










1 Ludwig-Maximilians-Universität München, Department of Biology II, Human Biology and Bioimaging, 82152 Planegg-Martinsried, Germany
Academic Editor: Brian A. Mendelsohn
Abstract
Antibody drug conjugates (ADCs) are rapidly becoming a cornerstone in targeted therapies, especially for the treatment of cancer. Currently, there are 12 FDA-approved ADCs, eight of which have been approved within the last five years, with numerous candidates in clinical trials. The promising clinical perspective of ADCs has led to the development of not only novel conjugation techniques, but also antibody formats, linkers, and payloads. While the majority of currently approved ADCs relies on cytotoxic small molecule warheads, alternative modes of action imparted by novel payloads and non-classical antibody formats are gaining attention. In this review, we summarize the current state of the art of ADC technologies, as well as comprehensively examine alternative payloads, such as toxic proteins, cytokines, PROTACs and oligonucleotides, and highlight the potential of multi-specific antibody formats for the next generation of therapeutic antibody conjugates.
Keywords
- review
- ADC
- antibody drug conjugates
- targeted therapy
- cancer
- immunotherapy
- linker chemistry
- alternative payloads
- toxins
Though initially considered insurmountable, the challenge of cancer therapy has been steadily eroded throughout the past decades. The development of chemotherapeutics and radiotherapy have allowed the curing of cancer patients and proven to be a milestone in medical history. However, the systemic toxicity imposed by traditional therapy has prompted an urgent focus towards the development of targeted therapies. Identifying differential expression profiles between cancer cells and normal cells brought about specific agents such as targeted small molecules, and later, antibody-based therapeutics. With the quickfire approval of Rituximab and Trastuzumab in the 1990s for leukemia and breast cancer respectively, antibodies were introduced as highly potent and versatile new weapons in the fight against cancer [1, 2]. Antibody-based technologies have continued development since then, with an array of agents such as monoclonal antibodies (mAbs), immune checkpoint inhibitors, and antibody-drug conjugates (ADCs) constituting the foundations of modern-day cancer therapy. ADCs, which consist of cancer-specific antibodies equipped with payloads selected to kill cancer cells (Fig. 1), have seen a particularly recent boom in popularity. With Mylotarg being the first approved ADC in 2000 for the treatment of myeloid leukemia, a further 11 ADCs are currently on the market as of 2022 - eight of these having been approved within the previous five years (Table 1 (Ref. [3, 4, 5, 6, 7, 8, 9, 10, 11, 12, 13, 14]); reviewed in depth by Fu et al. and Tong et al. [15, 16]).
 Fig. 1.
Fig. 1.General Structure of Antibody-Drug Conjugates (ADCs). ADCs are based on three major elements: the antibody, the drug and the linker. Each of these elements contributes to the overall characteristics of the assembled molecule. The antibody (1) constitutes the core element that most importantly defines binding specificity and facilitates the targeted delivery of the drug. The drug/payload (2) is the element to be delivered and to manipulate the targeted cells. Most attached drugs are cytotoxic agents that induce cell death. But many alternative payloads have been explored to specifically manipulate certain cells. The third element is the linker (3) which physically connects the drug(s) and the antibody. Depending on its design it defines the site of drug attachment and the drug to antibody ratio (DAR). Additionally, it can be used to introduce elements that, for example, increase hydrophobicity and serum stability or introduce drug release mechanisms.
| Agent | Commercial Name | Target | Payload | Indications | Approval year |
| Gemtuzumab ozogamicin [3] | Mylotarg (Pfizer) | CD33 | Calicheamicin | AML | 2000/2017 (reapproved) |
| Brentuximab vedotin [4] | Adcetris (Seagen) | CD30 | MMAE | HL & ALCL | 2011 |
| Ado-trastuzumab emtansine [5] | Kadcyla (Roche) | HER2 | DM1 | Breast cancer | 2013 |
| Inotuzumab ozogamicin [6] | Besponsa (Pfizer) | CD22 | Calicheamicin | ALL | 2017 |
| Moxetumumab pasudotox [7] | Lumoxiti (AstraZeneca) | CD22 | PE38 | HCL | 2018 |
| Polatuzumab vedotin [8] | Polivy (Roche) | CD79B | MMAE | DLBCL | 2019 |
| Enfortumab vedotin [9] | Padcev (Seagen) | Nectin-4 | MMAE | Urothelial carcinoma | 2019 |
| Fam-trastuzumab deruxtecan [10] | Enhertu (Daiichi Sankyo) | HER2 | DXd | Breast cancer | 2019 |
| Sacituzumab govitecan [11] | Trodelvy (Immunomedics) | Trop-2 | SN38 | TNBC | 2020 |
| Belantamab mafodotin [12] | Blenrep (GlaxoSmithKline) | BCMA | MMAF | Multiple myeloma | 2020 |
| Tisotumab vedotin [13] | Zylonta (ADC Therapeutics) | CD19 | PBD Dimer | LBCL | 2021 |
| Loncastuximab tesirine [14] | Tivdak (Genmab/Seagen) | TF | MMAE | Cervical cancer | 2021 |
| AML, Acute Myeloid Leukemia; ALCL, Anaplastic Large-Cell Lymphoma; ALL, Acute Lymphocytic Leukemia; HCL, Hairy Cell Leukemia; HER2, Human Epidermal Growth Factor Receptor 2; CD, Cluster of Differentiation; DLBCL, Diffuse Large B-Cell Lymphoma; TNBC, Triple-Negative Breast Cancer; LBCL, Large B-Cell Lymphoma; BCMA, B-Cell Maturation Antigen; TF, Tissue Factor; MMAE, Monomethyl Auristatin E; MMAF, Monomethyl Auristatin F; PBD, Pyrrolobenzodiazepine. | |||||
ADCs impose various requirements on the antibody: First, due to the exceptional potency of the attached toxins, the used antibody must show high specificity towards its target, because off-target binding can lead to severe side effects. Ideally, antigens should be homogenously and exclusively expressed on cancer cells, but not on healthy ones. For cancers where no exclusively expressed cancer antigen is known (which usually is the case), overexpressed antigens are the option of choice. Since downregulation, mutation, and alternative splicing of the target by the cancer cell are major mechanisms of immune evasion leading to survival of cancer cells that can cause a relapse, the antigen should also be necessary for cancer survival, thereby reducing the chance of resistance. Additionally, the administration of mAbs and ADCs represents an introduction of a large non-self molecule to the patient’s immune system. To reduce immunogenicity, the field has moved from murine antibodies at the conceptualization, then to chimeric and finally to humanized and human antibodies and frameworks over the last decades [17].
