

1 Beijing Institute of Dental Research, Beijing Stomatological Hospital & School of Stomatology, Capital Medical University, 100050 Beijing, China
Abstract
Tumor immunity is a cycle that begins with the release of antigens from tumor cells and ends with the destruction of tumor cells. High mobility group box 1 (HMGB1) is a nonhistone protein widely present in the nucleus of mammalian cells and can be released by immune cells or tumor cells. As a proinflammatory mediator or alarm protein, the activity and function of HMGB1 are determined by the environment, binding receptors, redox status and posttranslational modifications (PTMs), and HMGB1 plays a key role in inflammation and tumor immune processes. In this review, we summarize in detail the current studies on the dual role of HMGB1 in tumor immunity, focusing mainly on immunosuppressive effects, such as regulatory T cells (Tregs), myeloid-derived suppressor cells (MDSCs) and tumor-associated macrophages (TAMs), as well as antitumor immunoenhancement effects, such as immunogenic cell death (ICD). Finally, we discuss the potential and challenges of HMGB1 in antitumor immunotherapy.
Keywords
- HMGB1
- tumor immunity
- MDSCs
- Tregs
- TAMs
- ICD
The burden of cancer incidence and mortality is rapidly increasing and is becoming a serious threat to human survival and quality of life, especially in developing countries [1]. Immunotherapy is gaining increasing attention as a powerful clinical strategy for combating cancer. However, these therapies also have serious side effects, including autoimmunity and nonspecific inflammation, preventing the further application of cancer immunotherapy [2]. Therefore, it is especially important to further investigate the related mechanisms of tumor immunity. Chen DS et al. [3] proposed that the cyclic process of tumor immunity can be divided into seven major steps, beginning with the release of antigen by tumor cells (step 1) and finishing with the killing of tumor cells (step 7). Antigen-presenting cells (APCs), such as dendritic cells (DCs), recognize cancer-associated antigens (step 2), initiate and activate cytotoxic T lymphocyte (CTL) responses (step 3 and step 4), and infiltrate the tumor stroma to recognize and kill tumor cells (step 5 and step 6). Then, T-cell-mediated cytotoxic responses release new tumor antigens and promote the tumor-immune cycle. However, there is negative feedback to impede the above steps during cancer development. Therefore, it is crucial to study the related mechanisms and processes activating the tumor-immune cycle, including immunogenic cell death (ICD), APC maturation, T-cell response activation, immune cell infiltration, attenuation of immunosuppression, and enhancement of effector T-cell activity [4].
High mobility group box 1 (HMGB1), a nonhistone protein, plays an important role in cancer immunity. HMGB1 consists of three distinct structural domains, including two crosstalk HMG box structural domains (A and B boxes) and one 30 amino acid-long acidic C-terminal tail [5]. Among these, the B box regulates cytokine activity and induces the secretion of proinflammatory cytokines by macrophages [6], which can be antagonized by the A box [7]. The C-tail can bind both the A and B boxes and contribute to the regulation of HMGB1 binding to DNA [8]. The functions of intracellular HMGB1 (as a DNA binding protein) and extracellular HMGB1 (as a cytokine) are very different. Intracellular HMGB1, a highly conserved nuclear protein, maintains nucleosome and telomere homeostasis, regulates DNA repair and gene transcription by binding transcription factors, and plays the role of a tumor suppressor gene [9, 10, 11]. For example, studies have shown that HMGB1 exerts an antitumor role during K-Ras-driven pancreatic carcinogenesis and chemotherapy in breast cancer [12, 13]. Extracellular HMGB1 acts as an inflammatory factor and exerts pro- or antitumor effects in different ways. For instance, released HMGB1 not only contributes to ICD-induced anticancer immunity in early chemotherapy but also promotes the survival and recurrence of residual cancer cells in late chemotherapy. In this review, we focus on the Janus-faced roles of HMGB1 as an immunomodulator in tumor immunity and then provide additional possibilities for targeting HMGB1 in tumor immunotherapy. Specifically, we focus on the functions of secreted HMGB1 in regulatory T cells (Tregs), myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs) and ICD.
