
 , Jing Li 1,*
, Jing Li 1,*1 Heart Center & Beijing Key Laboratory of Hypertension, Beijing Chaoyang Hospital, Capital Medical University, 100020 Beijing, China
2 Department of Nephrology, Beijing Chaoyang Hospital, Capital Medical University, 100020 Beijing, China
†These authors contributed equally.
Abstract
Background: The SARS-CoV-2 vaccine has been implemented in response to the 2019 Coronavirus Disease (COVID-19) pandemic worldwide. Dysregulation of gut metabolite is associated with COVID-19 patients. However, the effect of vaccination on the gut metabolite remains unknown, and it is critical to investigate the shifts in metabolic profiles following vaccine treatment. Methods: In the present study, we conducted a case-control study to assess the fecal metabolic profiles between individuals who received two doses of intramuscular injection of an inactivated SARS-CoV-2 vaccine candidate (BBIBP-CorV) (n = 20), and matched unvaccinated controls (n = 20) using untargeted gas chromatography and time-of-flight mass spectrometry (GC-TOF/MS). Results: Significant different metabolic profiles were observed between subjects receiving SARS-CoV-2 virus vaccines and the unvaccinated. Among a total of 243 metabolites from 27 ontology classes identified in the study cohort, 64 metabolic markers and 15 ontology classes were dramatically distinct between vaccinated and unvaccinated individuals. There were 52 enhanced (such as Desaminotyrosine, Phenylalanine) and 12 deficient metabolites (such as Octadecanol, 1-Hexadecanol) in vaccinated individuals. Along with altered metabolic compositions, multiple functional pathways in Small MoleculePathway Database (SMPDB) and Kyoto Encyclopedia of Genes and Genomes (KEGG) varied between groups. Our results indicated that urea cycle; alanine, aspartate, and glutamate metabolism; arginine and proline metabolism; phenylalanine metabolism and tryptophan metabolism were abundant after vaccination. Additionally, correlation analysis showed that intestinal microbiome was related to alteration in metabolite composition and functions. Conclusions: The present study indicated the alterations in the gut metabolome after COVID-19 vaccination and the findings provide a valuable resource for in-depth exploration of mechanisms between gut metabolite and SARS-CoV-2 virus vaccines.
Keywords
- gut metabolite
- COVID-19
- SARS-CoV-2
- vaccine
- BBINP-CorV
The 2019 coronavirus disease (COVID-19) is a highly infectious which has become a threat to global health [1]. The number of infections and deaths from COVID-19 is rising alarmingly and on 12 September 2022, there are more than 605,912,418 confirmed cases and 6,491,649 deaths worldwide according to World Health Organization (WHO) COVID-19 Dashboard [2]. In face of such a severe epidemic, vaccination is the best way to control the pandemic. BBIBP-CorV, an inactivated vaccine, is from China National Biotec Group Co. and Beijing Institute of Biological Products, whose safety and performance have been assessed by randomized, double-blind, controlled trials among different groups [3, 4, 5].
The human intestinal microbiota consists of 10
Evidence from clinical and animal studies indicated that composition and functions of intestinal flora is critical in modulating the immune response to vaccines [14, 15]. Ng SC et al. [16] found that a higher abundance of B. adolescentis was observed in CoronaVac (inactivated COVID-19 vaccine) high-responders, which is related to the immune protection of the enriched carbohydrate metabolic pathways. While, to date, whether inactivated SARS-CoV-2 virus vaccines influences the fecal metabolome profiles has not been determined. Therefore, in present study, adults who have received inactivated vaccine (BBIBP-CorV) to examine molecular alterations in the gut metabolome using untargeted metabolomics based on time-of-flight mass spectrometry (GC-TOF/MS).
In the current study, 40 healthy adults, aged between 18–59 years old, were enrolled in Beijing Chaoyang Hospital, and 20 of them were vaccinated since 1 January 2021 to 1 April 2021. The inclusion criteria were as follows: healthy adults who have completely received two doses of intramuscular injection of BBIBP-CorV (as the vaccinated group, n = 20) and 20 unvaccinated individuals (as the unvaccinated group, n = 20). The exclusion criteria were that, participants with cancer, previous heart failure, renal failure, stroke, peripheral artery disease, and chronic inflammation disorders; with previous SARS-CoV-2 exposure or infection; and those who received statin, aspirin, insulin, metformin, antibiotics, or probiotics treatment within the last two months. This study was performed in accordance with the Helsinki declaration, and was approved by the Medical Ethics Committee from Beijing Chaoyang Hospital. Written informed consent was obtained from all study participants prior to enrollment.
The fresh middle section of the fecal samples was collected from all the
participants. And for participants among the vaccinated group, the samples were
harvested within 1 month of the second dose of vaccine. All the stool samples
were transported to the laboratory and frozen at –80 °C until use. The
sample preparation procedures were performed according to the previously
published methods [17, 18]. Briefly, 5 mg of samples were weighted and placed
in a microcentrifuge tube, mixed with 25 mg of prechilled zirconium oxide beads
and 10
The untargeted metabolomics profiling was performed on XploreMET platform, a
GC-TOF/MS system (7890B, Pegasus HT, Leco Corp., St. Joseph, MO, USA) with an
Agilent 7890B gas chromatography and a Gerstel multipurpose sample MPS2 (Gerstel,
Muehlheim, Germany). A Rxi-5 ms capillary column (30 m
The raw data generated from GC-TOF/MS were processed with ChromaTOF software for automated baseline denosing, smoothing, peak picking, deconvolution and peak alignment. The identification for compound was carried out by comparing both mass spectrometry (MS) similarity and fatty acid methyl esters (FAMEs) retention index distance with the referenced standards in JiaLib database. Briefly, metabolite was annotated by blasting the retention indices and mass spectral data with the previously generated and known structures (as reference standards) in JiaLib metabolite database, which comprises over 1200 mammalian metabolites and has become one of the most comprehensive metabolite libraries. The platform Imap(1.0, Metabo-Profile, Shanghai, China) was used for subsequent statistical analyses.
PCA was known as an unsupervised method for modeling, frequently applied to determine data outliers, clustering, and classification trends prior to knowledge of the sample group. The principal components (PCs) such as principal component 1 (PC1) and principal component 2 (PC2) derived from the data set, were orthogonal to one another and could reflect the reducing levels for the variation in the data set. PLS-DA is a generalized multiple regression modeling method dealing with multiple collinear mass spectral data and classes variables. PLS-DA as a multi-class classifier, is utilized to visualize the distinctions in global metabolic profiles between groups. PLS-DA provided more valuable information beyond PCA. The Orthogonal PLSDA (OPLS-DA) is a modification of PLS method, its algorithm decomposed the raw data set into systemic variations, orthogonal/unrelated information, and residual. Statistical algorithms of PCA, PLS-DA and OPLS-DA were adapted from the statistical analysis software packages in R studio (http://cran.r-project.org/, Version 3.3.3, Auckland, New Zealand).
Continuous variables were presented as median with interquartile range, while
categorical variables were indicated as numbers and percentages. Wilcoxon rank
test and chi-squared tests were applied to determine continuous and categorical
variables, respectively. Student’s t-test or Mann-Whitney Wilcoxon test
was applied to examine the significant difference between groups. Z score in
heatmaps was calculated by subtracting the mean and dividing the standard
deviation. Z-score would be negative when the raw value was below the average
level and positive when it was higher than the mean. Volcano plot was used to
describe the significantly disparate variables between groups. The analyses were
performed with SPSS, Version 22.0 (IBM Corp., Armonk, NY, USA). The p
values were two-tailed, and a p value
The current study consisted of 20 healthy controls with no history of SARS-CoV-2 virus vaccines vaccination, and 20 individuals who have completed the two doses intramuscular injection of BBIBP-CorV. The clinical information including primary demographics, physiology, and biochemistry characteristics of the participants have been described in Supplementary Table 1. Compared with unvaccinated controls, there was no significant difference detected in the vaccinated subjects, either in clinical parameters of sex, body mass index, systolic and diastolic blood pressure levels, fasting blood glucose, total cholesterol, triglyceride, High-Density Lipoprotein Cholesterol (HDLC), Low-Density Lipoprotein Cholesterol (LDLC), uric acid or White Blood Cell (WBC).