The key technology to potentiate the very concept of ADCs is the linkage of drug to antibody. Most currently approved ADCs rely on stochastic chemical conjugation to endogenous amino acids, namely lysines or cysteines. This strategy led to inherent problems such as batch heterogeneity, molecule stability, and off-target toxicity in vivo. To this end, modern-day linker technology is fine-tuned to improve stability (using more robust linkers), counteract hydrophobicity (incorporating hydrophilic elements such as polyethylene glycol; PEG), and provide more uniform batch production and optimized drug-to-antibody ratios (DAR) via site-specific conjugation methods.
Aside from optimization of linker technologies, payload discovery and selection are an integral facet of ADC design. The current landscape of ADCs is geared towards targeted cytotoxicity, given their capacity to vastly widen the therapeutic window. By designing ADCs to target tumor-associated antigens (TAAs) or neoantigens, the ability to deliver toxic warheads to cancer cells while sparing healthy cells is conferred. Not only does this largely avoid the off-target effects of chemotherapy (fulfilling a crucial criterion to Paul Ehrlich’s “magic bullet”), but also enables the use of much stronger therapeutic agents. As such, extremely toxic drugs such as DNA-targeting agents, microtubule inhibitors, and topoisomerase inhibitors constitute the near entirety of payloads employed by marketed ADCs. Although employment of small molecule cytotoxins has proven clinically and commercially successful, expansion beyond these agents as ADC payloads has gained recent attention. As our understanding of the field grows, the future of ADCs is therefore likely to involve an ever-increasing presence of alternative payloads such as protein toxins, protein targeting chimeras (PROTACs) and oligonucleotides.
Taken together, the burst of recent ADC approvals is a reflection of the exceptional technological developments made in a short space of time. The landscape continues to expand rapidly, with a wide breadth of research dedicated to addressing the many remaining challenges of ADCs and prospective therapeutic strategies. In this review, we look towards the future of ADC development, summarizing the latest innovations in antibody formats and linker technologies, and comprehensively examining novel payloads.
Most current ADCs make use of the traditional IgG structure composed of two pairs of heavy and light chains interlinked via disulfide bonds. The IgG scaffold is made of the constant heavy (CH) and constant light (CL) domains, with the antigen-binding site determined by the variable heavy (VH) and variable light (VL) domains in concert. The domains CH2 and CH3 make up the Fc region. The classic IgG is the most prevalent antibody type in the immune response and as such has multiple interaction points with the immune system. The antibody type can not only act as a “transporter” for the payload but confer additional cytotoxicity via antibody-dependent cellular cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC) by activating immune cells. On top of that, full-length antibodies circulate in the blood for long time before being cleared, decreasing the number of doses needed to achieve therapeutic concentration levels. However, the large size of an IgG molecule (~150 kDa) hampers tissue penetrance, rendering the effective treatment of solid tumors with traditional ADCs a significant challenge (Fig. 2). The large size additionally leads to higher variability during drug conjugation via stochastic methods - although this has been somewhat alleviated by newer site-specific techniques. Furthermore, the existence of full antibodies as covalently linked multimeric proteins requires expensive expression in eukaryotic systems to produce correctly folded and glycosylated antibody molecules. The production of structurally simpler and more penetrative antibody derivatives has therefore become a thriving field of research.
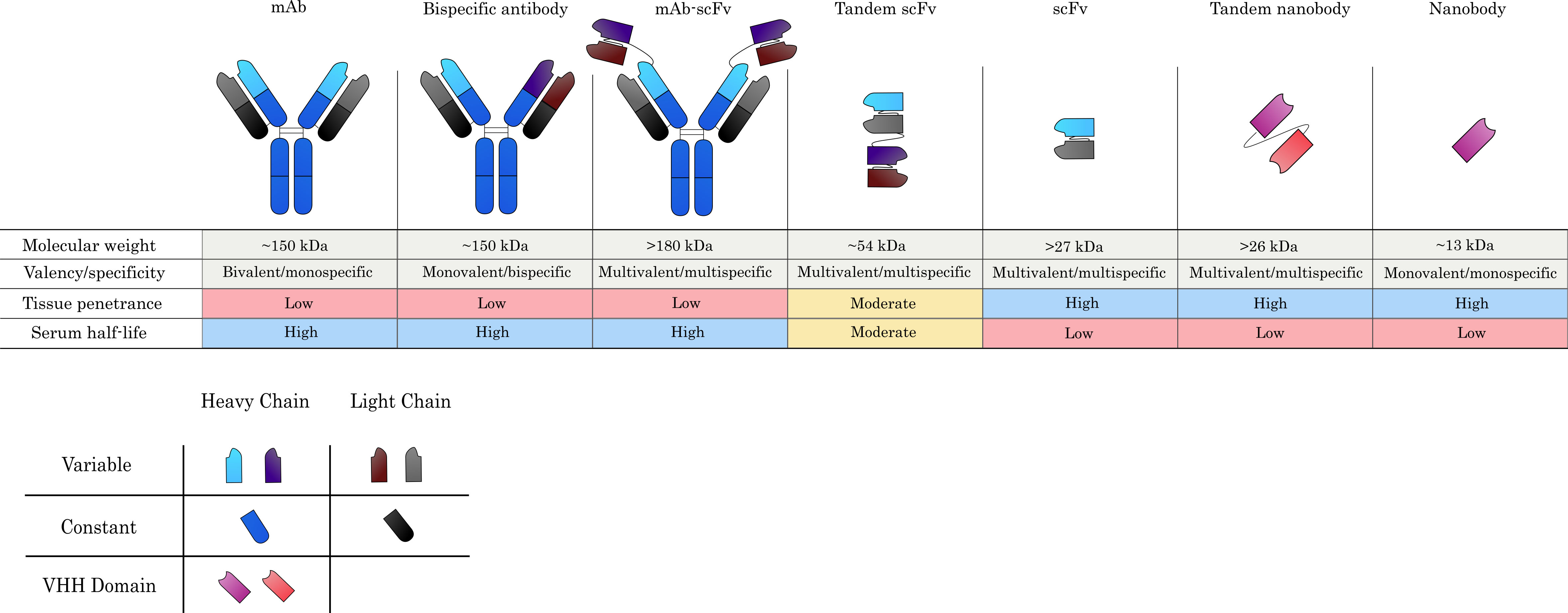 Fig. 2.