HMGB1 is normally transferred to the extracellular environment as an alarm protein in response to multiple stimuli (inflammation, hypoxia and stress). However, due to the absence of signal peptide sequences, HMGB1 is not secreted by the classical protein secretion pathway but is translocated from the nucleus to the cytoplasm and then secreted via secretory lysosome-mediated exocytosis [14]. There are usually two secretory pathways. On the one hand, as one of damage-associated molecular patterns (DAMPs), HMGB1 is passively and directly released out of cells during apoptosis or necrosis and can become an adjuvant or danger signal for the immune system [15]. On the other hand, many cancer cells and immunoreactive cells also actively secrete HMGB1, which can be detected in cancer patient serum [16]. Furthermore, multiple forms of posttranslational modifications (PTMs) can also regulate the secretion of HMGB1, such as phosphorylation, methylation, acetylation, glycosylation and oxidation. For example, methylation alters the conformation of HMGB1, attenuating its ability to bind to DNA [17]. Additionally, extracellular HMGB1 delivers danger signals to surrounding cells and performs a variety of pathological and physiological functions by binding to classical receptors including receptor for advanced glycation end products (RAGE) and Toll-like receptor 2/4 (TLR2/4) [18].
HMGB1 can be passively released after various types of cell death, including apoptosis, necrosis, ferroptosis, pyroptosis, and other types of cell death, in response to various stimuli or injuries. The passive release of HMGB1 can be influenced by various factors and conditions, such as poly ADP-ribose polymerase 1 (PARP1), receptor-interacting serine-threonine kinase 3 (RIPK3), cathepsin, antioxidant enzymes, caspases, and deoxyribonucleases (DNases) [19].
Studies have reported that DNA-damaging drugs can induce HMGB1 release from
necrotic cells by activating PARP1. In contrast, knockout of PARP1 in mouse
fibroblasts attenuated the release of HMGB1 during the above process [20, 21]. In
addition, RIPK1, a key promoter of necrosis, plays an important role in the
formation of necrosomes. In RIPK3
Immune cells or cancer cells can actively secrete HMGB1 in response to
endogenous or exogenous stimuli, such as lipopolysaccharide (LPS),
interferon-
First, ROS not only affect the passive release of HMGB1 but also regulate its
active secretion. For example, paclitaxel can upregulate the levels of ROS and
activate p38 mitogen-activated protein kinase 1 (MAPK1)-nuclear
factor-
MDSCs, also known as immature myeloid cells, are heterogeneous cells derived
from bone marrow (BM) and play a pivotal role in immunosuppression. MDSCs can
impair protective T-cell immunity in cancers and participate in tumor immune
escape, as well as promote tumor angiogenesis and invasion and enhance tumor cell
stemness [38, 39]. The expansion and activation of MDSCs are controlled by
multiple growth factors and cytokines derived from the stroma, tumor cells and
immune cells. Among them, granulocyte macrophage colony stimulating factor
(GM-CSF), vascular endothelial growth factor (VEGF) and granulocyte colony
stimulating factor (G-CSF) are the most critical cytokines for MDSC expansion
[40]. In addition, interleukin 6 (IL-6) and tumor necrosis factor-
Secreted HMGB1 is an essential cytokine in the TME and can also affect the
differentiation, activation and survival of MDSCs. Hubert P et al. [44]
examined tissue specimens of human invasive breast, lung and cervical cancers and
found that HMGB1 was mostly observed in the cytoplasm, while the nucleus was
stained in normal tissues, suggesting that malignant tumors may secrete HMGB1.
Targeting extracellular HMGB1 significantly reduced MDSCs and inhibited
reactivated antitumor immune responses. Li J et al. [45] provided
evidence that HMGB1 may mediate renal cell carcinoma immune escape by promoting
MDSC proliferation. The authors found that downregulation of HMGB1 significantly
inhibited tumor growth and prolonged survival in Renca-bearing mice treated with
Tumor-derived exosomes are extracellular vesicles that can send signals to
immune cells in the TME, leading to immune escape and metastasis of tumor cells
[52]. HMGB1 is also expressed on exosomes, and exosomes carrying HMGB1 are also
involved in the regulation of MDSCs. Hepatocellular carcinoma (HCC) cell
exosome-derived HMGB1 promotes the activation of B cells and the expansion of
T-cell Ig and mucin domain (TIM)-1
| Cancer types | Immune cells | HMGB1 effect | HMGB1 inhibitor | Signaling pathway | Ref |
| Renal cell carcinoma | MDSC proliferation and recruitment | Immune escape | Gr-1 antibody | - | [45] |
| Colorectal cancer | MDSC recruitment | Peritoneal metastasis | A Box | - | [46] |
| Lewis lung carcinoma | Inhibition of M-MDSC migration and facilitation of M-MDSC differentiation | Improved prognosis | Resveratrol | Apoptosis | [47] |
| Melanoma | Attracted and activated MDSCs | Tumor metastasis | - | - | [48] |
| Mouse tumors | Enhanced the differentiation of BM progenitor cells toward MDSCs | Tumor progression | A Box | RAGE | [50] |
| HCC | Activation of B cells and expansion of TIM-1 |
Tumor immune escape | - | TLR2/4 and MAPK pathways | [53] |
| Mouse thymoma | Breg differentiation | Tumor growth | MyD88-NF- |
[54] | |
| Gastric cancer | Activated neutrophils | Tumor metastasis | TLR4/NF- |
[55] |
HMGB1, high mobility group box 1; MDSC, myeloid-derived suppressor cells; BM, bone marrow; HCC, hepatocellular carcinoma; TIM, T-cell Ig and mucin domain.