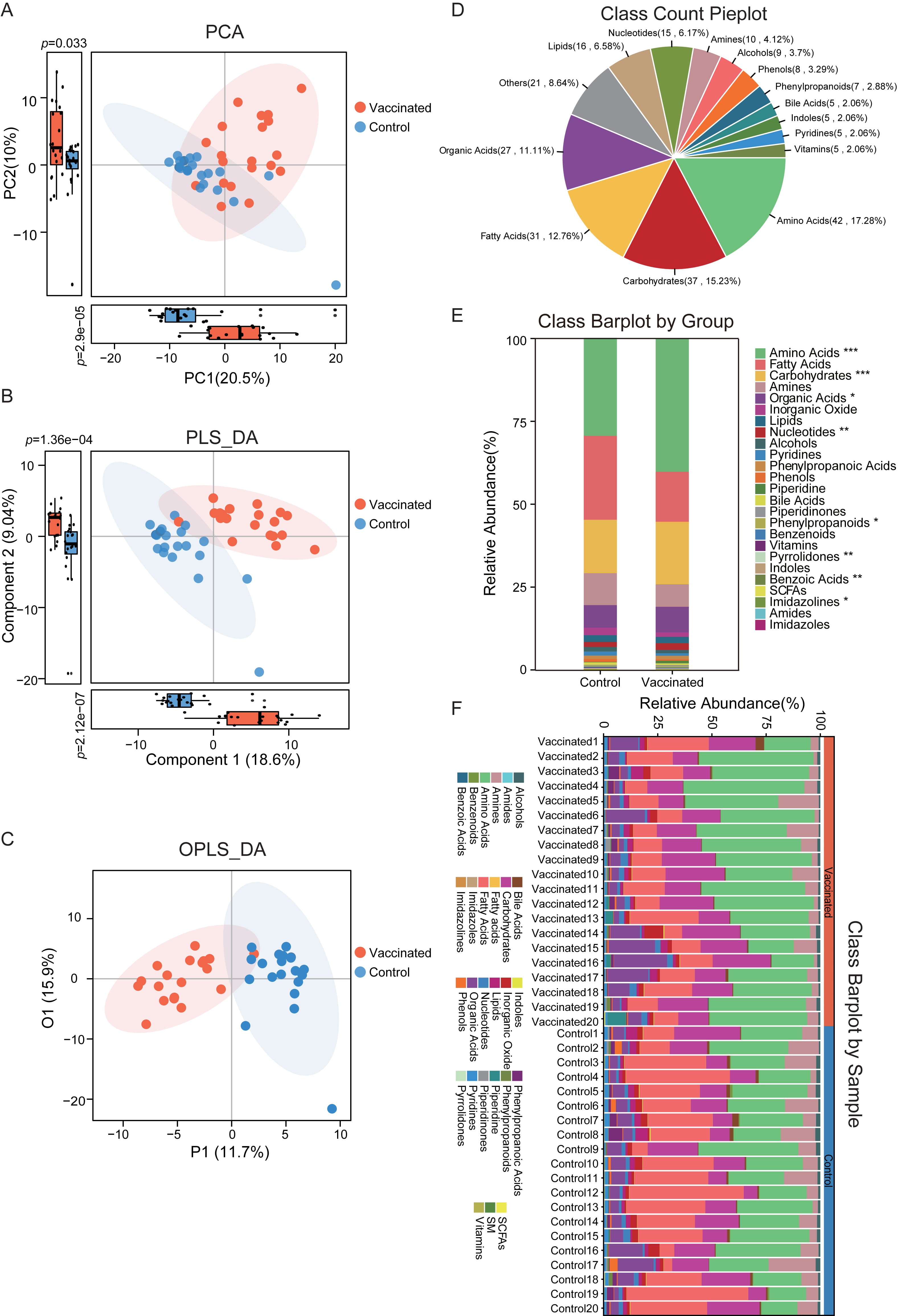
According to the untargeted metabolomics data based on GC-TOF/MS, we obtained the fecal metabolic characteristics of BBIBP-CorV vaccinated subjects. Clustering analysis of PCA, PLS-DA and OPLS-DA was performed respectively to identify the separation and classification trends. In the PCA plots, vaccinated individuals were observed to be roughly distinguished from the untreated controls (Fig. 1A). There was dramatic difference in both PC1 and PC2 of the PCA plots, with vaccinated subjects displaying much higher score. The PC1 accounted for 20.5% of the whole variance, and PC2 is responsible for 10% of the variance between groups. Similarly, in the PLS-DA scatterplots, we detected clearer clusters of vaccinated and unvaccinated group, respectively (Fig. 1B). An obvious separation between groups was obtained by PLS-DA, and significant distinctions in component 1 and component 2 were confirmed. For component 1, it could explain 18.6% of the difference between groups, and component 2 accounted for 9.04%. Additionally, prominent disparity between individuals who received BBIBP-CorV and the untreated controls was further validated in OPLS-DA analysis (Fig. 1C).
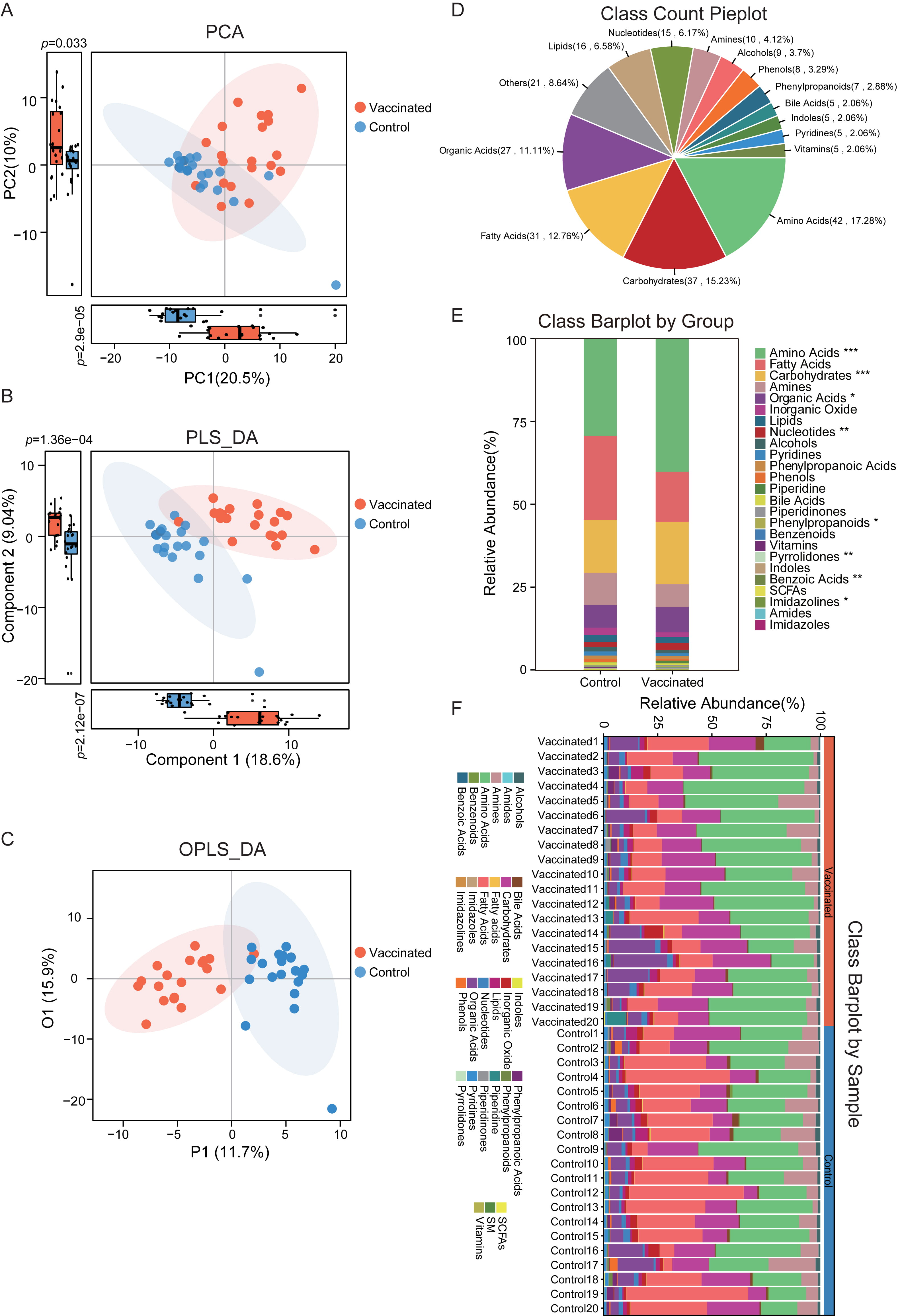 Fig. 1.
Fig. 1.The discrimination of metabolic profiles between vaccinated and
control group. (A) All the samples in vaccinated and control group were
distinguished within the PCA plots according to metabolite annotation data. There
was bits of overlap between groups, and significant difference was observed in
PC1 and PC2, respectively. p
Considering the distinguished metabolic features in vaccinated participants based on multivariate statistical analyses, we further carried out the metabolite annotation. The detailed metabolic composition was obtained, and the top 15 most abundant classes identified were depicted in Fig. 1D. Here we detected 42 amino acids (accounted for 17.28% of total identified metabolites), 37 carbohydrates (15.23%), 31 fatty acids (12.76%), 27 organic acids (11.11%), 16 lipids (6.58%), 15 nucleotides (6.17%), 10 amines (4.12%), 9 alcohols (3.7%), 8 phenols (3.29%), 7 phenylpropanoids (2.88%), 5 bile acids (2.06%), 5 indoles (2.06%), 5 pyridines (2.06%) and 5 vitamins (2.06%).
In addition, there were a total of 27 metabolic classes identified in the study cohort, and the relative abundance and proportion of fecal metabolite classes in each group and individual samples were shown in Fig. 1E,F. The 25 metabolic classes included amino acids, fatty acids, carbohydrates, amines, organic acids, inorganic oxide, lipids, nucleotides, alcohols, pyridines, phenylpropanoic acids, phenols, piperidine, bile acids, piperidinones, phenylpropanoids, benzenoids, vitamins, pyrrolidones, indoles, benzoic acids, short-chain Fatty Acids (SCFAs), imidazolines, amides, imidazoles. It was interesting that the abundance of amino acids, carbohydrates, organic acids, nucleotides, pyrrolidones, benzoic acids and imidazolines were dramatically distinct between vaccinated and unvaccinated subjects, and most of these classes were augmented in vaccinated group.
