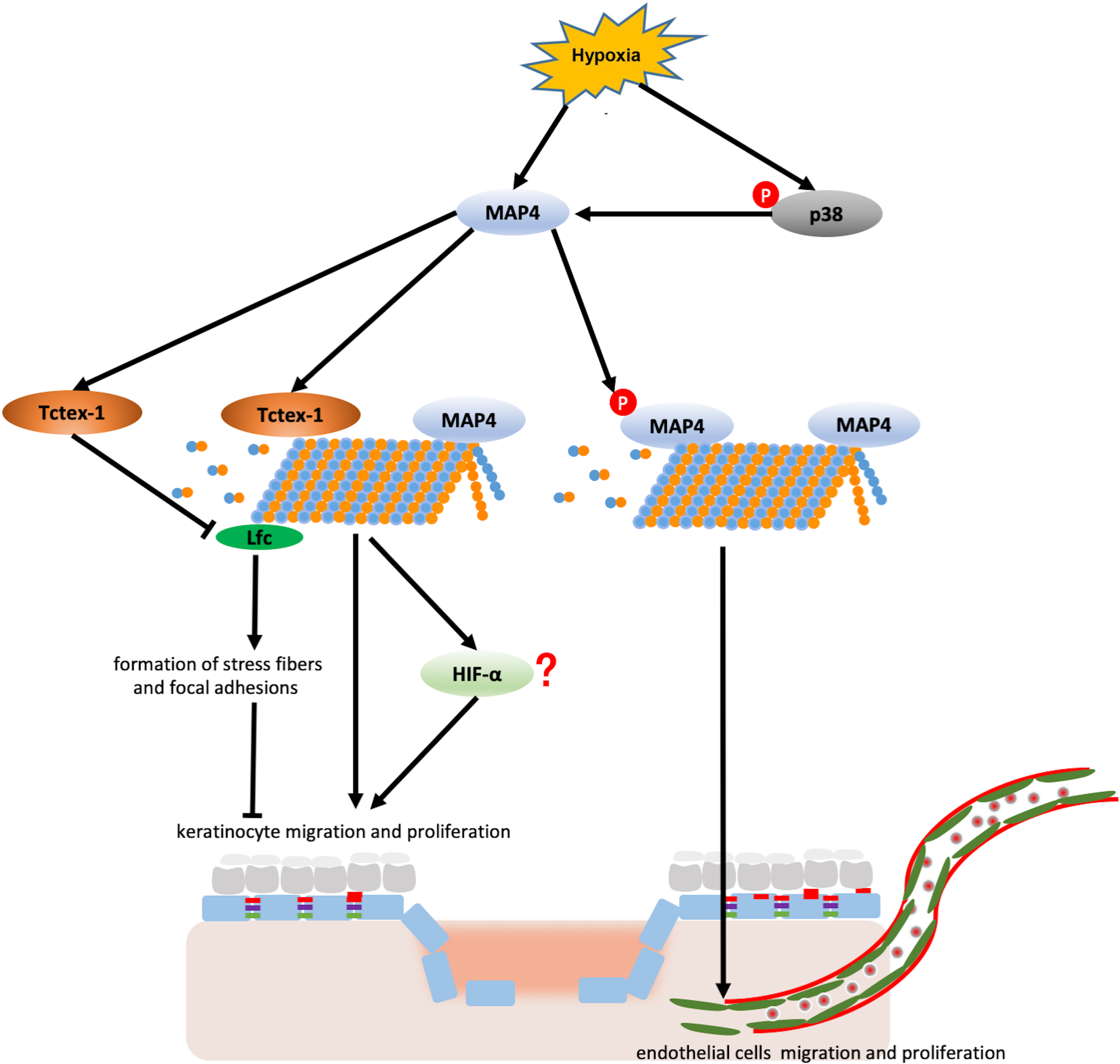 Fig. 2.
Fig. 2.The role of MAP4 play in wound healing under hypoxia. One side,
hypoxia increase MAP4 expression. MAP4 increasing Tctex-1 expression. Tctex-1
induce MT depolymerization, but also inhibit the formation of stress fibers and
focal adhesions by inhibiting the function of Lfc; Lfc, the
murine isoform of ARHGEF2 (also known as GEF-H1 in human) is a MT-associated
guanine nucleotide exchange factor (GEF). HIF-1
Based on the function of MT network, its role in cardiocytes has been paid attention to for a long time, and MAP4 has been deeply studied in the pathogenesis of various cardiac diseases, including cardiac hypertrophy, hypoxic myocardial injury, and myocardial infarction (MI) induced heart failure.
As is well-known, cardiac
hypertrophy due to systolic pressure overloading usually leads
to contractile dysfunction, but the exact underlying mechanism remains unclear.