Fig. 2.Commonly used antibody formats in ADC design. Monoclonal antibodies (mAbs) serve as the main scaffold from which alternative formats are derived and confer specificity against a single target, whilst maintaining bivalency due to the presence of two antigen-binding sites. Antigen-binding arms can be engineered to produce bispecific antibodies, which can recognize two target antigens in a monovalent capacity. Single-chain variable fragments (scFvs), which consist only of the variable heavy (VH) and variable light (VL) chains connected by a flexible linker, can also be grafted onto mAbs to yield large multispecific and/or multivalent constructs. Much smaller constructs, such as tandem scFvs and scFvs alone are also commonly investigated for ADC development. Nanobodies, which constitute the VHH domains of camelid heavy-chain-only antibodies, constitute the smallest possible natural antibody derivatives and are one of the novel antibody formats currently being explored for ADC-based applications. Generally, larger constructs tend to yield poorer tissue penetration, while smaller derivatives are superior in this respect. Serum half-life is reduced, however, when lower molecular weight agents are used or they do not contain an Fc domain.
Antigen-binding fragments, or Fab fragments, consist of an antibody light chain
linked by a disulfide bond to the VH and CH1 domains (Fig. 2). These constructs
are monovalent and lack an Fc region, with a total size of ~55
kDa. Fab fragments have been shown to penetrate tumors more effectively and are,
unlike full-length antibodies, not strictly limited to expression in mammalian
expression systems. The lack of an Fc region in these constructs presents a
double-edged sword from a clinical perspective. On the one hand, the absence of
Fc
The most prevalent derivative of full-length IgG is the single-chain variable fragment (scFv). An scFv is composed of just the VH and VL domains interconnected by a recombinant linker, constituting the smallest possible derivative of natural antibodies. Smaller still is the single-domain antibody, or nanobody, that has been derived from camelid heavy chain-only antibodies (HcAbs) which lack a light chain. As a result, the antigen-binding site of an HcAb molecule consists of a single variable (VHH) domain that can be cloned and expressed to yield a fully functional nanobody (Fig. 2). Similar to Fab fragments, thes absence of an Fc region has clinical implications. Therapeutics based on these formats such as Blinatumumab need frequent or even continuous administration to the patient due to serum clearance, despite the appealing prospect of reduced immunogenicity. The following subsection highlights the challenges and progress made with the development of scFv and nanobody-based ADCs, herein referred to as single-chain ADCs.
The use of single-chain antibody derivatives in cancer therapy has been an appealing prospect due to their improved solid tumor penetrance, ease of production and manipulation. Several agents, such as TAA-specific scFvs and nanobodies, are currently in clinical trials [21]. While the idea of conjugating single-chain constructs to cytotoxins to produce miniaturized ADCs is not a new one, the field is still quite nascent in a practical sense. Among the historical roadblocks to the development of novel antibody fragments is their production. Given their usual lack of an Fc region, protein A-based purification (the conventional method of purification for full-length antibodies) is incompatible. Furthermore, many antibody derivatives are comparatively less stable and more prone to physicochemical limitations (such as aggregation) than their full-length counterparts [22]. These issues have had to be addressed through the engineering of recombinant pulldown tags [23, 24, 25], as well as fine-tuning amino acid compositions to increase the constructs’ stability in solution [26, 27, 28]. These findings resolve major obstacles in the production of single-chain antibodies, however, problems can still be encountered on an individual basis.
A 2014 study by Chen et al. [29] highlighted the difficulties associated with such constructs by producing a hepatoma-specific scFv conjugated to a DNA-intercalating ADM payload. Despite achieving an extremely high DAR of over 14 and the superiority of the scFv-ADM conjugate in vitro, mouse xenograft models could only produce modest tumor growth inhibition. The discrepancy between in vitro and in vivo data for such small constructs is not uncommon and likely has to do with unfavorable biodistribution patterns. The renal molecular weight cutoff value in mammals is roughly 40 kDa, often rendering such small constructs therapeutically suboptimal due to their rapid clearance rates [30]. Furthermore, despite the demonstrably superior tissue penetration of smaller constructs, other factors such as the binding-site barrier often limit biodistribution within solid tumors. In 2016, Fang et al. [31] produced similar findings using a nanobody-drug conjugate (NDC) specific to Major Histocompatibility Complex II for the treatment of B-cell lymphoma. Once again, nanomolar efficacy was achieved in vitro with relatively mild tumor inhibition in xenograft models, despite observing high tumor uptake. However, an interesting finding in this study was the dramatic reduction of cancer dissemination in a metastasis model, falling in accordance with the increased accessibility of cancer cells in the bloodstream and consequent ease of elimination by NDCs.