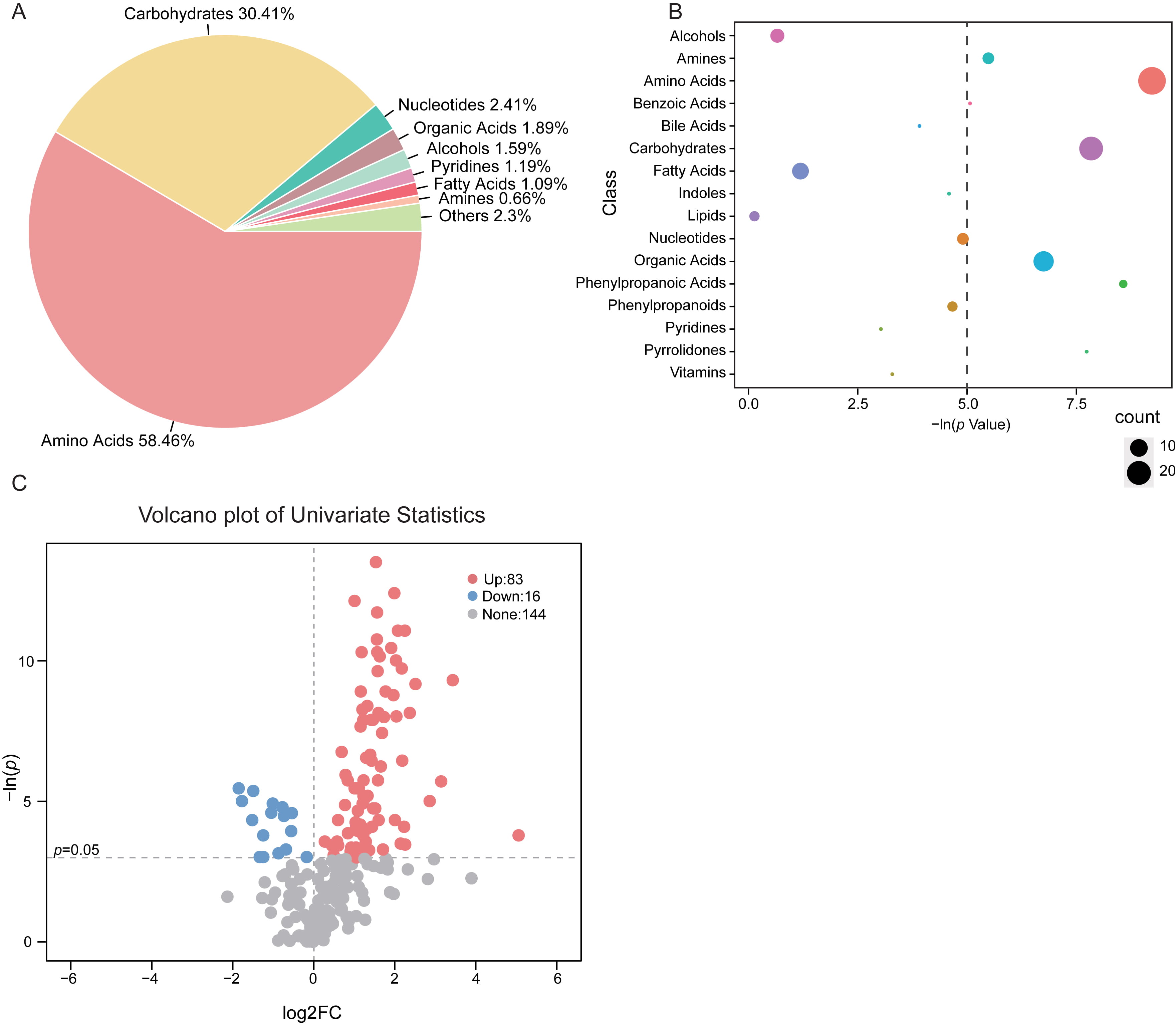
The composition and percentage of varied fecal metabolite class in vaccinated
participants indicated that, the majority of the altered metabolites was mainly
consisted of amino acids (58.46%) and carbohydrates (30.41%) (Fig. 2A). As
compared with the untreated controls, there were 26 metabolites within the amino
acids class and 20 from the carbohydrates class that are discrepant in vaccinated
groups (Fig. 2B). Notably, for the total of 243 identified fecal metabolites in
the subjects, we observed 99 features significantly different between groups
based on univariate analysis (p
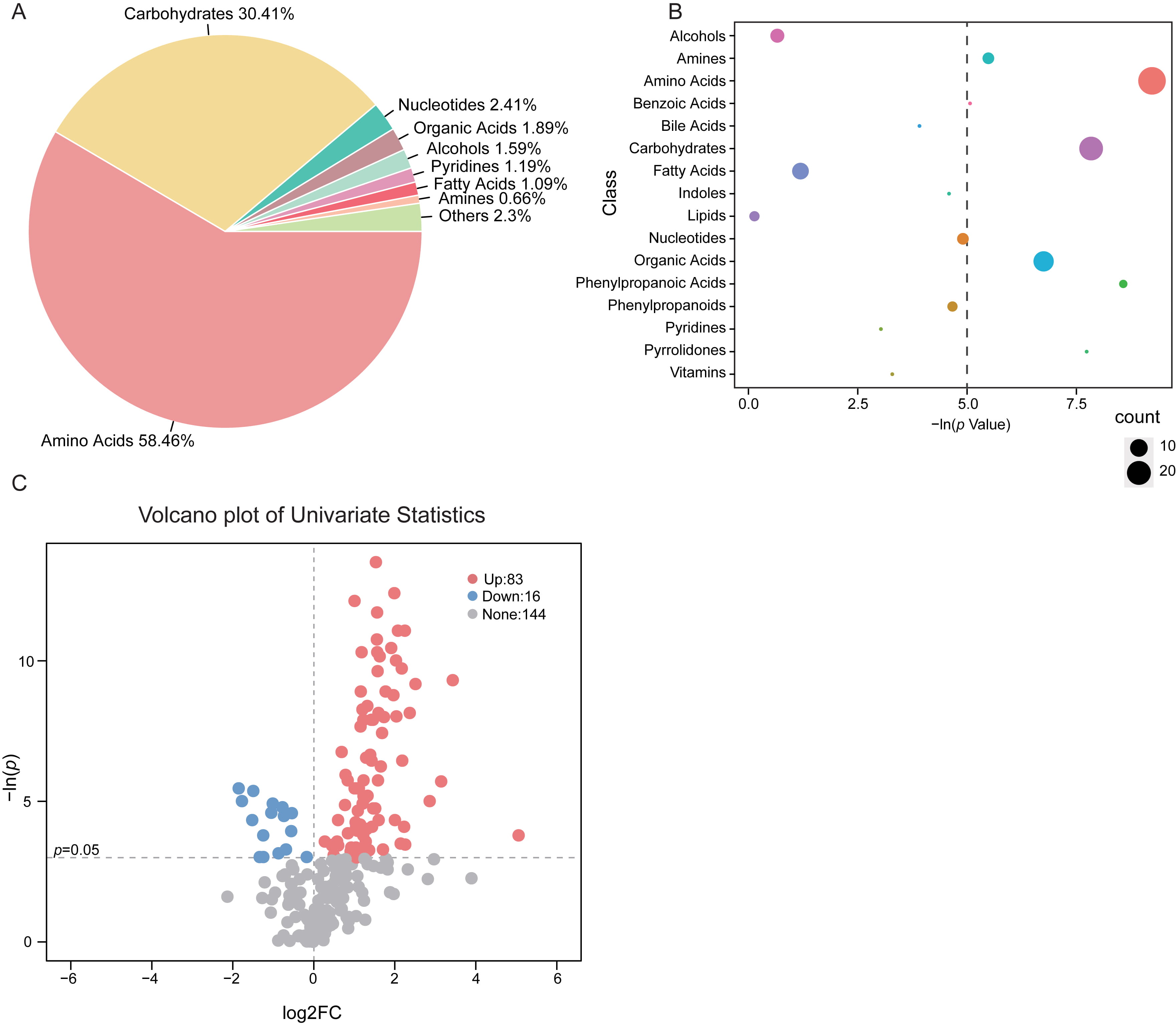 Fig. 2.
Fig. 2.Shifts of gut metabolic classes in
vaccinated subjects. (A) Proportion of the altered metabolite classes in
vaccinated group was shown in pie plot. (B) Enrichment of the varied metabolite
class in stool samples of vaccinated group. The size of the bubble represented
the number of metabolites within the class. The dashed line indicated p
value at 0.05. (C) Volcano plot depicting the fold change (FC) and p
value of each identified metabolite. The threshold value for significantly
distinct metabolites selection was p
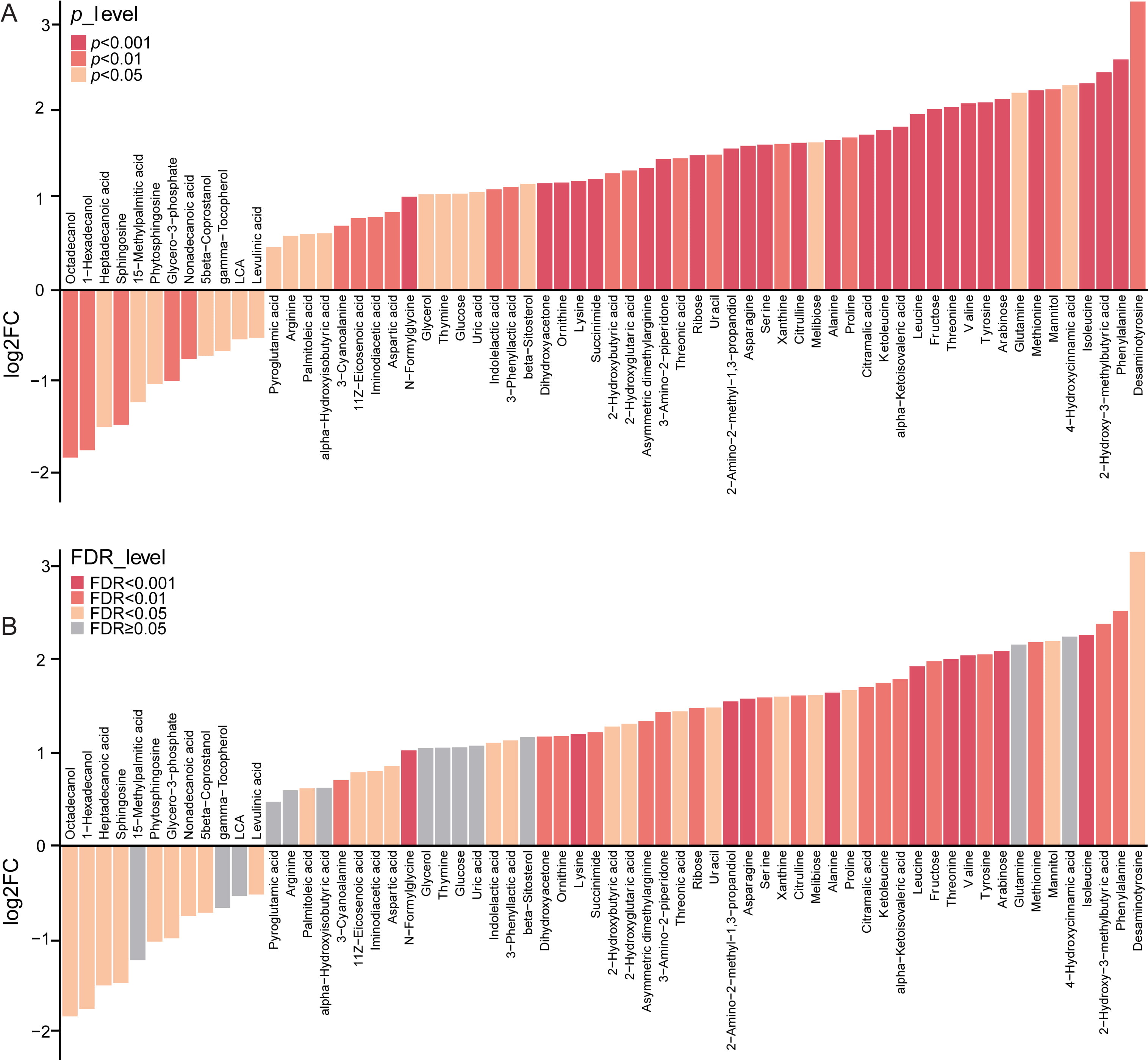
Next, we further focused on the 64 altered
metabolic markers in the stool of vaccinated samples compared with the controls,