MT network is one of important constitute cytoskeletal
elements of cardiomyocytes and plays an indispensable role in the regulation of
heart beating. The abnormality of MT may play
an important role in the pathogenesis of cardiac hypertrophy. According to
quantifying the MTs and measured sarcomere motion during MT
depolymerization in a model of pressure-hypertrophied
myocardium, Tsutsui et al. [66] found that stress
loading increased density of the MT component of the
cardiomyocytes cytoskeleton, which induced the entirety of the
cellular contractile dysfunction in pressure-overload cardiac
hypertrophy. Subsequent studies of this team found that
the increased MT network density imposes a viscous load on
active myofilaments during contraction, and the increased MT network density is a
persistent feature of severely pressure overloaded, hypertrophied and failing
myocardium [67, 68]. In addition, the same team further revealed MAP4 is markedly
upregulated in hypertrophy than in control hearts, and the
overexpression of MAP4 caused a shift of
tubulin dimers to the polymerized fraction and formation of a
dense and stable MT network in adult cardiomyocytes [69]. It has been known that
active transport of mRNAs and structural protein synthesis of
cardiomyocytes are important for a fully compensatory growth response to
hemodynamic overloading of the heart. MTs not only serve as
tracks for structural protein (such as receptors), but also for mRNAs. It has
been found that MAP4 decoration of MT inhibits of
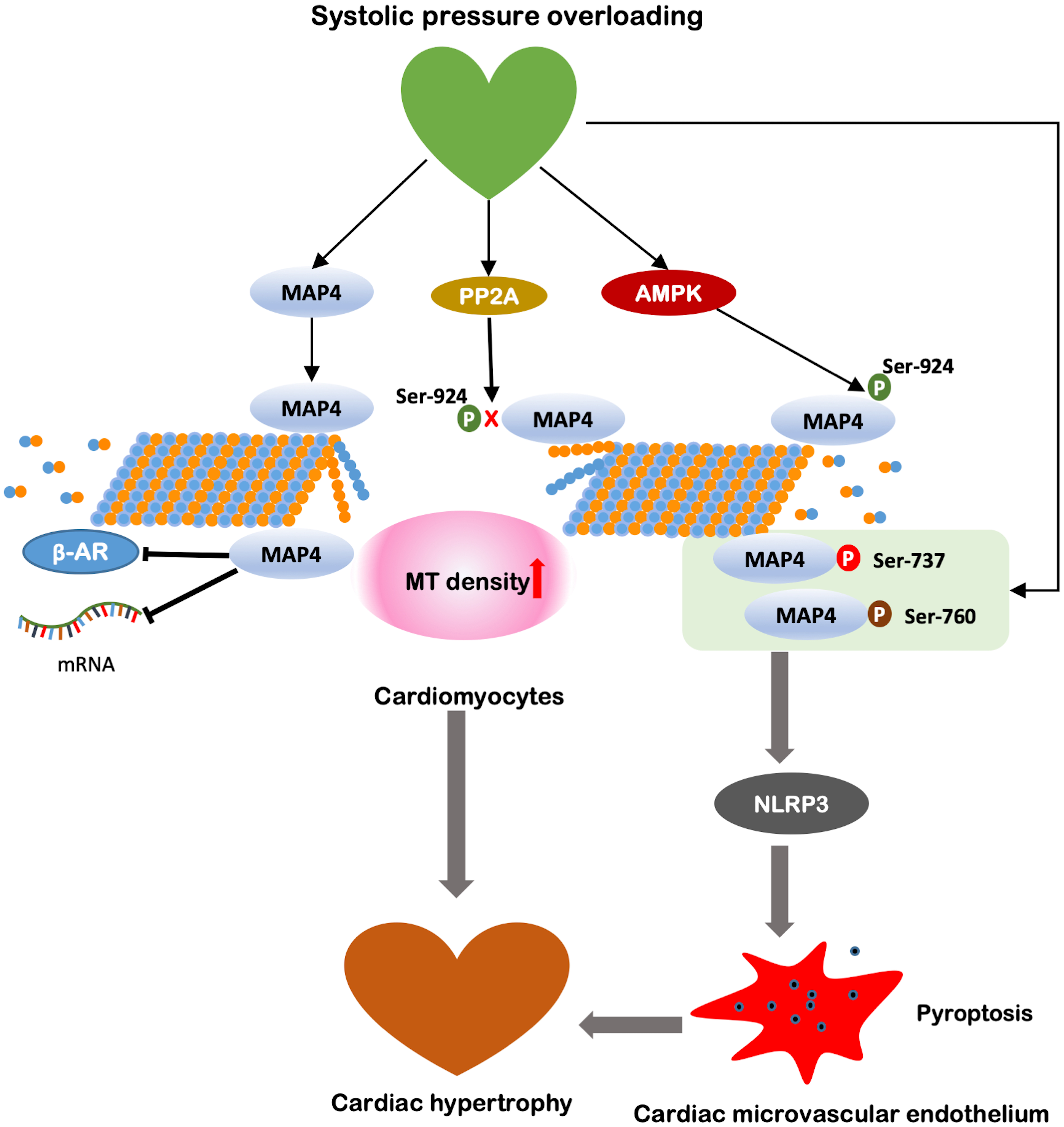
In addition, site-specific MAP4 dephosphorylation at Ser-924 induced by Ser/Thr protein phosphatases types 1 (PP1) and 2A (PP2A), which is the major determinant of MAP4-MT affinity, was prominent in hypertrophied myocardium and causes MT network densification in pressure overload cardiac hypertrophy [49, 50]. Conversely, another team found that activation of adenosine 5‘-monophosphate (AMP)-activated protein kinase (AMPK), a member of