Given the standout limitation of renal clearance, the stabilization of
single-chain ADCs is an increasingly popular strategy. Albumin is often exploited
for this purpose, due to its high molecular weight (roughly 66 kDa in humans) and
consequently higher retention time. NDCs equipped with platinum-derived
chemotherapeutic agents have demonstrated vastly superior physiological retention
and tumor accumulation when fused to an albumin-binding nanobody at the
C-terminus [32]. This strategy was furthered by Crescendo Biologics using an
engineered human scAb (VH domain) against prostate-specific membrane antigen
(PSMA) conjugated to a more potent DNA-alkylating agent and fused to an albumin
binder [33]. A notable finding was the inverse trend between monovalent and
biparatopic NDCs; biparatopic constructs were superior in vitro, while
the monospecific ones produced significantly higher tumor regressions in mice.
This hinted at the complex dynamics imposed by solid tumors, wherein the improved
internalization imparted by biparatopic binders does not necessitate increased
therapeutic efficacy. The authors further demonstrated the possibility of a
binding-site barrier phenomenon caused by the sequestration of fast-internalizing
nanobodies at the tumor periphery. This represents a potentially valuable insight
for the use of single-chain ADCs, suggesting that their improved tissue
penetrance can only be capitalized upon if internalization rates are modulated to
circumvent the binding-site barrier. In 2021, a highly similar approach was
undertaken by Xenaki et al. [34] using a HER2 model. Once again, a
monovalent nanobody against the receptor was recombinantly fused to an albumin
binder and conjugated to the microtubule inhibitor auristatin F. Addition of the
albumin binder resulted in a nearly 15-fold increase of serum half-life and
visibly improved tumor accumulation over time. Xenograft studies on a HER2
In terms of clinical advancement, however, the scFv-based Moxetumumab pasudotox is at the forefront of single-chain ADCs. Moxetumumab pasudotox deviates from the traditional definition of an ADC, which generally refers to the attachment of a small molecule payload and is instead recombinantly fused to a truncated Pseuodomonas exotoxin protein. Technically classed as an immunotoxin (see section 5.1 for in-depth review), Moxetumumab pasudotox was originally designed in 1997 against the lymphoma TAA CD22 [35]. The excellent preclinical performance of the agent has led to its deployment in clinical trials under the licensing of Medimmune and eventual FDA approval in 2018 for the treatment of hairy cell leukemia [7]. Moxetumumab pasudotox’s successes highlight the advantages associated with using genetically amenable constructs such as scFvs and nanobodies. The ability to express these genes along a single open reading frame and lack of post-translational assembly render it simple to insert protein fusion partners to generate recombinant antigen-specific therapeutics. Accordingly, many other immunotoxins based on single-chain antibodies are being evaluated clinically and experimentally, which will be expanded upon further below.
Another example of creative recombinant ADC design was the recent production of
an anti-CD38 nanobody fused to the GALA peptide by Chen et al. [36].
GALA is a short pH-sensitive peptide which, upon activation in acidic conditions,
can insert itself into lipid bilayers and aid endosomal escape due to its
amphipathicity. The group proposed that the conjugation of a drug, in this case
MMAE, to a nanobody-GALA fusion would result in higher endosomal escape after
internalization and hence improved efficacy compared to the anti-CD38 nanobody
without GALA. The study’s in vitro assays showed this hypothesis to be
true, where the EC
A standout example of the wide modularity afforded by antibody engineering is the use of bispecific antibodies (bsAbs). In this format, antigen-recognition domains in bivalent antibodies are designed to have a different specificity on each arm. BsAbs have quickly become relevant in a therapeutic context, with most clinical applications having been made using immune cell-stimulating constructs (reviewed comprehensively in [37]). Multispecific constructs can also be engineered to ensure antibody binding only when multiple antigens on the same cell are encountered simultaneously. In this scenario, on-target off-tumor effects may be reduced by designing agents which recognize more than one target among a cancer cell population, but does not bind sufficiently if only one target is encountered [38]. Strides have also been made using bispecific ADCs. The rationale behind bispecific ADCs generally falls into either of three schools of thought: employing biparatopic antibodies to improve antigen binding dynamics, enhancing lysosomal trafficking for more efficient drug delivery, and/or including moieties specific to alternative TAAs in order to widen the target range.
In a 2016 study, a biparatopic antibody targeting two separate HER2 epitopes was
constructed by grafting a trastuzumab scFv onto a full-length antibody [39].
Analysis of HER2 dynamics revealed this bsAb to significantly boost HER2’s
capacity for internalization and visibly improved its localization to the
lysosome in comparison to Trastuzumab, hinting at the potential effectiveness of
a biparatopic strategy. This hint was confirmed when, upon conjugation to a
microtubule-inhibiting payload, the biparatopic antibody outperformed T-DM1
(Kadcyla) in HER2 positive cancers both in vitro and in vivo.
Furthermore, the biparatopic antibody retained activity against the
T-DM1-resistant cell line JIMT-1 and maintained EC50 values in picomolar ranges
against HER2
A biparatopic antibody against mesenchymal-epithelial-transition (MET) receptors for the treatment of MET blocker-refractory lung cancer, conjugated to the microtubule inhibitor M114 and dubbed METxMET-m114, was superior to a monospecific MET receptor antibody conjugated to MMAE in in vitro assays [41]. A further testament to this strategy was the complete regression of MET receptor-overexpressing xenografts in mice treated with 5 mg/kg METxMET-M114 a dose at which the monospecific ADC resulted in relapse. Most notably, METxMET-M114 eliminated tumors that were refractory to monospecific ADC treatments, once again indicating the potential viability of biparatopic ADCs for treatment of resistant cancers. Similarly to the biparatopic anti-HER2 bsAb MEDI4276, naked METxMET has previously been documented to rapidly internalize and localize to the lysosomal compartment- a likely key factor in its superior efficacy [42].