which showed variable importance in projection (VIP)
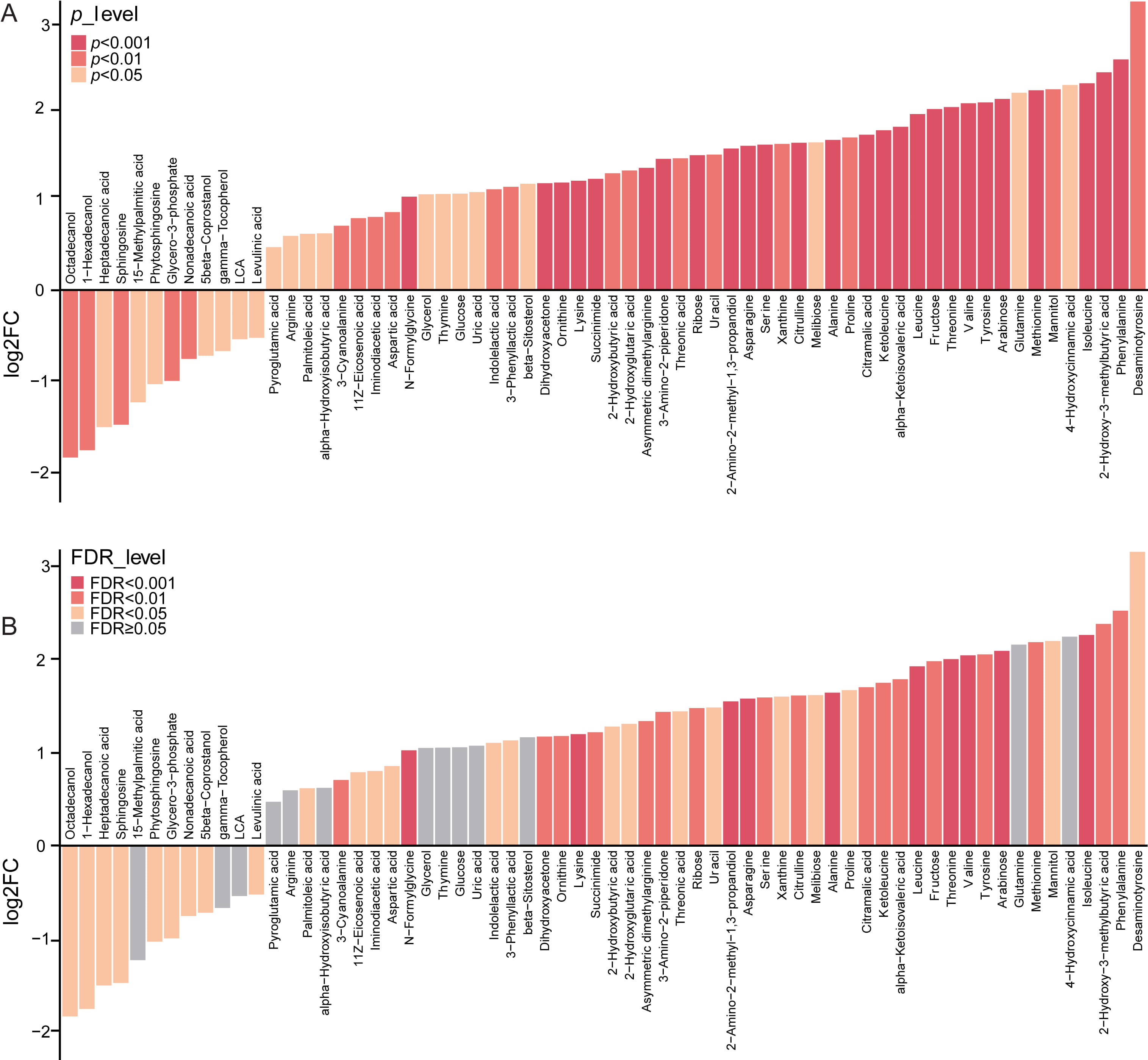 Fig. 3.
Fig. 3.The alteration of fecal metabolites between vaccinated and un
vaccinated group. (A) 64 significantly altered metabolic markers in the stool of vaccinated individuals as compared with the controls,
with VIP
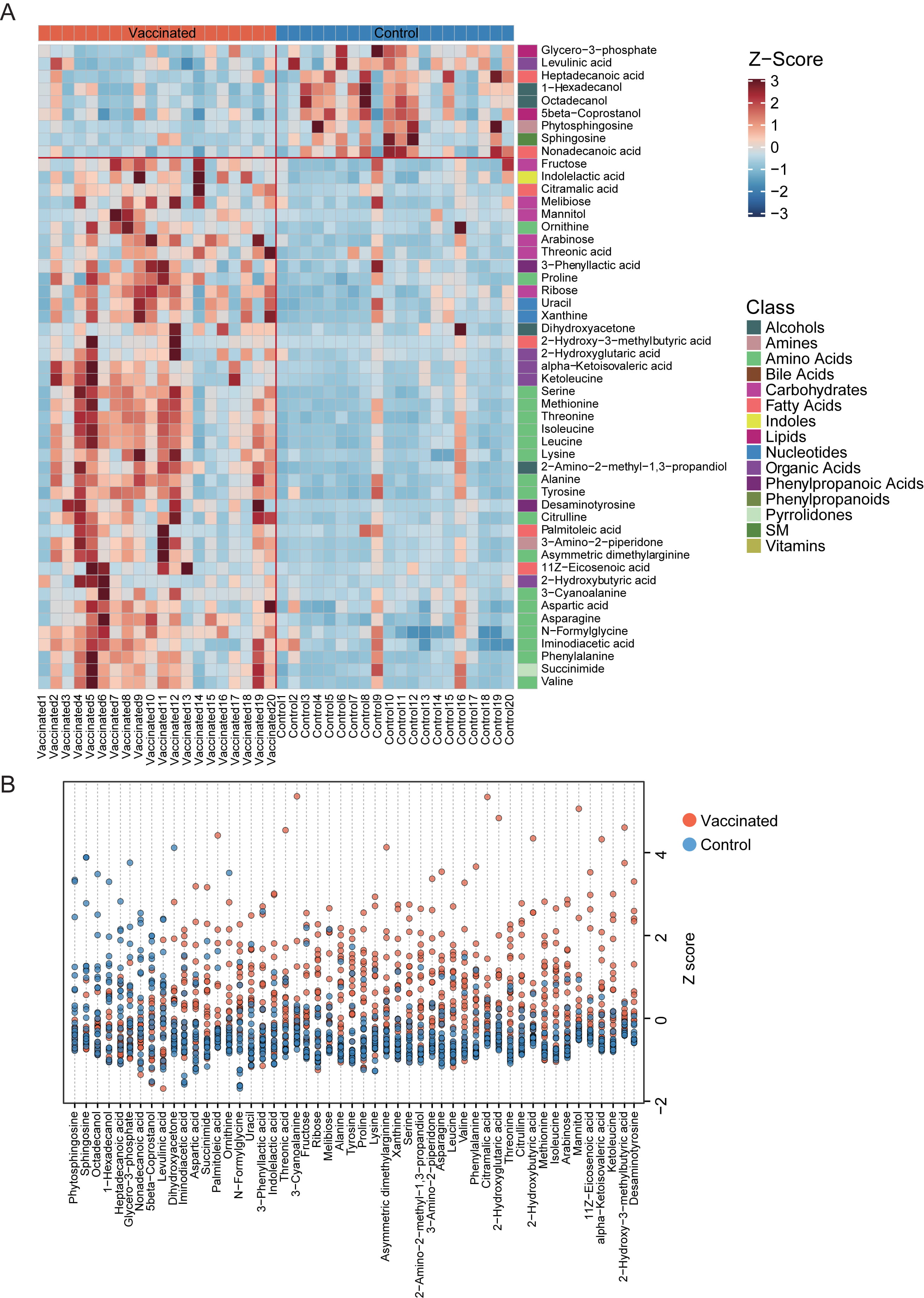
Relative abundance of the varied fecal metabolites in each vaccinated and unvaccinated individuals were shown in detail with clustering heatmap (Fig. 4A,B). The 9 metabolic markers reduced in vaccinated populations included glycero-3-phosphate, levulinic acid, heptadecanoic acid, 1-hexadecanol, octadecanol, 5beta-coprostanol, phytosphingosine, sphingosine, nonadecanoic acid, which were from lipids, phenylpropanoic acids, organic acids, fatty acids, alcohols, amines and SM, respectively (Fig. 4A). It was interesting that, those enhanced metabolites in stool samples of vaccines-treated participants were primarily from amino acids, carbohydrates and organic acids etc., especially essential amino acid. Among them, we found 3-phenyllactic acid, which could be produced by Bifidobacterium species via aromatic lactate dehydrogenase, has been previously suggested as contributor to the antiobesity function of green tea polyphenols [19, 20].
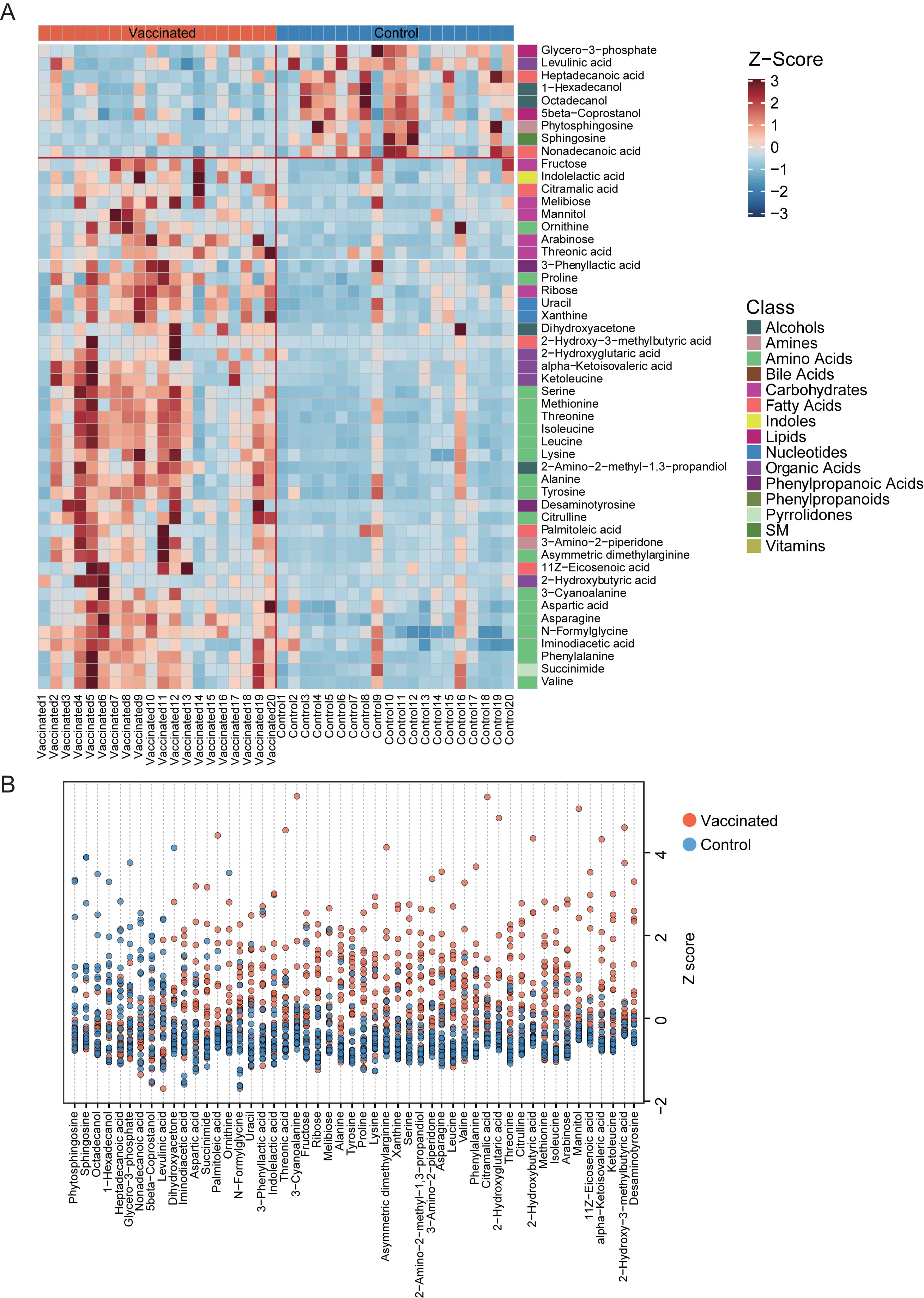 Fig. 4.