the MARK/PAR kinase subfamily, increases MAP4 phosphorylation at the analogous site, resulting in limiting accumulation and densification of MT that occurs in response to hypertrophic stress, which indicates that AMPK plays a role in counteracting the effects of PP1/PP2A in cardiac hypertrophy [73]. Tetralogy of Fallot (TOF) is a rare congenital condition caused by a combination of four heart defects that are present at birth, characterized by low arterial oxygen saturation and right ventricular hypertrophy [74]. It is a good model to study the effect of hypoxia and cardiac hypertrophy (induced by pressure overload). We recently found remarkable increased level of MAP4 (Ser-737 and Ser-760) phosphorylation in the hypertrophic right ventricular tissues in patients with TOF. With the phosphorylated MAP4 (at Ser-667A, Ser-737E and Ser-760E) knock in (KI) mouse that mimicked the phosphorylated form of MAP4 in human, we found that MAP4 phosphorylation at these sites induces MT disassembly and age-dependent pathological cardiac hypertrophy [75]. Besides, we noted that the cardiac microvascular density in the myocardium of the MAP4 KI mice is remarkably decreased. Furthermore, we demonstrated that MAP4 phosphorylation reduces cardiac microvascular density through NLRP3-related pyroptosis of cardiac microvascular endothelium, preliminarily revealing the mechanism of MAP4 phosphorylation-induced cardiac hypertrophy [76].
In general, according to the available research results until now, the expression level of MAP4, phosphorylation and dephosphorylation at specific sites of it, can induce different changes in MT stability and density, leading to its diverse roles in the pathogenesis of cardiac hypertrophy (Fig. 3). Further studies are needed to elucidate the comprehensive regulatory network of MAP4 and MTs in the cardiac hypertrophy pathogenesis.
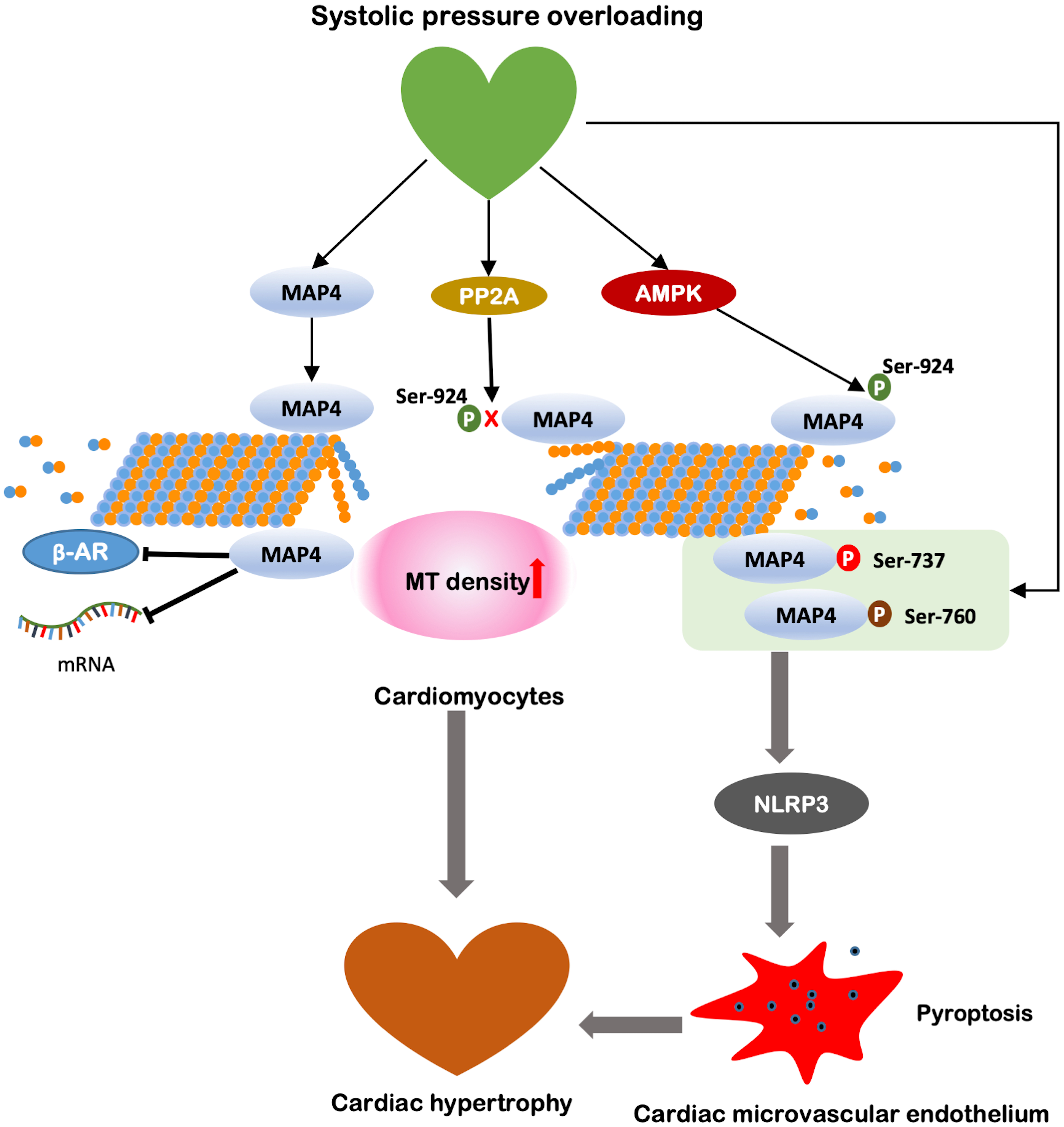 Fig. 3.
Fig. 3.The mechanism of MAP4 in systolic pressure
overloading induced cardiac hypertropghy. Under the systolic pressure