In a different approach to optimizing intracellular trafficking, bispecific
modalities have been adapted to target lysosome-associated receptors alongside
the target antigen. DeVay et al. [43] demonstrated the promise of this
strategy by producing an ADC specific to HER2 and APLP2 (HER2xAPLP2). The latter
was identified as a receptor which is rapidly internalized and processed by the
lysosomal pathway. By simultaneously binding APLP2, HER2 could more efficiently
be redirected to the lysosomal pathway in a “piggybacking” mechanism. By
conjugating HER2xAPLP2 to the microtubule inhibitor MMAD, EC50 values in
picomolar ranges were obtained upon treatment of both HER2
Multispecific antibodies also present an appealing solution to intratumor heterogeneity. Cancer cells will often evolve to shed, express, and alter their surface antigens over the course of disease progression. Targeting a single antigen could, therefore, leave antigen-negative populations unharmed and subsequently select for ADC-resistant cells. Multispecific constructs designed based on tumor expression profiles (i.e., multi-target constructs) have thus been proposed to eliminate different tumor cell populations using a single agent. Experiments in this direction have so far been mostly limited to immunomodulatory and directly pathway-disruptive agents [21], with little progress having been made with multi-target ADCs. The most advanced agent of this kind is the innovative DT2219: a bispecific construct composed of two scFv domains complementary to the B-cell lymphoma markers CD19 and CD22 [46]. Genetically fused to a cytotoxic diphtheria toxin moiety, DT2219 is technically classified as an immunotoxin as opposed to a traditional ADC. This construct performed well in preclinical studies both in vitro – exhibiting picomolar IC50 values - and in vivo – producing significant tumor regression in mice. While DT2219 has already made it to clinical trials [47], concerns over immunogenicity of the diphtheria toxin have prompted efforts to produce an artificially deimmunized version of the construct [48].
Returning to more familiar small molecule-equipped ADCs, a method has recently
been developed by Yamaguchi et al. [49] to simultaneously target HER2
and the folate receptor (FR) or the
ADCs are almost exclusively designed based on the IgG class, which represents
the class-switched form of antibodies that facilitate most humoral immune
responses in a natural setting. IgG can be further divided into four different
subclasses: IgG1, IgG2, IgG3, and IgG4. These mainly differ in the nature of
their Fc and hinge regions as well as Fc-gamma receptor (Fc
IgG2, which has four inter-chain disulfide bonds instead of two and therefore
higher DAR potential in cysteine-based conjugation chemistry, has seen
encouraging recent progress. ADCs for the treatment of renal cell carcinoma
(phase II [52]), lymphomas (phase I [53]) and c-Met-expressing cancers (phase I
[54]) have produced favorable outcomes. These candidates demonstrated notably low
toxicities, which has been attributed to the lack Fc
Native IgG4 exhibits the unorthodox behavior of interchain disulfide bond
reduction and subsequent exchange of heavy chains in vitro. The result
of this is the assembly of “mismatched” antibody arms, often resulting in
monovalent bispecific products [55]. In order to rectify this, IgG4 has been
engineered with a serine-to-proline substitution at the hinge region (mimicking
that of IgG1), which serendipitously improved tissue distribution and serum
stability [56]. Engineered IgG4 has therefore been in use for ADC development,
and is indeed already represented in the current ADC market by Mylotarg and
Besponsa [3, 6]. In terms of Fc
Before we move towards the various cargos that are being explored for ADCs, we will briefly summarize recent advances in conjugation techniques and linker designs. Both aspects play a key role in ADC stability, pharmacokinetic and pharmacodynamic [58, 59]. Relevant parameters for the conjugation strategy are the site-of-attachment, the drug-to-antibody ratio and stability in biological matrices (Fig. 3). While stability is an important design feature of linkers, further aspects like solubility, release mechanisms and multivalency are being considered as relevant for the potency and safety of ADCs. In this section we highlight how researchers have implemented these features in recently developed conjugation methods and linker designs. For a comprehensive overview of site-selective modification strategies for ADCs we would like to bring your attention to a very recent review by Walsh et al. [60] and a detailed and comprehensive review of cleavable linkers can be found in a 2019 review by Bargh et al. [61].