Fig. 4.The distribution of fecal metabolites with significant difference between vaccinated and control group. (A) Heatmap for the significantly shifted metabolites in feces samples of individuals receiving vaccines. The abundance of each metabolite was normalized into Z score. The class of each metabolite was shown in the right of the heatmap. (B) Z-Score plot of varied metabolic biomarkers between groups. The z-score value of 9 metabolites (phytosphingosine, sphingosine, octadecanol, 1-hexadecanol, heptadecanoic acid, glycero-3-phosphate, nonadecanoic acid, 5 beta-coprostanol, levulinic acid) was abundant in the control group, and the z-score of 42 metabolites was enriched in the vaccinated group.
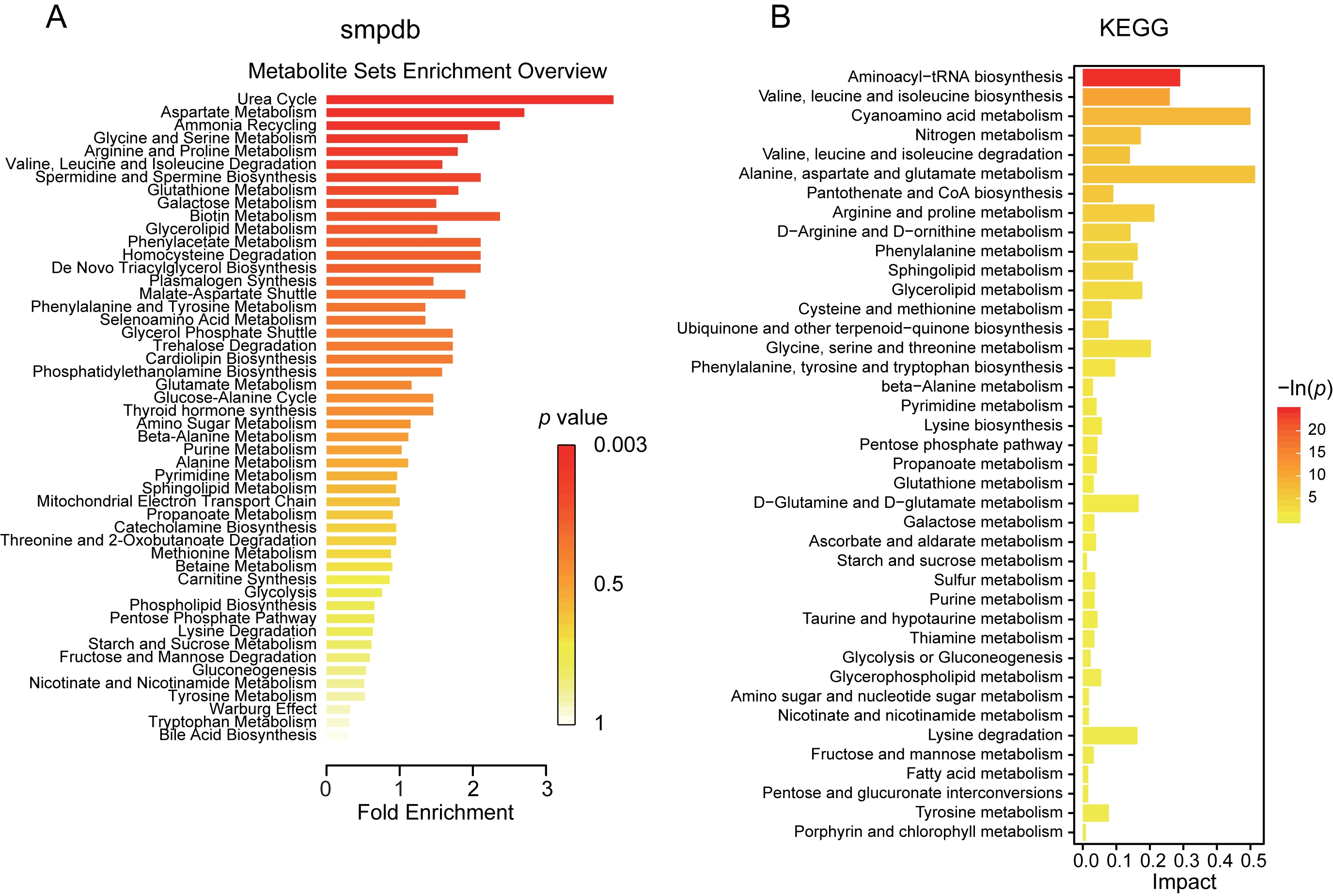
Next step, we examined the potentials of metabolic and functional pathways in the gut of vaccinated subjects via Small MoleculePathway Database (SMPDB) and Kyoto Encyclopedia of Genes and Genomes (KEGG). The varied fecal metabolite mainly functioned in the process of urea cycle, ammonia recycling, metabolism of aspartate, glycine, serine, arginine, proline, glutathione, galactose, glycerolipid, phenylacetate, phenylalanine and tyrosine, degradation of valine, leucine, isoleucine and homocysteine, biosynthesis of spermidine, spermine and plasmalogen, as indicated by SMPDB (Fig. 5A). The potential metabolic capacities were further confirmed through KEGG pathway analysis, and functions in valine, leucine, isoleucine and lysine degradation, alanine, aspartate, glutamate, arginine, proline and tyrosine metabolism, pentose phosphate pathway were validated (Fig. 5B). These observations demonstrated striking alteration of metabolism in subjects with vaccination.
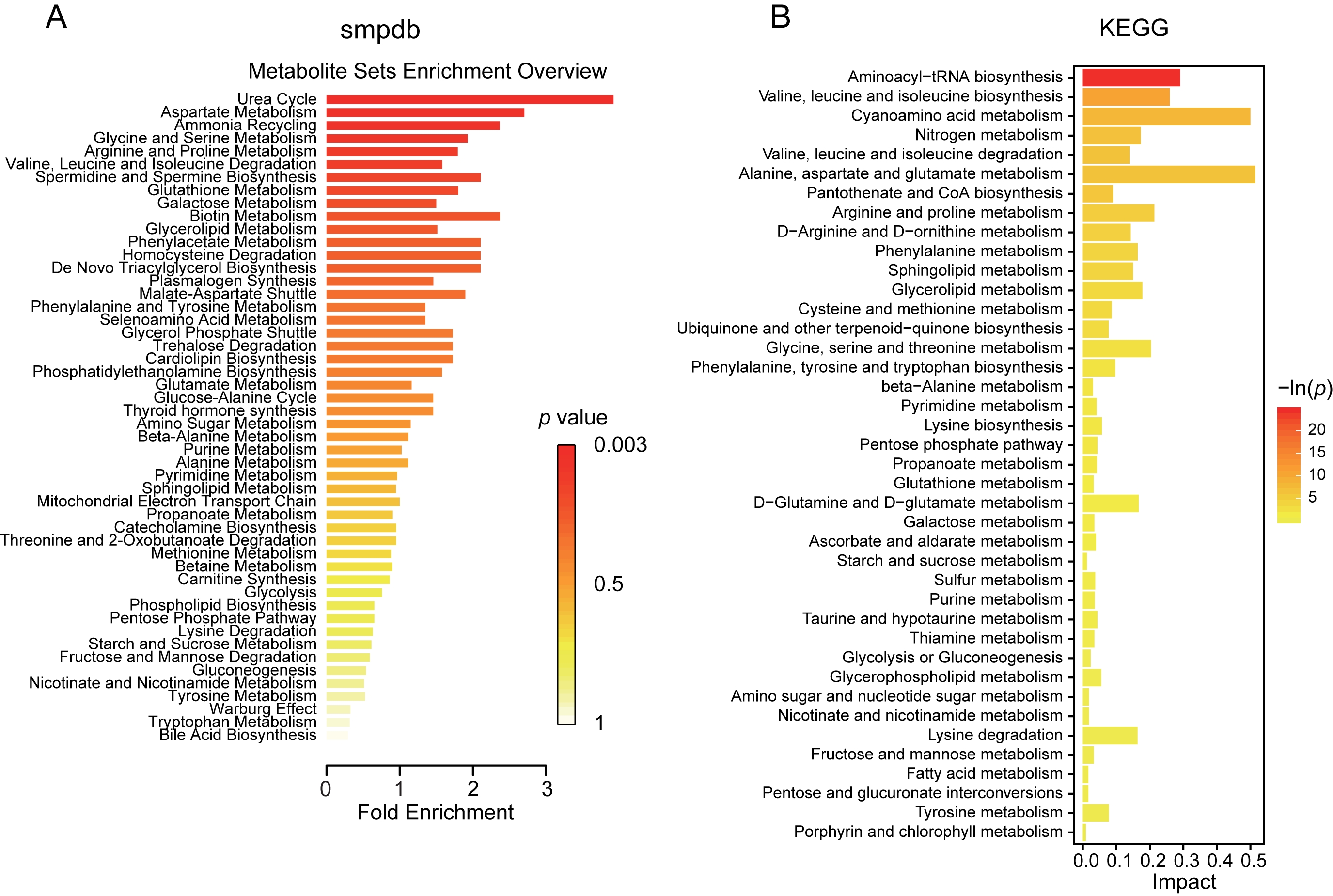 Fig. 5.