overloading, MAP4 expression increases and promotes tubulin dimers to be
polymerized. MT becomes more stable, and the MT density of
cardiomyocytes increases. Besides, MAP-4 decoration of MTs
inhibits transport of mRNAs and
Hypoxic myocardial injuries, mainly caused by hypoxia or ischemia, are related
with coronary artery disease, myocardial infarction, hypertensive heart disease,
cardiomyopathy, obstructive sleep apnea, or severe burns [77]. The mechanism of
hypoxia-induced myocardial injury is what we have been focusing on for a long
time. More than a decade ago, our group discovered that the hypoxia (1% O
As disturbances of energy metabolism play important roles in structural and
functional damage of cardiomyocytes induced by hypoxia, and
hypoxia-inducible factor (HIF)-1
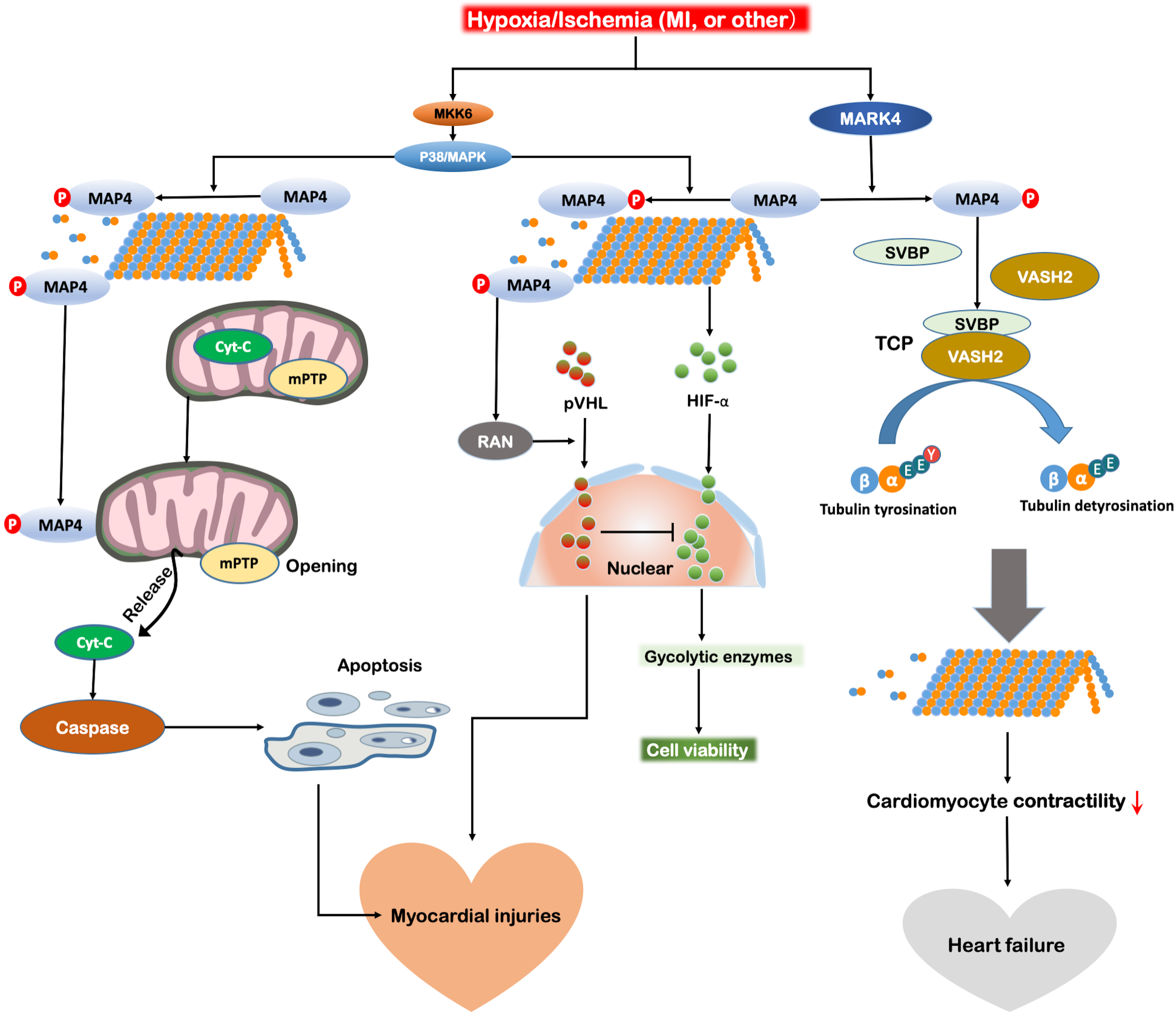
Mitochondria, the major energy sources of the cells, are more sensitive to hypoxia and could not be ignored in the mechanism of myocardial hypoxic injury [82]. MT network stability is crucial to the structure and function of mitochondria. We had reported in the early stages of cardiomyocyte hypoxia, a transient overexpression of MAP4 temporarily reinforce MT networks by increasing the synthesis and polymerization of tubulin, which effectively inhibits hypoxia-induced mitochondrial permeabilization and enhances the cell’s tolerance to hypoxia, being vital for cellular survival in cases of temporary severe hypoxia [83]. Variation of mitochondrial function is an initiating factor of cell apoptosis, and apoptosis induced by physiological events such as hypoxia is another important mechanism of hypoxic myocardial injuries [84]. Whether MAP4 play roles in cardiomyocyte apoptosis under hypoxia condition caught our interest. After research, we found that phosphorylation of MAP4 in cardiomyocytes treated with hypoxia make them translocated from cytosol to mitochondria, which affects the MT stability and leads to mitochondrial permeability