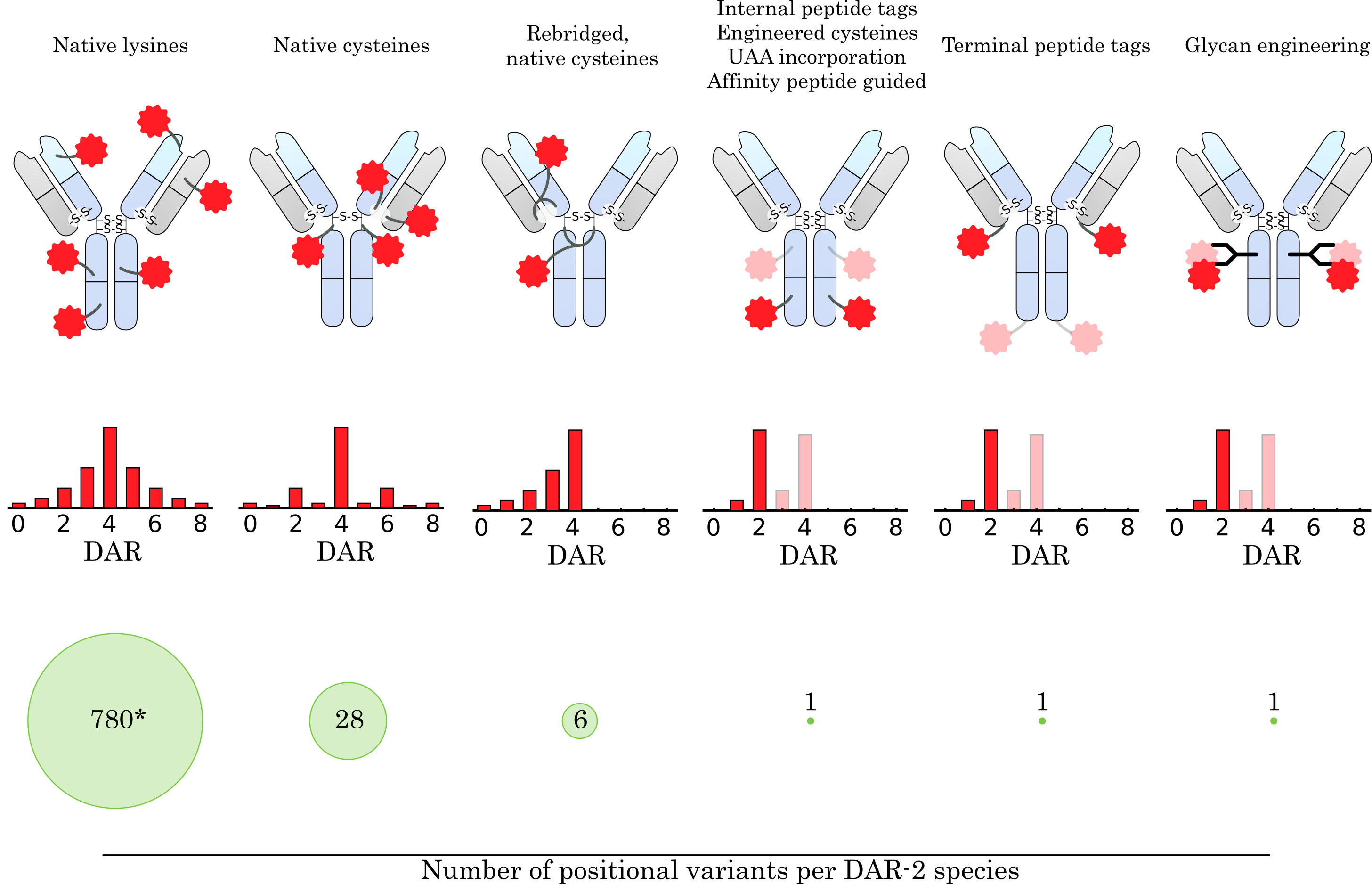 Fig. 3.
Fig. 3.Comparison of conjugation concepts for drug attachment. How the drug is conjugated to the antibody has a major impact on the resulting ensemble of conjugate species. The average drug-to-antibody ratio (DAR), DAR distribution, and number of positional variants for a given DAR species are highly dependent on the conjugation method and linker design. The targeting of native amine groups of surface-exposed lysines leads to a broad DAR distribution, which is mainly determined by stochastic effects and the biophysical stability of high DAR species. Moreover, the large number of surface-exposed lysine residues on an IgG molecule leads to a large heterogeneity within a DAR species (* 780 theoretical variants for DAR-2, assuming 40 surface-exposed lysine residues addressable for conjugation). Conjugation to native cysteines narrows the DAR distribution, limits the maximum DAR to 8, and greatly reduces the number of positional variants. Using a linker design that rebridges the native disulfide bonds further narrows the DAR distribution, reduces the number of positional variants, and restores the stabilizing cysteine linkage. Site-specific conjugation methods that rely on the introduction of bioorthogonal handles (e.g., peptide tags, unnatural amino acid (UAA) incorporation, affinity-guided conjugation or glycan engineering) provide even greater control over DAR distribution and eliminate the presence of positional variants for DAR-2 species. In addition, they allow the introduction of two (or more) different drugs at specific positions on the antibody. The number of attached drug molecules can be precisely controlled by the introduced handle and linker design.
Currently approved ADCs exclusively use amine- or thiol-directed conjugation for drug attachment. In these cases, drugs are attached to surface exposed lysine residues or reduced intrachain-disulfides, respectively. By targeting endogenous residues, no engineering of the antibody is necessary. However, the site of attachment and the number of attached molecules is hard to control, leading to a heterogenous product containing multiple ADC species of varying DARs. Furthermore, cysteine conjugation via maleimides poses the risk for hydrolysis and thiol-exchange to endogenous proteins in biological matrices [62, 63]. These aspects are considered to negatively affect the potency and safety of the conjugates [63, 64, 65]. To address these issues researchers have developed stability-improved maleimides [66, 67, 68] like self-hydrolyzing maleimides [64], N-phenyl maleimides [69] or N-aryl maleimides [70] and alternative thiol-reactive moieties with improved characteristics like N-Methyl-N-phenylvinylsulfonamides [71] or Ethynylphosphonamidates [72, 73]. One side-effect of thiol conjugation is the destruction of the intra-chain disulfide bonds stabilizing the antibody’s quaternary structure. In this context bis-functional thiol-reactive groups have been described to re-bridge the reduced disulfides upon toxin conjugation [74, 75, 76, 77]. Besides stability, researchers also addressed conjugate heterogeneity resulting from the presence of eight thiol groups after reduction by preferentially targeting the Fab intrachain disulfides [78]. By introducing cysteines at alternative positions in the antibody sequence, precise control over position and number of attached molecules can be achieved. Several approaches based on engineered cysteines, like Genentech’s Thiomab [79, 80], have been described for ADC generation [81, 82] including high throughput screening of cysteine mutation sites [83]. These studies further reinforced the importance of conjugation site and respective molecular microenvironment for conjugation efficacy, ADC solubility and stability and drug release.