Fig. 5.Potential functional metabolic pathways of the altered metabolites between vaccinated and control group. (A) Pathway enrichment analysis and bar plot based on all the altered metabolites was performed using pathway-associated metabolite sets SMPDB. (B) The KEGG pathway terms identified according to the varied metabolites in the vaccinated group were described in the bar plot. KEGG, Kyoto Encyclopedia of Genes and Genomes; SMPDB, Small Molecule Pathway Database.
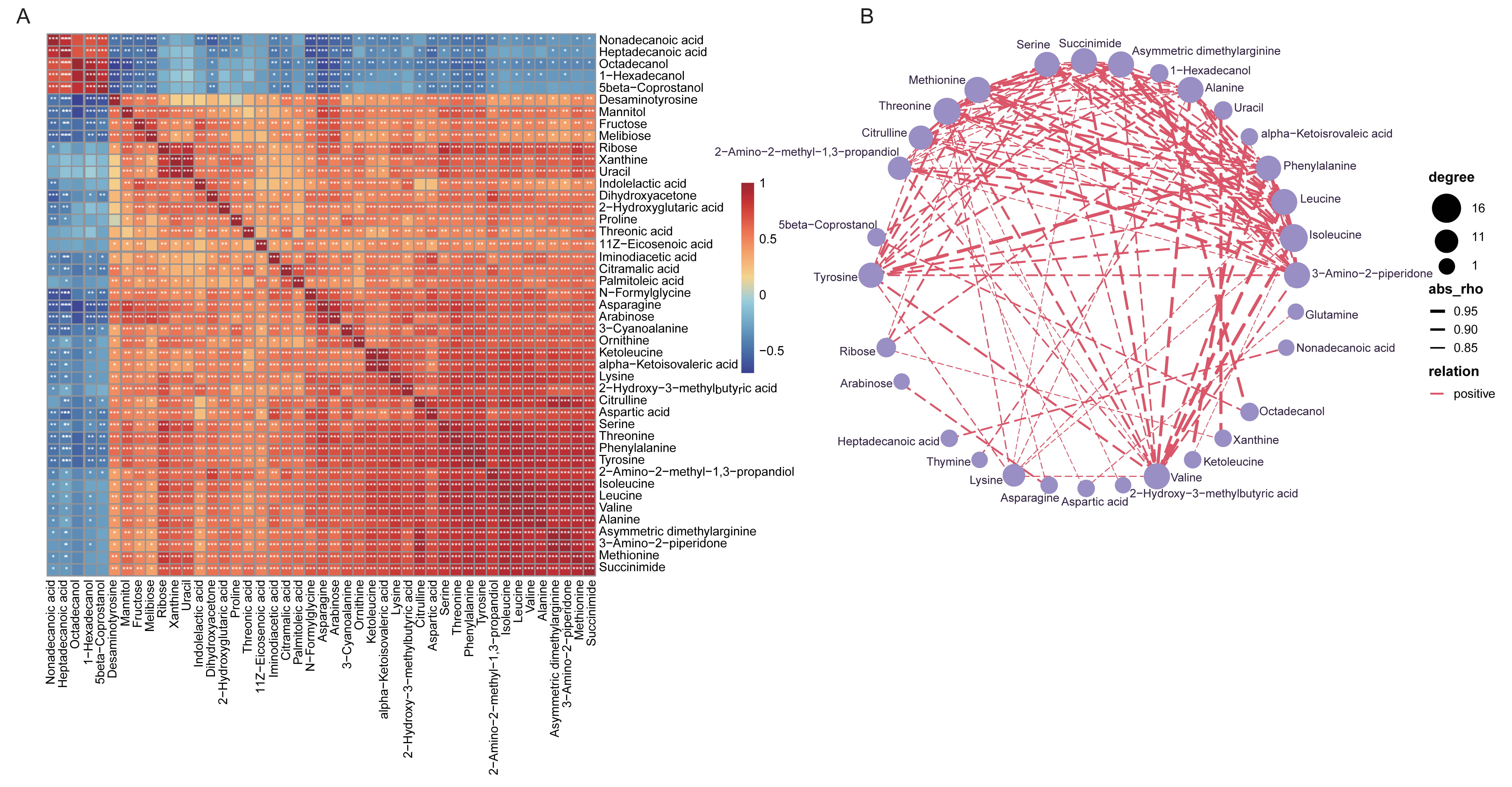
The significant correlations between different metabolic compositions were determined, and Fig. 6A described the positive or negative relationship of each metabolites. Extremely apparent relevance was detected in the majority metabolites with each other, suggesting the shifts by vaccines treatment might be collaborative. The co-occurrence network of all the varied fecal metabolites with Spearman’s correlation coefficients higher than 0.8 was further identified, and all the association were positive (Fig. 6B). For instance, 2-hydroxy-3-methylbutyric acid was positively linked to isoleucine, leucine, phenylalanine, alanine, succinimide and so on; and 4-hydroxycinnamic acid was positively associated with phenylalanine, leucine and isoleucine.
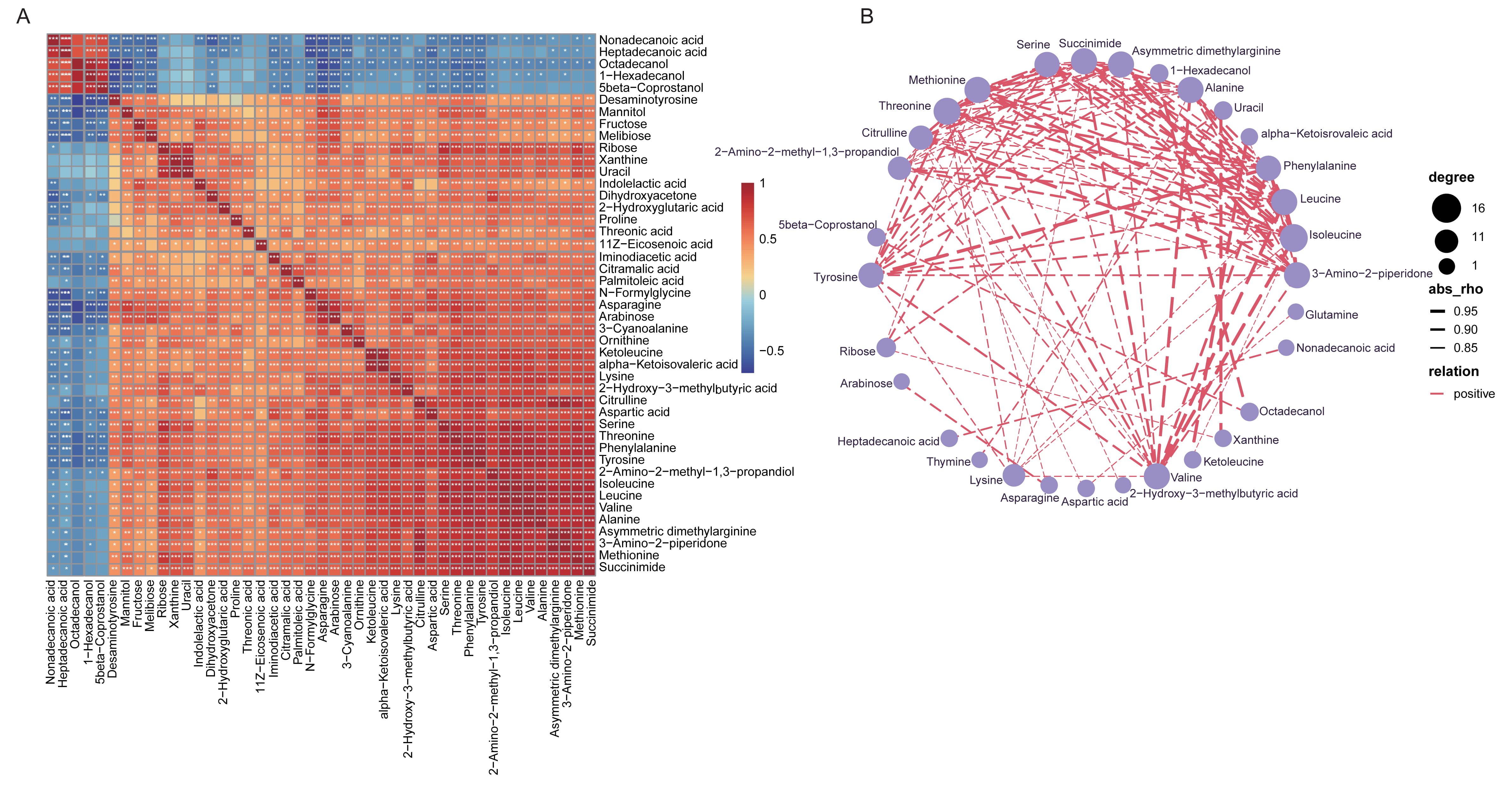 Fig. 6.