transition pore (mPTP) opening and cytochrome c (cyt-c) release, activating caspase pathway and promotes mitochondrial apoptosis [85]. These results confirm that MAP4 is a vital regulator for the structural stability and function of mitochondrial. What’s more, we recently discovered that mitochondrial translocation of phosphorylated MAP4 and increased cardiomyocyte apoptosis together with MT disassembly was detected prior to the onset of cardiac remodeling, indicating a non-negligible role MAP4 in the pathogenesis of cardiac diseases [75]. Still and all, the potential mechanism of MAP4 phosphorylation-induced mitochondrial dysfunction involves many regulatory factors and a complex regulatory network, needs to be gradually analyzed by more studies [86].
Myocardial infarction (MI), a leading cause of cardiovascular
disease and death in humans, is caused by myocardial ischemia.
MI induces substantial cardiomyocytes death and the loss of
their contractility function in the remaining viable cells,
which leads to heart failure [87]. The MT cytoskeleton can
transmit mechanical signals and resist compression in contracting cardiomyocytes,
it has identified that cardiomyocyte contractility is impaired by
MT detyrosination, and suppressing MT detyrosination improves
contractility of cardiomyocyte in human heart failure [88, 89]. In a recent study, the deficiency of Microtubule-Affinity Regulating Kinase
4 (MARK4) substantially strengthens cardiomyocyte contractility in an acute MI
mice model, and the further mechanism study revealed that
MARK4 regulates cardiomyocyte contractility through promoting MAP4
phosphorylation, the later facilitates the access of Vasohibin
2 (VASH2), which then couples to a small vasohibin-binding
protein (SVBP), and forms tubulin carboxypeptidase (TCP) that
induces detyrosination of MT
In summary, mainly based on these findings, the regulatory mechanism of MAP4 in hypoxic myocardial injuries and MI induced heart failure is becoming clear (Fig. 4).