HMGB1 not only affects the activation and differentiation of MDSCs, but also
regulates MDSC survival through autophagy. Autophagy is a basic lysosomal
catabolic pathway that prolongs MDSC survival and is a key pathway for
MDSC-mediated immunosuppression [56]. Parker KH et al. [57] cultured
MDSCs under starvation conditions and treated them with an autophagy antagonist
(bafilomycin) or HMGB1 antagonist (ethyl pyruvate) and found that HMGB1 promoted
autophagic MDSC survival and modulated the autophagic flux of MDSCs. However,
autophagy in the TME also attenuated the inhibitory activity of MDSCs on
CD4
Tregs represent another important immunosuppressive cell type in the TME. In the
1970s, Tregs were originally defined by Gershon et al. [59] and
described as suppressor T cells that functionally inhibit the immune response by
impacting the activity of other cells [60]. Among these, classical Tregs are
thymus-derived CD4
| Cancer types | Immune cells | HMGB1 effect | HMGB1 inhibitor | Ref |
| HNSCC | Promoted Tregs migration | Counteracted anticancer immuninty | - | [63] |
| Mouse breast and lung cancer | Promoted IL-10 production by Tregs | Inhibited anticancer immunity | - | [64] |
| Mouse breast cancer | Tumor-associated Tregs activation | Inhibited immunotherapy | - | [65] |
| Neuroblastoma | Tregs differentiation | Suppression of antitumor immunity | Anti-HMGB1 antibody | [66] |
HNSCC, head and neck squamous cell carcinoma; IL, interleukin.
Macrophages are common immune cells and can be activated into proinflammatory
macrophages (M1) by the classical pathway and pro-tumor macrophages (M2) by the
nonclassical pathway [69]. M1 and M2 subtypes are dynamic and modifiable. TAMs,
high jacked by tumors, are a major component of the TME and play an important
role in tumor growth, metastasis, angiogenesis and immunosuppression. It has been
reported that TAMs resemble M2 macrophages but are not completely M2 isoforms and
consist of different proportions of M1 and M2 macrophages at different stages
[70]. In many solid tumors, TAMs have been reported to be associated with poor
patient prognosis and to facilitate tumor progression [70, 71, 72]. In epithelial
ovarian cancer (EOC), HMGB1 overexpression is associated with TAM infiltration
and lymph node metastasis, and in vitro HMGB1 may combine with TAMs and
promote lymphangiogenic potential [73]. A retrospective study found that the
perineural HMGB1 levels in HCC positively correlated with TAM infiltration and
may have important prognostic value [74]. In another study, the HMGB1/TLR2/NADPH
oxidase 2 (NOX2)/autophagy axis was reported to activate M2 macrophages in HCC
[75]. Chen H et al. [76] found that cancer-released HMGB1 may act on
CD68- and CD163-positive M2-like macrophages to promote tumor progression by
staining human fibroblast sarcoma tissues. In esophageal adenocarcinoma, HMGB1
binding to TLR2 activated M2 macrophages in vitro [77]. Additionally,
HMGB1 activated RAGE signaling to enhance the anti-inflammatory and protumor
effects of M2-like TAMs, which was different from activating the classical
NF-
HMGB1 is also closely associated with TAM-mediated treatment tolerance. In
cervical cancer, septin 9 (SEPT9) can interact with HMGB1 to modulate macrophage
polarization and promote irradiation resistance [11]. In prostate cancer, HMGB1
is essential for enzalutamide-resistant TAM activation and neuroendocrine
differentiation. HMGB1-activated macrophages secrete IL-6, which directly
promotes HMGB1 transcription by STAT3 signaling [83]. In pancreatic ductal
adenocarcinoma (PDAC), TAMs mediate drug resistance by upregulating HMGB1
expression [84]. In colorectal cancer, activated HIF1
| Cancer types | Immune cells | HMGB1 effect | Effect of HMGB1 inhibitor | Signaling pathway | Ref |
| HCC | Activated M2 macrophages | Induced poor prognosis | - | TLR2/NOX2/autophagy | [75] |
| Fibroblast sarcoma | M2-like macrophages | Tumor progression | - | - | [76] |
| Esophageal adenocarcinoma | Activated M2 macrophages | Tumor progression | - | TLR2 | [77] |
| Glioblastoma | Enhanced M1-like polarization of TAMs | Enhanced temozolomide sensitivity | - | RAGE-NF- |
[81] |
| Mouse bladder cancer | Increased M1-like TAMs | - | Glycyrrhizin, antitumor | - | [82] |
TAM, tumor-associated macrophages; M1, proinflammatory macrophages; M2, pro-tumor macrophages.