Fig. 6.Spearman’s correlation of the top 50 most dominant fecal
metabolites shifted in vaccinated individuals and the network relationship. (A)
Heatmap showing Spearman’s correlation analysis for the co-occurrence profiles of
the top 50 most correlated fecal metabolites altered between groups. The
correlation coefficient was expressed in different colors. Red, positive
correlation; Blue, negative correlation. *, p
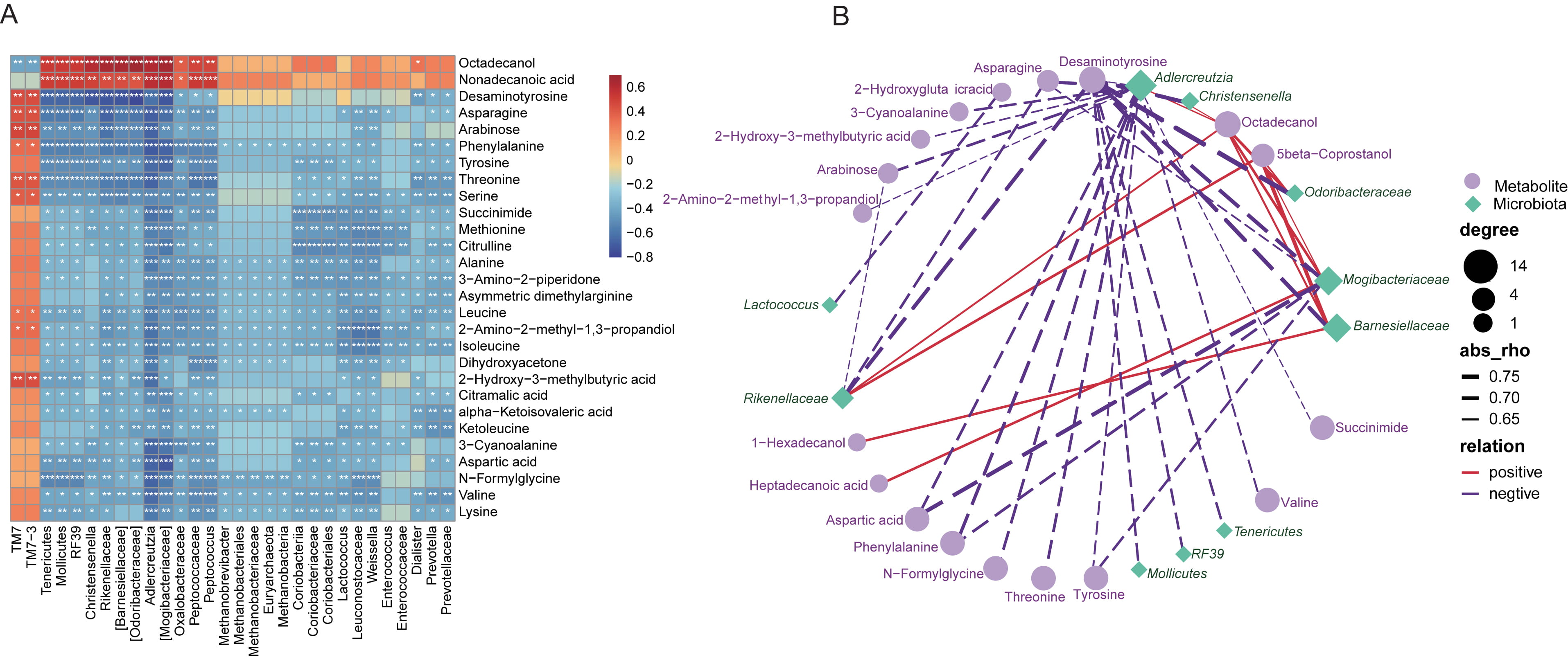
As many fecal metabolites have been indicated to provide functional readout of
intestinal microbiome, we thus performed Spearman’s correlation analysis between
the altered metabolites identified in the present work, and the varied gut
microbiota. Here, it was found that, most of the metabolites were negatively
related with the microbial taxonomic compositions, except for octadecanol and
nonadecanoic acid, TM7 and TM7-3 (Fig. 7A). Again, the
co-occurrence profiles of fecal metabolome and gut microbiome, with correlation
coefficients higher than 0.6, were further indicated in Fig. 7B.
Odoribacteraceae was identified to be negatively associated with
desaminotyrosine, and Lactococcus was negatively linked to asparagine.
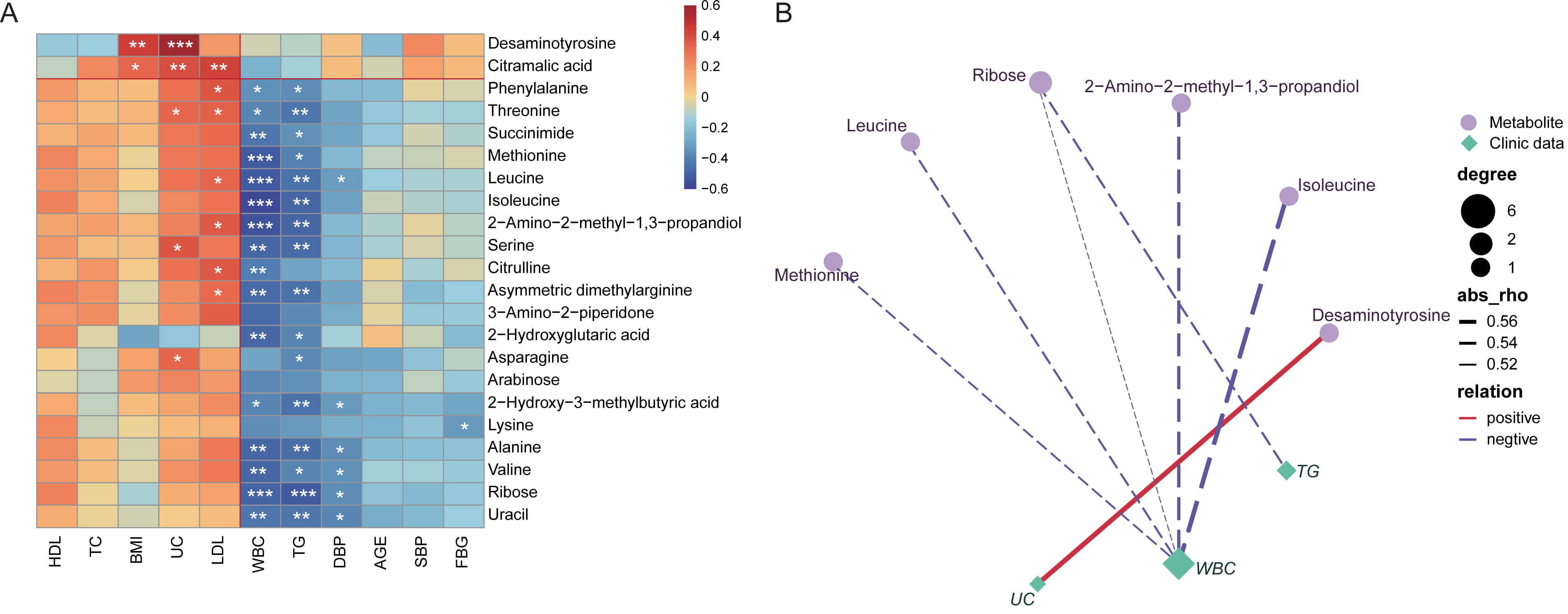
Moreover, almost all the altered fecal metabolites were negatively related to
host levels of triglyceride (TG), white blood cell (WBC) and systolic blood
pressure (SBP), but positively to low-density lipoprotein cholesterol (LDL) (Fig. 8A). Also here, obvious correlations was identified, including positive
association between desaminotyrosine and uric acid (UC), negative relationship
between WBC and 2-Amino-2-methyl-1,3-propandiol, Isoleucine, Leucine, Methionine
and Ribose, respectively. Ribose was observed to be negatively related with TG
levels (Fig. 8B, correlation coefficients
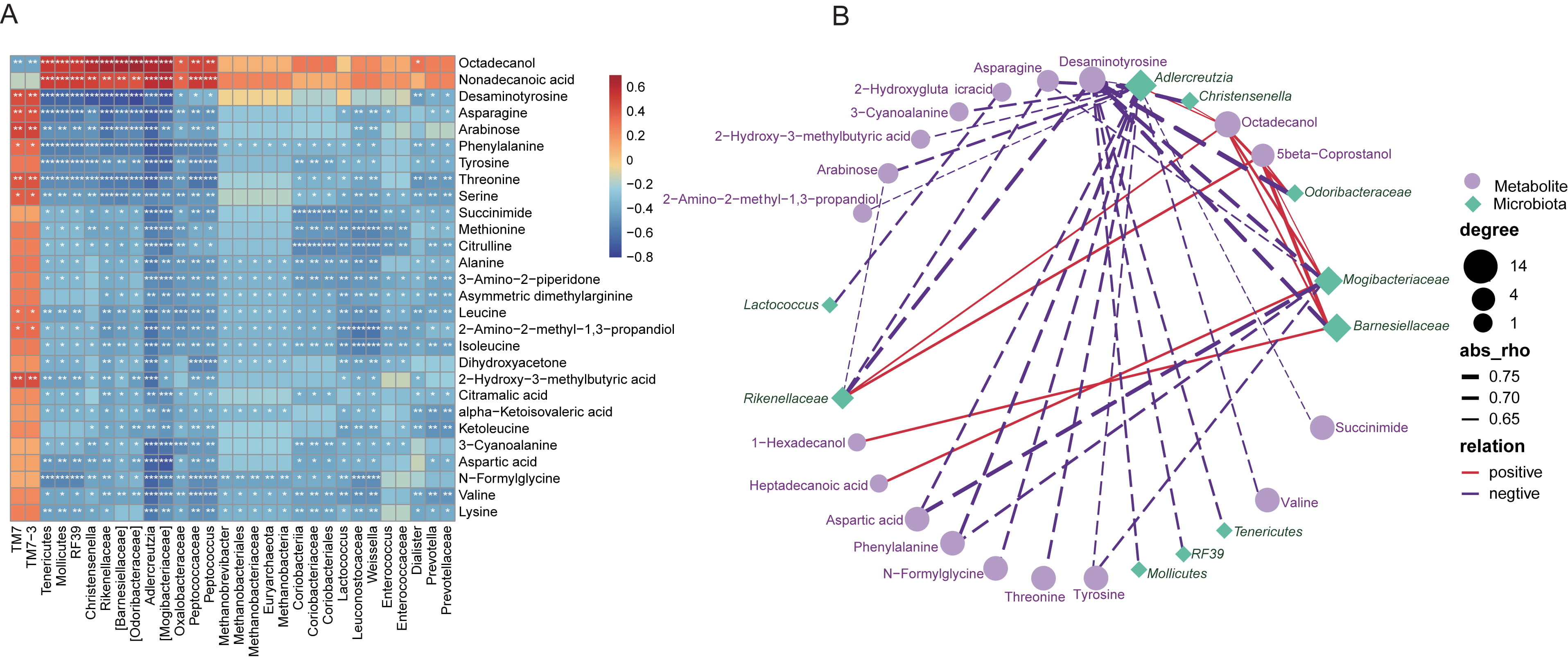 Fig. 7.
Fig. 7.Vaccines-associated fecal metabolites correlated with the gut
microbiota differentiated in vaccinated individuals. (A) Heatmap describing
Spearman’s correlation for the association between the top 30 altered metabolites
and the top 30 most correlated gut microflora shifted in vaccinated subjects. The
correlation coefficient was expressed in colors. Red, positive correlation; Blue,
negative correlation. *, p
 Fig. 8.
Fig. 8.Correlation analysis between the altered fecal metabolites and
clinical characteristics of the study cohort. (A) Spearman’s
correlation analysis of the association between the varied metabolites (most
correlated 22 metabolites) and the clinical characteristics of each participants.