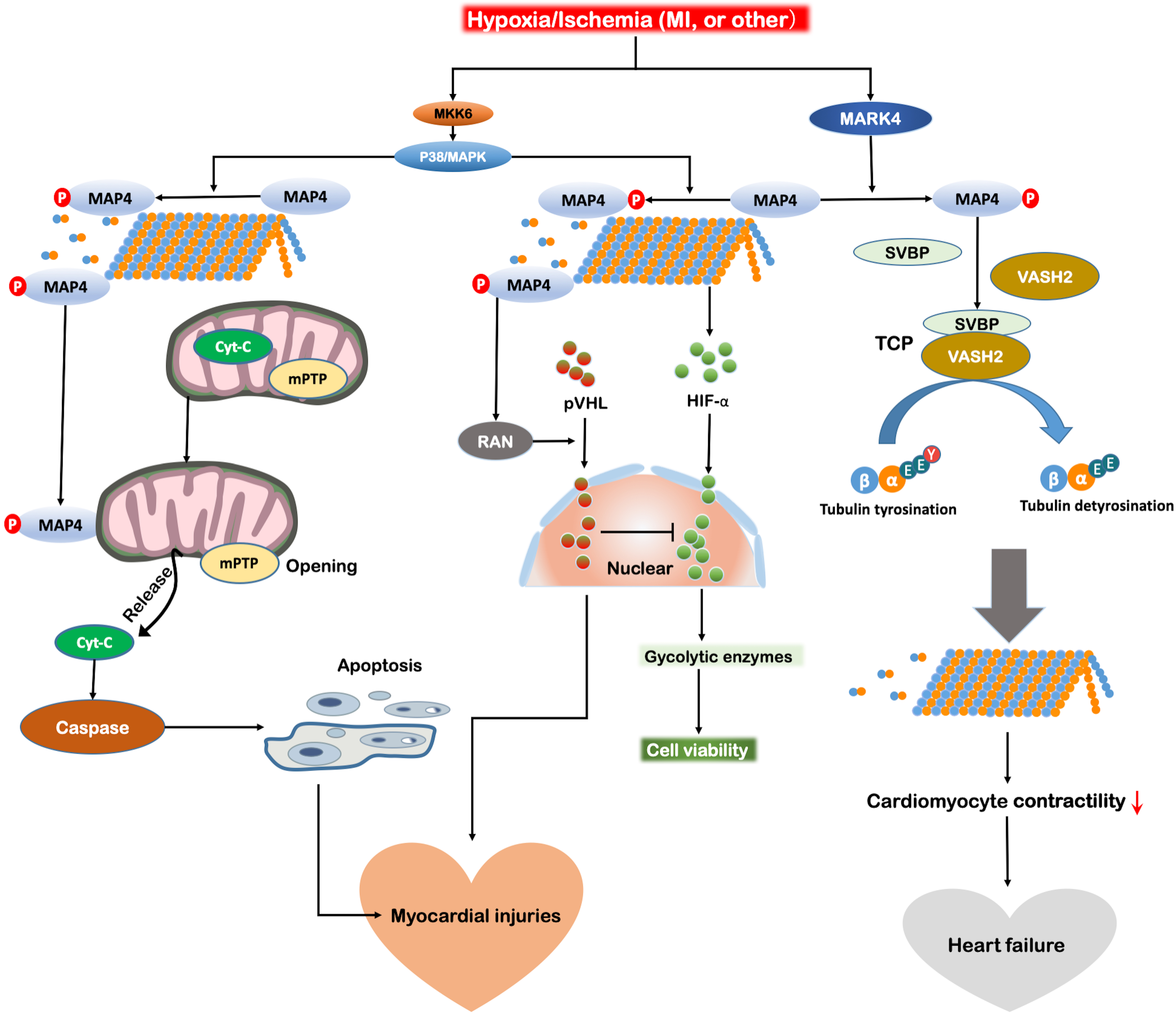 Fig. 4.