ICD is one of the main approaches to kill tumor cells in tumor therapy.
Specifically, ICD is a specific functional type of regulated cell death (RCD)
that activates an adaptive immune response to antigens expressed by dying cells.
ICD can be triggered by viral infections, radiotherapy, chemotherapy and
photodynamic therapy (PDT). The molecular hallmark of ICD is the release of DAMPs
from dying cancer cells, which are recognized by innate pattern recognition
receptors (PRRs), such as TLRs, thereby activating an antitumor-specific immune
response [86]. HMGB1, as an important immunostimulatory DAMP, plays an important
role in ICD released from the nucleus to surrounding dead cells. However, the
functions of intranuclear HMGB1 and released HMGB1 are completely different. Vaes
RDW et al. [87] indicated that upregulated levels of intranuclear HMGB1
may perform an important function in radiotherapy-induced resistance and
therefore cannot serve as a suitable marker for radiotherapy-induced ICD.
However, many studies have shown that chemoradiotherapy leads to the release of
HMGB1 in the TME, and released HMGB1 is essential for ICD immunogenicity [88]. A
prospective study of patients with advanced lung cancer showed that systemic
anticancer treatment (specifically platinum-based chemotherapy) increased the
serum levels of HMGB1 in patients, possibly slowing tumor growth by promoting ICD
[89]. Bains SJ et al. [90] showed that increased HMGB1 expression during
neoadjuvant chemotherapy was positively associated with distant metastasis-free
survival (DMFS) in colorectal cancers. In esophageal squamous cell carcinoma
(ESCC), chemoradiotherapy elevated HMGB1 levels in patient serum and increased
HMGB1 expression in the TME, thereby promoting patient survival. The improved
prognosis may be due to the ability of HMGB1 to mature DCs and further induce
tumor antigen-specific T-cell responses [91]. Similarly, it has been reported
that overexpression of mitochondrial translation initiation factor 2 (MTIF2) in
HCC cells reduces the release of HMGB1, inhibites DC maturation and CD8
However, in some other studies, HMGB1 was released following tumor treatment (radiotherapy or chemotherapy) and played the converse role. For example, serum HMGB1 levels were evaluated during chemoradiotherapy, but were not associated with the prognosis of patients with non-small cell lung cancer (NSCLC) [99]. In a mouse colon cancer model, serum HMGB1 levels were evaluated in mice after doxorubicin treatment, which promoted tumor cell regeneration and metastasis that could be inhibited through treatment with anti-HMGB1 antibody [100]. In another study, HMGB1 was negatively associated with chemotherapy-induced antitumor responses by interacting with DC-specific T-cell immunoglobulin domain and mucin domain 3 (TIM-3) (Table 4, Ref. [91, 92, 93, 94, 101]). The combination of a STAT3 inhibitor (stattic) and doxorubicin increased HMGB1 levels in melanoma and colon cancer cell media and promoted the secretion of the cytokine IL-12 in DCs, a marker of DC maturation and induction of the Th1 response, compared to monotherapies in vitro. This result suggests that inhibiting STAT3 promoted doxorubicin-induced ICD [102].