The correlation coefficient was expressed in colors. Red, positive correlation;
Blue, negative correlation. *, p
In the present study, we observed that the fecal metabolome profiles were disparate significantly between subjects receiving SARS-CoV-2 virus vaccines and those unvaccinated. Among a total of 243 metabolites from 27 ontology classes identified in the study, 64 metabolic markers and 15 ontology classes were dramatically distinct between vaccinated and unvaccinated individuals. There were 52 enhanced such as Desaminotyrosine, Phenylalanine, and 12 deficient metabolites such as Octadecanol, 1-Hexadecanol in the vaccinated individuals. Furthermore, correlation analysis indicated that intestinal microbiome was related to alterations in metabolite composition and functions.
Currently, BBIBP-CorV has received emergency use from the WHO, meanwhile it is approved for use in more than 45 countries worldwide [21]. In China, BBIBP-CorV, as one of the major inactivated COVID-19 vaccines, has been shown to be generally safe and elicit effective antibody responses [22, 23]. As of now, accumulating evidence show that the gut microbiome plays an essential role in the progression of COVID-19 [10, 24]. Yeoh YK et al. [9] found that the gut microbiome of COVID-19 patients was strongly disturbed compared with healthy controls, with depletion of beneficial commensal bacteria including Faecalibacterium prausnitzii, Eubacterium rectale and enrichment of conditional pathogenic bacteria such as Clostridium hathewayi, Bacteroides nordii in the gastrointestinal tract. Most recently, investigators indicated that gut microbiome ecology was stratified well with COVID-19 severity. With availability of COVID-19 vaccines, researchers have identified the alterations in oral and intestinal microbiomes of vaccinated individuals [16, 25]. However, the effect of BBIBP-CorV vaccination on metabolites thought to be downstream of gut microbiota has not been determined. Here we for the first time explored the shifts of gut metabolites after vaccination.
Overall, the metabolite profiles of gut microbiota in vaccinated individuals
were significantly different from those within unvaccinated subjects.
Specifically, we found that the expression levels of several metabolites, such as
desaminotyrosine, glutamine, isoleucine, were prominently abundant in vaccine
recipients. Previous study showed that desaminotyrosine could elicit protection
against the influenza virus [26]. Desaminotyrosine was further confirmed by Steed
et al. [27] to suppress influenza and reduce lung immunopathology. In
addition, glutamine is an L-
In addition, in the present work, we have identified that several metabolites were depressed in the gut of vaccinated individuals in comparison with unvaccinated subjects, especially for Heptadecanoic acid etc. Heptadecanoic acid as a kind of odd chain saturated fatty acids, has been known to play an essential role in various important biological and nutritional process [34]. The EPIC-Norfolk prospective study showed that Heptadecanoic acid abundance was negatively associated with coronary heart disease risk [35]. Furthermore, recent observation based on the EPIC and INTERACT studies indicated that Heptadecanoic acid also exhibited a strong inverse correlation with type II diabetes [36]. Based on our findings of suppressed Heptadecanoic acid after SARS-CoV-2 vaccination, it was speculated that the reduction of protective Heptadecanoic acid in response to vaccines might be related to some typical side effects of BBIBP-CorV vaccines.
Previously, study has compared the serum metabolic profiles of pre-vaccination samples with post-vaccination samples from the same individual upon Sinovac COVID-19 vaccines but not BBIBP-CorV vaccines [37]. He M and colleagues [37] examined sera samples from 30 individuals before SARS-CoV-2 vaccination (Sinovac) and 29 thereinto after two-dose vaccination. Under untargeted liquid chromatography-mass spectrometry/mass spectrometry analysis, several metabolites such as L-glutamic acid, gamma-aminobutyric acid, succinic acid, and taurine showed increasing shifts from post-vaccination to pre-vaccination. Moreover, metabolites associated with two-dose vaccination mainly participated in butanoate metabolism and glutamate metabolism. Whereas there remains limited knowledge of alterations in fecal metabolome before and after BBIBP-CorV vaccination, and further attempts to clarify this should be a priority in future studies.
Along with altered metabolic compositions, multiple functional pathways in SMPDB and KEGG were detected to be varied dramatically between groups. Our results indicated that urea cycle; alanine, aspartate, and glutamate metabolism; arginine and proline metabolism; phenylalanine metabolism and tryptophan metabolism were more enriched after vaccination. The urea cycle (ornithine cycle) is a critical amino acid metabolism pathway for waste nitrogen dispose in mammals [38]. Previous studies showed that urea cycle dysregulation plays a crucial role in regulating various metabolic processes, leading to hyperornithinemia, hyperammonemia and gyrate atrophy in humans [39]. Besides, urea cycle dysfunction serves as a biofluids biomarker in cancer patients and is implicated inenhanced response to immune checkpoint therapy [40]. It was quite interesting that, Li T et al. [41] found that among severe COVID-19 patients, the urea cycle is also significantly aberrant. Even in the early stages of severe COVD-19, patients also exhibited abnormal profiles in three amino acid pathways including alanine, aspartate, and glutamate metabolism; arginine and proline metabolism; phenylalanine metabolism. However, when compared with unvaccinated controls, vaccinated subjects showed enrichment in these amino acid pathways mentioned above. The possible reason for this phenomenon might be that the inactivated vaccines enable the adaptive immune system to form a lasting memory of viral component [42], which in turn enabled vaccinated subjects less susceptible to SARS-CoV-2 invasion and more resistant to COVID-19.
As previously reported, carbohydrate and amino acid metabolism are essential for
immune cells to survive, proliferate and function [42]. Our results indicated
that fructose and arabinose of the carbohydrates was positively correlated with
serine and succinimide within the amino acids. Fructose is indicated to be an
important cause of obesity and obesity-related cardiometabolic complications
[43, 44]. Evidence suggested that fructose-induced obesity showed higher level of
chronic inflammation, and accumulation of macrophages in adipose tissues, which
are responsible for the production of TNF-
In the present study, it was found that the majority altered fecal metabolites were negatively related to intestinal microbiota. Enterococcus is an important opportunistic pathogen causing a wide variety of infections [50], which is extremely enriched in various diseases such as coronary artery disease [51] and infective endocarditis [52]. Previous study has detected negative correlation between Enterococcus and isoleucine, and total amino acids in animal models [53]. We also found that Enterococcus was negatively linked with several essential amino acid metabolites that were elevated in the vaccinated group, such as Isoleucine, Leucine and Methionine. It was speculated that upon COVID-19 vaccines, shifts of intestinal flora might directly or indirectly affect the profiles of metabolites and thus play a role in immune regulation.
Branched-chain amino acids including Leucine, Isoleucine and Valine, exert a vital role in modulating body weight and muscle protein synthesis [54]. Ma Q et al. [55] suggested that Leucine, Isoleucine or their combination in drinking water significantly decreased relative white adipose tissue weight, serum TG and free fatty acid compared with mice fed with high fat diet. These observations are in line with our findings of the negative relationship between TG and Leucine, Isoleucine, Valine, respectively. Furthermore, previous study indicated that desaminotyrosine exhibited obvious toxicities to the kidney at a higher dose [56]. Oral administration of high-dose desaminotyrosine significantly affected the renal function indexes by elevating blood urea nitrogen and creatinine in animals. Moreover, histopathological analysis uncovered severe granules and vacuolar degeneration of renal tubular epithelial cells and interstitial edema in response to desaminotyrosine [56]. It is well known that enhanced level of UC is important risk factor for kidney disease [57, 58]. Here we demonstrate a positive correlation between UC and desaminotyrosine, the causality of which requires further confirmation.
In current study, we found that COVID-19 vaccines elicited alterations in multiple metabolites, which exerted potential roles in differential functional pathways, and might further correlated with blood antibody and cytokine response. The varied fecal metabolites were revealed to mainly function in the processes of amino acid metabolism. For example, succinic acid semialdehyde, L-glutamic acid, Gamma-Aminobutyric acid, succinic acid and oxoglutaric acid were inalanine, aspartate and glutamate metabolism, and L-glutamic acid, oxoglutaric acid were in D-Glutamine and D-glutamate metabolism. It has been documented that inactivated SARS-CoV-2 vaccines could produce rapid and strong antibody responses, such as IgG, making them critical during the pandemic of COVID-19 [5]. Previous evidence suggested that metabolites L-glutamic acid, succinic acid semialdehyde and succinic acid involved in the alanine, aspartate and glutamate metabolism, as well as D-Glutamine and D-glutamate metabolism pathways identified in our study were positively correlated with the levels of IgM, IgG or IgA [37]. This information facilitates us to speculate that the highlighted metabolites and metabolic pathways might be closely related to body’s immune responses after immunization.
Understanding how SARS-CoV-2 vaccines affect the composition of metabolites downstream of gut microbiota may provide important information regarding alterations within the gut environment following immune responses to SARS-CoV-2 infection. Nevertheless, there are several limitations in the current study. Firstly, the data interpretation might be limited due to relatively small sample size, and the conclusions need to be validated by larger studies. Additionally, in the present study, samples from a single time point after vaccination were analyzed, and the composition of intestinal metabolites might have varied at different time points after vaccination. Thus, further studies including sampling at different time points after vaccination could provide more reliable information on temporal function due to COVID-19, reveal the effect of COVID-19 vaccination on the metabolic composition of gut mcirobiota, and elucidate the underlying mechanisms.
The present study depicted the alterations in the gut metabolome after COVID-19 vaccination based on GC-TOF/MS methods. Our observations support an association between the gut microbiome, metabolite perturbation, and COVID-19 vaccination, and provide valuable resource for in-depth exploration of metababolic profiles in vaccinated individuals, understanding the complex mechanisms between gut metabolite and SARS-CoV-2 virus vaccines.
Data sets used and analysed during the current study are available from the corresponding author on reasonable request.
JL, MC, YD and YS—conceived the study, directed the project, designed the experiments. JL, YD and YS—interpreted the results and wrote the manuscript. PW, JJ, YS, MC—recruited and collected the clinical details from the subjects. JL and YD—analyzed the data. JL, MC, YD and YS—revised the manuscript. All authors contributed to editorial changes in the manuscript. All authors read and approved the final manuscript.
This study was performed in accordance with the Helsinki declaration, and was approved by the Medical Ethics Committee from Beijing Chaoyang Hospital. Written informed consent was obtained from all study participants prior to enrollment (ethical number: 2021-ke-440).
Not applicable.
This research received no external funding.
The authors declare no conflict of interest.
Supplementary material associated with this article can be found, in the online version, at https://doi.org/10.31083/j.fbl2804065.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.