Fig. 4.Mechanism of MAP4 in
hypoxic myocardial injury and MI induced heart failure. Myocardial hypoxia
activate MKK6/p38/MAPK pathway, which induces MAP4 phosphorylation and the
depolymerization of MT. The phosphorylated MAP4 detaches from
MT and translocates to mitochondria from cytosol, leading to mPTP opening and
cyt-c release, which activates caspase pathway and promotes mitochondrial
apoptosis and lead to myocardial injuries. In addition, the phosphorylated MAP4
induced MT network breakdown upregulated Ran-mediated pVHL nuclear accumulation
and promoted HIF-1
Acute lung injury, usually caused by infection or
inflammation, is a disorder of acute inflammation that causes disruption of the
lung endothelial and epithelial barriers, frequently resulting in significant
morbidity and high mortality [91]. MT stability in vascular endothelium are
crucial for the regulation of endothelial barrier function. Our team was the
first to study and report the MAP4 phosphorylation-dependent MT disassembly and
its regulatory mechanism in inflammation-induced acute lung injury. In our study,
we use lipopolysaccharide (LPS) and tumor necrosis factor-
In these two years, the role of MAP4 in renal disease has gradually begun to gain attention. In a recently reported study, through proteomics analysis in animal kidney tissues and cells and in the urine of patients with CKD, MAP4 was identified as a potential marker for renal parenchymal injury and candidate contributors to chronic kidney disease (CKD) pathophysiology [95]. Therefore, the specific role of MAP4 in the pathogenesis of kidney disease is the most anticipated. After analyzing the protective effects of troxerutin, a flavonoid compound with potential anti-cancer, antioxidant, and anti-inflammatory activities, against cisplatin-induced kidney injury. A latest study shows that troxerutin may enhance MAP4 expression via activating Phosphoinositide 3 kinase/Protein kinase B (PI3K/AKT) pathway, thus exerting the effects of attenuating cellular apoptosis, alleviating oxidative stress and inflammatory response [96]. In addition to the effects of its overexpression, its phosphorylation is also an important regulatory mechanism. Our latest study reported the role of MAP4 phosphorylation in proteinuria in diabetic nephropathy (DN) and its possible mechanisms [97]. The content of MAP4 phosphorylation significant elevated not only in diabetic patients’ urine samples, but also in the kidney of streptozocin (STZ)-induced diabetic mice. Mice with MAP4 mutation mimic MAP4 phosphorylation showed robust DN pathology as compared to wild-type mice and their kidney showed more susceptible to STZ injury, and high glucose (HG) could induce elevation of MAP4 phosphorylation, following rearrangement of MTs and enhanced cell permeability, accompanied with dedifferentiation and apoptosis of podocytes, and these effects were rectified by MAP4 dephosphorylation. What’s more, all HG-induced pathological alterations were reinstated with pretreatment of p38/MAPK inhibitor SB203580, indicating that activation of p38/MAPK pathway induced the MAP4 hyperphosphorylation. For the first time, the detailed mechanism map of MAP4 in the pathogenesis of DN is clearly described. However, the existing studies on MAP4 in renal diseases are limited. More studies are needed to explore the detailed mechanism of MAP4 in different renal cells and different types of renal disease.
MTs and their dynamics and stability play key roles in the process of mitosis and cell division, which makes MTs be a main target for cancer cell invasion/migration regulating [98]. As a key regulator of MT stability, MAP4 is considered as an important effector in a variety of tumors and cancers, including bladder cancer [45], esophageal squamous cell carcinoma (ESCC) [99], lung adenocarcinoma (LADC) [100], ovarian cancer [101, 102], and osteosarcoma [103].
In bladder cancer, the cyclic adenosine monophosphate/protein kinase A (cAMP/PKA) signaling pathway disrupts MT cytoskeleton by the phosphorylation of MAP4, which potently inhibits the invasion and the migration of cancer cells [45]. In ESCC, it has been revealed that MAP4 promotes cell invasion and migration by activating the ERK-c-Jun-vascular endothelial growth factor (VEGF) signaling pathway [99]. MAP4 may be a good biological marker of tumors and cancers. It has been found that MAP4 expression was associated with tumor stage, lymph node metastasis and shorter survival of the ESCC patients, indicating MAP4 as an independent prognostic indicator for ESCC [99]. Similarly, MAP4, significantly correlated with the differentiation, pathological T stage, TNM stage and overall survival of LADC, representing as a prognostic biomarker for LADC, what’s more, overexpression of MAP4 efficiently promotes the migration and invasion of LADC cells [100]. MAP4 also may be a potential biomarker for ovarian cancer. There is a study showed that MAP4 expression was significantly associated with ovarian cancer histological subtype stage, grade and residual tumour, but not with overall survival, indicating it may be involved in early stages of tumour spread [102]. Besides, MAP4 could interact with some oncogene and act as an regulator in tumor. It has been reported that the funtion of bone morphogenetic protein/retinoic acid inducible neural-specific 3 (BRINP3), which acts as an oncogene and promotes the proliferation and invasion of osteosarcoma cell lines, can be suppressed by MAP4 overexpression [103]. In addition, a novel fusion of the microtubule-associated protein 4 (MAP4) gene and the MALT1 gene was reported in diffuse large B-cell lymphoma, but its function remains to be elucidated [104].