Engineered cysteines were the first step towards site-specific conjugation of antibodies in the sense that a new reactive group was introduced into the antibody that could be addressed selectively for conjugation. With cysteines, the distinct molecular microenvironment of the thiol group renders its nucleophilic character different from the antibodies’ native disulfide bridged cysteines. Based on this general principle of introducing a selectively addressable site in the antibody molecule in combination with bio-orthogonal conjugation reactions, several strategies have been described over the recent past for the generation of site-specifically modified ADCs.
Following a similar strategy as with cysteine engineering, researchers described tyrosine engineering by combining the insertion of a tyrosine residue at a surface exposed position with conjugation of a tyrosine-reactive moiety for the generation of ADCs [84, 85]. Proteinogenic amino acids can be introduced into antibodies by straight forward genetic mutations but do only offer limited orthogonality in their reactive groups. By expanding the genetic code through methods like amber suppression the door was opened for a new class of orthogonally reactive groups that have been known to chemists but were previously hardly usable for protein engineering. The incorporation of unnatural amino acids carrying reactive handles like aldehydes [86] azides [87], strained-alkenes [88] or selenocysteine [89] has been successfully used to generate site-specifically conjugated ADCs.
An alternative to amino acid side chains as an attachment site are the N-glycans linked to asparagine297 in the CH2 domain. Several strategies to attach drugs to these residues have been described including the conjugation to native glycans [90], introduction of modified sugars during antibody production by metabolic engineering [91], the incorporation of bioorthogonal handles by exploiting the substrate tolerance of natural glycosyltransferases [92] or the engineering of glycotransferases [93, 94]. While targeting the N-glycans does not require changes in the amino acid sequence of the antibody and is thus universally applicable to all antibodies without the need for mutagenesis, the alteration of the glycosylation pattern is being discussed as a risk for increased immunogenicity [95].
Alternatively, enzymatic incorporation of bioorthogonal handles has been used to conjugate to amino acid recognition motifs. A widely used strategy is based on microbial transglutaminases (mTG) [96, 97] to either conjugate endogenous glutamines (Q295) [98, 99] or an engineered recognition motif (Q-tag) [59]. While initial attempts used direct enzyme catalyzed conjugation of modified substrates [98], later studies showed the incorporation of universal bioorthogonal handles and subsequent modular attachment of toxins for ADC generation [97].
An alternative strategy uses a formylglycine-generating enzyme (FGE) to oxidize a terminal cysteine in a recognition motif (CxPxR) to an aldehyde-containing formylglycine [100]. In a second reaction step the aldehyde serves as a bioorthogonal handle to attach the payload of choice [101, 102]. Initially used at the C-terminal position, recently an internal recognition sequence (LCTPSR) was inserted at 436 positions throughout the antibody sequence and screened for suitability of toxin conjugation [103].
The transpeptidase sortase A has also been used for the generation of site-specific ADCs. It catalyzes the cleavage between Tyr and Gly in a recognition motif (LPxTG) and the re-ligation of a glycine-containing substrate [104]. The sortase based strategy allows N- and C-terminal labeling of antibodies and has been used for ADC generation [105, 106, 107]. Another recently developed terminal labeling strategy is based on the C-terminal ligation of tyrosine derivatives to a human recognition sequence (Tub-tag) by a tubulin tyrosine ligase (TTL) [108, 109, 110]. Notably, the TTL has a broad substrate tolerance and thus allows the incorporation of a variety of bioorthogonal handles [111] for subsequent site-specific toxin-conjugation.
Apart from these well-established methods several more site-specific conjugation concepts, like pi-clamp [112] or CD38 domain-tag [113], and conjugation to native lysines by using affinity peptides, e.g., as guides [114, 115] have been published in the context of ADC generation and are comprehensively covered elsewhere [60, 116]. Furthermore, very recently a strategy combining two site-specific conjugation methods was described for the attachment of two distinct cytotoxic drugs [117].
Not only the conjugation method but also the linker itself is an integral and customizable element of ADCs. Apart from physically linking drug and antibody it also has the potential to contribute to characteristics like linkage stability, drug release and aggregation behavior [118]. Currently approved ADCs utilize either non-cleavable, chemically cleavable or cathepsin-cleavable linkers for intracellular liberation of cytotoxins. Non-cleavable linkers liberate the attached drug upon proteasomal degradation of the internalized antibody [119]. In this case the drug remains attached to the conjugated amino acid which can affect its bio-distribution and potency [120]. Cleavable linkers on the contrary allow triggered release. Currently approved ADCs are designed to exploit the distinct environment in lysosomes to trigger drug release. For example, pH sensitive or thiol-containing reduction-labile linker elements, found in gemtuzumab ozogamicin (Mylotarg [121]) and inotuzmab ozogamicin (Besponsa [122]), exploit the acidic and reductive milieu in lysosomes. But the majority harbors cleavage motifs (CV, CA or GGFG) sensitive to lysosomal proteases of the cathepsin family, especially to cathepsin B. Consequently, all currently approved ADCs rely on internalization of the ADC and trafficking to the lysosome for triggered intracellular liberation of the drug. Depending on its membrane-permeability the liberated drug can additionally act on non-targeted neighboring cells exerting a “bystander killing” effect [123]. Linkers that allow triggered traceless release have been a strong focus in recent ADC development [61]. Along this line, alternative release mechanisms have been described. One example is chemically triggered drug release as described recently with bioorthogonal release by click-cleavable elements [124]. Chemical-induced release of drugs allows for sequential targeting and release and does not necessarily rely on internalization of the ADC for drug release. A similar idea is followed up by using light induced cleavage of linkers. Instead of a chemical trigger, local irradiation is used to release the drug from the ADC [125, 126, 127]. This approach allows not only sequential targeting and release but in addition spatial release at a certain location in the body. Furthermore, the presence of reactive oxygen species in the tumor environment has been utilized for triggered release and may prove suitable for ADC drug release [128, 129, 130].