| Cancer types | HMGB1 effect | Effect of HMGB1 inhibitor | Signaling pathway | Ref |
| ESCC | Matured DCs and induced tumor antigen-specific T-cell responses | - | - | [91] |
| HCC | - | MTIF2 overexpression, impaired 5-Fluorouracil-induced ICD | Reduced the release of HMGB1 | [92] |
| Rectal cancer | Inducing T-cell immune responses | - | TLR4 | [93] |
| - | DC maturation | Neutralizing Abs | RAGE, MAPK and NF- |
[94] |
| Mouse Lewis lung cancer, colorectal adenocarcinoma | Diminishing the immunogenicity from dying tumor cells | - | Interacting with TIM-3 | [101] |
DC, dendritic cells; ESCC, esophageal squamous cell carcinoma; ICD, immunogenic cell death.
These contradictory results may be related to the different levels of HMGB1 in patients after treatment, where HMGB1 levels may be elevated or decreased. For example, in a clinical study of 11 patients with advanced HNSCC, tumor recurrence was observed in patients with downregulated HMGB1 levels after radiotherapy, but not in patients with upregulated HMGB1 levels. Moreover, HMGB1 was negatively correlated with gross tumor volume during treatment and positively correlated with grade three toxicity (RTOG), infection and C-reactive protein (CRP) levels [103]. Additionally, different froms of cell death induced by HMGB1 may contribute to the opposite effects. For instance, it has been reported that reduced HMGB1 can bind to RAGE and induce Beclin1-dependent autophagy to further promote tumor tolerance to chemoradiotherapy. On the other hand, oxidized HMGB1 can increase drug cytotoxicity by inducing caspase-9/-3-dependent apoptosis [104].
Next, the redox status of HMGB1 may also be another important influencing factor. HMGB1, a redox-sensitive DAMP, contains three cysteines (Cys23, Cys45 and Cys106) located within the binding regions of the A box and B box , and the redox status of each of these residues is critical for the secretion and function of HMGB1. First, the fully reduced state of HMGB1 is defined as the presence of all three cysteines in a fully reduced state in inactive cells with thiol groups (all-thiol HMGB1). All-thiol HMGB1 can exert chemotactic effects by binding to C-X-C Motif Chemokine Ligand 12 (CXCL12) and enhancing C-X-C chemokine receptor type 4 (CXCR4, CXCL12 receptor) signaling [105]. Second, the mildly oxidized state of HMGB1 refers to the disulfide bond between Cys23 and Cys45 while maintaining the reduced form of Cys106 (disulfide HMGB1). Disulfide HMGB1 is the single redox form of HMGB1 that is able to bind to the myeloid differentiation factor 2 (MD-2, TLR4 coreceptor)/TLR4 receptor complex to exert cytokine effects [106]. For example, HMGB1 released by neurons is likely the disulfide form that induces inflammation and pain by TLR4 [107, 108]. Third, the fully oxidized state of HMGB1 is defined as the full oxidation of all cysteine residues into sulfonyl groups (sulfonyl HMGB1). Sulfonyl HMGB1 was often considered an inactive molecule in the past, but a recent study has shown that sulfonyl HMGB1 acts as a tolerance signal on antigen-presenting cells in a RAGE-dependent manner and is involved in the recruitment of immune cells that inhibit cytotoxic cells, thereby attenuating their ability to kill tumor cells. Further studies revealed that sulfonyl HMGB1 can promote the accumulation of Tregs and MDSCs, increase the ratio of M2/M1 macrophages, and enhance the tolerance of dendritic cells [44]. Thus, it can be concluded that the fully reduced form of HMGB1 in three cysteines acted as the chemoattractant, whereas the disulfide-bonded HMGB1 acted as a proinflammatory cytokine, and further oxidation of cysteine to sulfonate (mediated by ROS) inhibited immunostimulatory activity and promoted dead cell-induced immune tolerance [109, 110]. Additionally, the extracellular redox state in tumors is highly variable and different extracellular redox states might occur in different cancers or in different stages of the same cancer type [111]. Thus, these conflicting results were mostly observed in established tumors, probably due to the higher basal levels of ROS in tumor tissues than in normal tissues [112]. ROS induce the release of HMGB1 [113] and may oxidize it to the sulfonate form. This may be partly responsible for the suppression of antitumor immunity by HMGB1 in some cancers. However, there are still aspects that need to be further investigated, such as PTMs, which can likewise affect the activity and function of HMGB1.