MTs are important targets for anticancer therapy. The anti-MT drugs, such as vinblastine, colchicine, paclitaxel, epothilones and estramustine and so on, are important anticancer drugs. These drugs generally bind to soluble tubulin and/or directly to tubulin of the MTs and affect the polymerization dynamics of MT, thus, disrupting the proliferation of tumor cells or inducing he death of tumor cells [105]. Basing on its regulating function, MAP4 may play roles in affecting the sensitivity to anti-MT drugs. It has been reported that treating with breast cancer cells with doxorubicin to increase expression of p53, a famous tumor suppressor gene, can decrease MAP4 expression, thereby decreases MT polymerization, and ultimately improves the vinca alkaloid sensitivity of these cells [36]. However, a research reported a completely different mechanism that sequential knockdown of dual kinases could stabilize MTs by inhibiting phosphorylation of p38 and MAP4, on the contrary, it enhances the response of ovarian cancer cell lines and xenografts to paclitaxel [101]. These two pathways have diametrically opposite effects on MT stability, but both improve the sensitivity of cancer cells to anti-MT drugs, the underlying mechanism needs further research.
In addition to the above diseases, MAP4 may also be involved in the occurrence of other diseases. T cells are key effector cells in the development of autoimmune and allergic diseases, there is a study assessed the role of MAP4 in T cell activation, revealing MAP4 as a checkpoint molecule that balances positive and negative hallmarks of T cell activation [106], which dicates that MAP4 may be involved in the pathogenesis of these diseases. MAP4 is also found to be abundant in neuronal cells such as brain-specific cells, cerebellar Purkinje cells and hippocampal cone cells [23], and has been implicated in some neurological and psychiatric disorders. For example, it has been revealed that MAP-4 may be involved in the effect of antidepressant treatment, which is associated with MT assembly, neuron survival, and neural plasticity changes in the hippocampus during chronic stress [107], and MAP4 is identified as an autoantigen inducing autoantibodies that involved in the pathogenesis of Alzheimer’s disease (AD) and serves as a diagnostic biomarker [108]. Moreover, MAP4 has been shown to be involved in some congenital diseases. A homozygous MAP4 variant has been identified in a consanguineous family with two affected children with severe growth retardation and normocephaly, demonstrating that MAP4 mutations contribute to the clinical spectrum of centrosomal defects [109]. Surprisingly, MAP4 mediates the infection processes of human papilloma viruses (HPV). Guo and colleagues unraveled that oncoprotein E7 of HPV16 interfered Mps1-MAP4 signaling cascade, which increases the stability of MT polymerization and extends mitotic progression, thereby extends infection window and facilitates the persistent HPV16 infection [46].
Conclusively, MTs are fundamental to cellular structure and involve almost all cellular activities, which are finely regulated by MAP4, and MAP4 is widely expressed in non-neuronal cells and tissue. MAP4 has been demonstrated to play roles in pathogenesis of wound healing and a great deal of human disease, including various cardiac diseases (such as cardiac hypertrophy, hypoxic myocardial injury and MI induced heart failure), acute lung injury, tumors and cancers (such as bladder cancer, ESCC, LADC, ovarian cancer, osteosarcoma), renal disease (such as CKD and DN), depressant, AD, HPV infection and potential autoimmune and allergic diseases. Various upstream and downstream regulatory mechanisms of MAP4 have been reported. MAP4 is not only a key node in the pathogenesis, but also a biomarker for the clinical characterization of many diseases, making it an optimal target for precise treatment of these diseases.
Although some diseases share the regulatory mechanisms of MAP4, most are different in many diseases. Before formulating MAP4-targeted therapy, we need to further analyze the more detailed regulatory mechanisms of MAP4 in different diseases. The pathogenesis of many other diseases may also be related to MAP4, which also needs more extensive study in the future. Immune diseases and allergic diseases are the most common diseases in human beings, studies on MAP4 in this category of diseases are still lacking, which will be a broad field of future research. Currently, only drugs targeting MTs are available in the clinic, mainly in the field of cancer treatment, drug development based on MAP4 will be very meaningful and urgent.
QC and JZ contributed to formal analysis, methodology, investigation and writing original draft. ZS and YH participated in conceptualization, data curation, project administration and writing review & editing. YH contributed to supervision and funding acquisition. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript. All authors have participated sufficiently in the work and agreed to be accountable for all aspects of the work.
Not applicable.
Not applicable.
This work was supported by Key Project of the National Natural Science Foundation of China (NSFC) (No. 81430042), and National Key Research and Development Program (2017YFC1103302).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.