The identification of prognostic biomarkers is important to distinguish high-risk patients from others on individualized therapy. HMGB1 exerts both pro- and antitumor effects in tumorigenesis and immunotherapy, depending on the environment, binding receptors, redox states, and PTMs. Thus, the role of HMGB1 in the prognosis of tumor patients is complex. One meta-analysis of seven studies reporting six different tumor types showed that high expression of HMGB1 is associated with poor progression-free survival (PFS) in patients with different tumors, including HCC, nasopharyngeal carcinoma, HNSCC, bladder cancer, prostate cancer, and cervical cancer [114]. Other studies reported that HMGB1 expression is negatively associated with the prognosis in patients with gastric cancer, neuroblastoma, and fibroblast sarcoma [55, 66, 76]. In contrast, high HMGB1 expression is positively correlated with the prognosis of patients with glioblastoma, rectal cancer, esophageal squamous cell carcinoma, and Lewis lung carcinoma [81, 91, 93]. Since HMGB1 is not only located intracellularly, it can also function as a cytokine in the extracellular environment. Further studies revealed that elevated HMGB1 levels in patient serum are highly associated with advanced stages of cervical cancer, pancreatic cancer, and malignant pleural mesothelioma [115, 116, 117, 118]. This suggests that both HMGB1 expression in cancer tissue and cancer patient serum could be a sensitive prognostic factor.
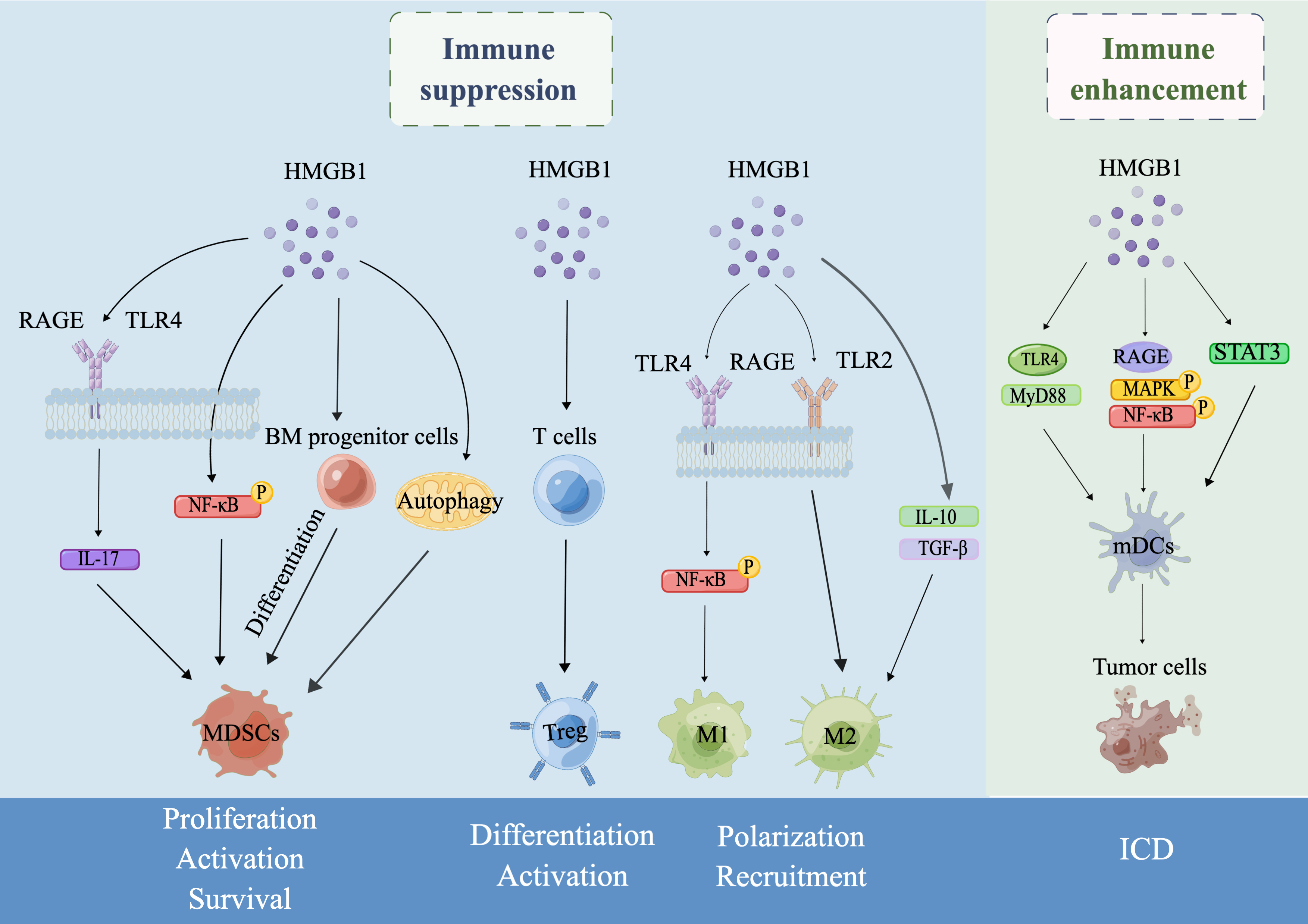
The TME is a highly complex and dynamic microenvironment that consists of tumor cells, innate cells, endothelial cells, lymphocytes and stromal cells [119]. The aim of tumor immunotherapy is to eliminate tumor cells in the TME by reactivating and maintaining the tumor-immune cycle and restoring host antitumor immune responses. Herein, we summarized the dual role of HMGB1 as an immunotherapeutic target. In brief, HMGB1 can exert immunosuppressive effects by promoting the proliferation, activation and survival of MDSCs, inducing the differentiation and activation of Tregs, and promoting the polarization and recruitment of TAMs. On the other hand, HMGB1 can also enhance antitumor immune responses in the host by participating in ICD produced by chemoradiotherapy, infections and PDT (Fig. 1). HMGB1 has been demonstrated to be a promising treatment target in experimental models for multiple inflammatory and infectious diseases thus far [120]. However, clinical trials targeting HMGB1 have been limited to glycyrrhizic acid (HMGB1 inhibitor) for the treatment of depression [121], and there are no clinical trials of drugs directly targeting HMGB1 for the treatment of cancers. Therefore, some main issues, which are also challenges for basic research or tumor immunotherapy, remain that need to be studied further. First, since the passive release and active secretion of HMGB1 can be adjusted at different levels, it is necessary to distinguish the active secretion mechanism and passive release mechanism of HMGB1 and whether they affect the role of HMGB1 in the tumor immunity. Second, it is unclear whether PTMs and redox states affect the role of HMGB1 in tumor immunity. Third, autophagy is a strictly catabolic pathway that plays an important role in cancer. However, it is unclear how HMGB1 is involved in the regulation of both MDSC survival and functional activity in the tumor microenvironment through autophagy. Therefore, the specific mechanism of autophagy in the regulation of MDSC survival and function through HMGB1 needs to be further investigated, which is important for understanding immunosuppression in tumorigenesis. Fourth, since HMGB1 localized at different locations plays different roles in the induction of Tregs, it is important to study the specific mechanisms involved separately. Fifth, it is unclear whether HMGB1 regulation of both M1 and M2 polarization in TAMs generated from the same cancer is associated with tumor stage. Sixth, the specific pathway of HMGB1, an important marker of ICD, that drives the occurrence and progression of ICD remains unclear. Since HMGB1 has a seemingly contradictory role in ICD, we speculate whether it is related to different cancer types or ICD inducers. The dosage of ICD-inducing agents is also a factor that needs to be focused on to avoid the occurrence of cytotoxicity. Therefore, the role of HMGB1 in ICD may be one of the main research directions for tumor immunotherapy in the future. Finally, considering that HMGB1 exerts different functions by binding to different partners or at different stages, the specific receptors and application conditions need to be taken into account when designing drugs targeting HMGB1.
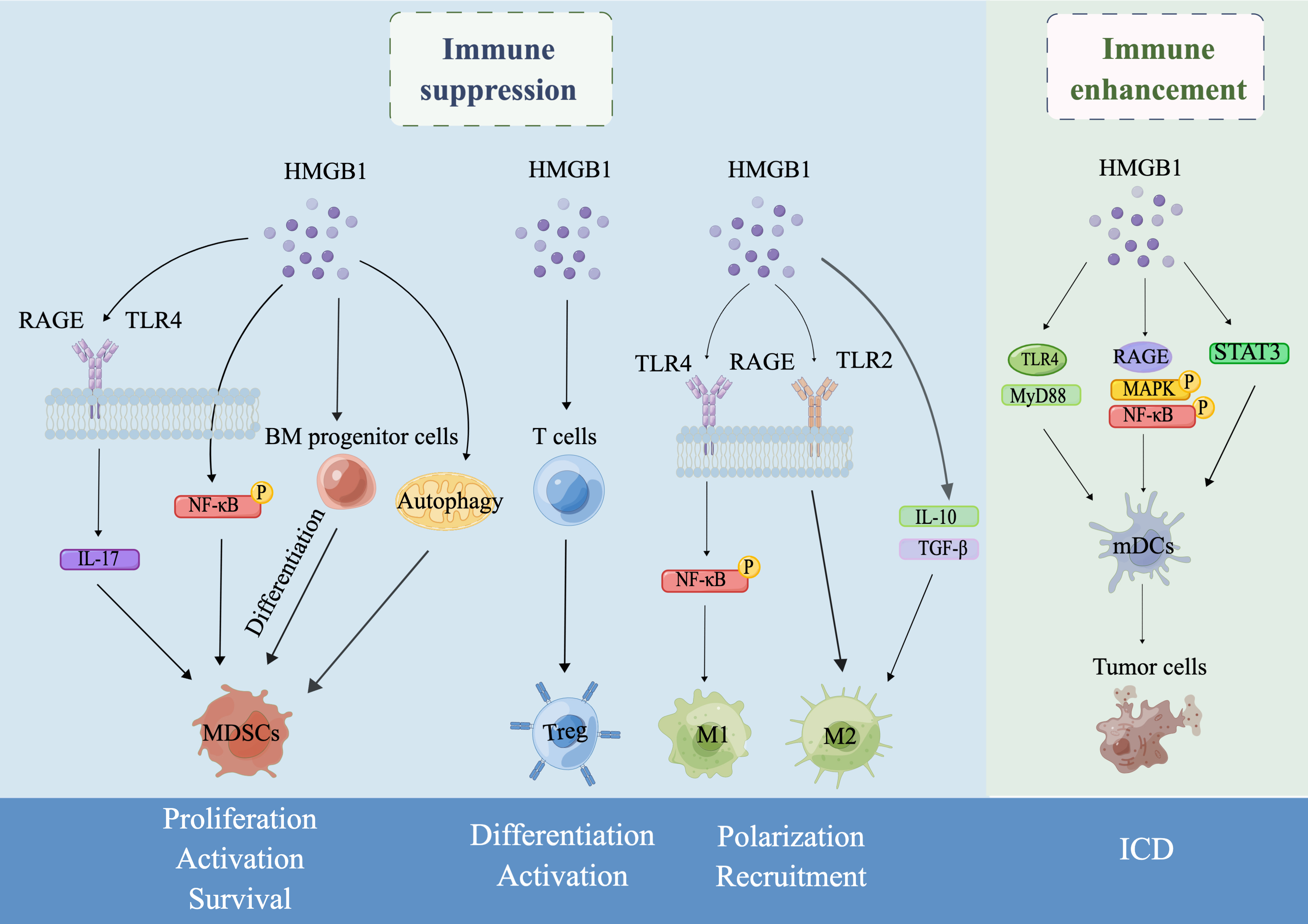 Fig. 1.
Fig. 1.Schematic illustration of the dual role of HMGB1 in tumor
immunity drawn by Figdraw. In the TME, HMGB1 can exert an immunosuppressive role
by regulating the proliferation, activation and survival of MDSCs, the
differentiation and activation of Tregs, and the polarization and recruitment of
TAMs. On the other hand, HMGB1 can also enhance antitumor immune responses by
inducing ICD. Specifically, first, secreted HMGB1 induces MDSC differentiation
from BM progenitor cells, increases MDSC proliferation by promoting IL-17
secretion by binding to the receptors RAGE or TLR4, enhances MDSC development by
activating nuclear factor-
TNF-
JW finalized the visualization, acquiring, analyzing, or interpreting data from the reviewed literature. XZ put forward the idea, supervised this study and revised the manuscript. All authors read and approved the final version of this manuscript.
Not applicable.
Not applicable.
This study was funded by Beijing (China) municipal Natural Science Foundation (7202057) and China National Nature Science Foundation (81772868).
The authors declare no conflict of interest.
References
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.