Cathepsin sensitive linkers have been used widely with the valine-citrulline (VC) dipeptide being the most prominent cleavage motif. However, modified VC motifs and alternative sequences have been described that exhibit improved solubility and rodent serum stability [10, 131, 132]. Besides cathepsin B researchers also explored other (lysosomal) proteases for triggered release [133]. Among them are (pyro-)phosphatases [134, 135], beta-glucuronidase [136, 137], beta-galactosidase [138, 139], sulfatase [140], caspase [141], and prostate-specific-antigen (PSA) [142]. Recently, the use of two cleavage motives in tandem has been shown to improve in vivo stability of ADCs [143]. While triggered release at a desired timepoint and environment mainly contributes to on-target efficacy and off-target integrity, other linker design aspects aim at improving the biochemical properties of ADCs. Most payloads used today are hydrophobic and increase the aggregation potential of the conjugate. Especially for ADCs with high DARs this effect can critically influence pharmacodynamic and -kinetic properties of an ADC [144]. While drugs can only be modified to a limited extent without influencing their potency, hydrophilic properties have been introduced into the linker to balance and counteract the hydrophobicity of the payload [118]. PEG-based elements have been explored for solubility enhancement and are employed in the recently approved ADCs sacituzumab govitecan-hziy (Trodelvy) and loncastuximab tesirine-lpyl (Zynlonta). Several PEG configurations have been designed ranging from linear PEG spacers between drug and antibody, to branching off PEG “side chains” [145, 146]. Modular assembly of PEG elements and payloads has become feasible by the development of universal branching elements [77, 147, 148]. In addition to PEG elements, biopolymer-based linkers like monodisperse polysarcosine [149] and dextran polymers (Dextramabs) [150] are also being explored for shielding drug hydrophobicity and the generation of highly loaded ADCs.
Hydrophilic linker designs are a promising approach to increase serum half-life, reduce unspecific uptake especially in the liver and pave the road for higher DARs. Combined with novel triggered release mechanisms they will further improve targeted delivery of cytotoxic drugs and alternative cargos to tumors and other disease associated sites.
With 12 approved drugs currently on the market (see Table 2), small molecule toxin ADCs have become a widely established strategy for the treatment of cancers. So much so, that the term “ADC” has become largely synonymous with the conjugate of an antibody to a small molecule toxin. Conceptually, ADCs allow the therapeutic use of extremely potent toxins that were otherwise impossible to administer systemically due to their lack of specificity. The antibody will guide the toxin towards the desired cells and tissues where the complex will be internalized, digested and the toxin released.
| Linker element | Functions | Designs/Mechanism | Examples |
| Reactive group | Conjugation to antibody | Targeting amino acid residues | Amine-reactive |
| Targeting glycans | Thiol-reactive | ||
| Targeting preinstalled bioorthogonal handles | Aldehyde-reactive | ||
| Cycloaddition-compatible (azides, alkines, tetrazines) | |||
| Protease cleavage site | Triggered drug release | Endo-/Lysosomal proteases | Cathepsin B, beta-galactosidase, beta-glucuronidase, sulfatases, phosphatases, caspase, matrix metalloproteases |
| Tumor microenvironment associated proteases | |||
| Non-protease triggered cleavage site | Triggered drug release | Chemical trigger | pH, disulfide reduction, oxidation, light |
| Physical trigger | |||
| Solubility enhancing element | Shield drug hydrophobicity | Linear or branched spacer | PEG, dextran, polysarcosine |
| Increase hydrophilicity | |||
| Self-immolative element | Traceless drug release | Cleavage induced electron cascade | para-amino benzyloxycarbonyl (PABC) |
| Branching element | Attachment of |
Branch valency | Branched polymers (PEG, dextran), |
| Position in the linker | Bis-ethynylphosphonamidates | ||
| Homo- or heterofunctional |
Efficient internalization after binding to the cell surface is a crucial characteristic for most ADCs, since most toxic payloads have to be delivered intracellularly and in a sufficient amount to kill the cell. Depending on the type of linker, the payload will be released by acidification of the endosome/lysosome, cleavage by endosomal/lysosomal proteases such as cathepsins or degradation of the antibody. Some toxins can pass across the lipid bilayer themselves, however, even toxins that show little membrane permeability such as MMAF are able to escape the lysosome as seen by their pronounced cytotoxic effect. Membrane-permeable toxins can also diffuse to and kill neighboring cells. This phenomenon has been termed the “bystander effect” and can improve the efficacy of the ADC in heterogenous tumors [151, 152, 153]. As a response to ADC treatment, cancer cells can interfere at many levels to reduce the effective load of functional toxin, such as altered internalization and endosomal trafficking, reduced acidification of the lysosome and the expression of multi-drug resistance proteins. Common resistance mechanisms are reviewed in-depth elsewhere [154, 155]. The resistance mechanisms pose a major challenge for ADC development and emphasize the need to develop novel toxins and linkers to circumvent resistance to currently used toxins.
Another defining factor of any ADC is the DAR. For example, small molecule toxin ADCs show higher efficacy in vitro the higher the DAR. However, as mentioned before, ADCs with high DAR are also more prone to aggregation, exhibit faster plasma clearance rates and stronger accumulation in the liver in vivo. Consistent DAR of the final product is of importance for reproducibility during manufacturing and safety for the patient. A variety of methods have therefore been described to evaluate DAR values to ensure batch-to-batch consistency, which are discussed elsewhere [156, 157]. Studies suggest a DAR of roughly 3–4 to hit the sweet spot between efficacy and clearance for current small molecule toxin ADCs [158, 159, 160] and thereby maximizing the therapeutic index. This is in accordance with the DARs of currently approved ADCs such as ado-trastuzumab emtansine (Kadcyla), which exhibits an average DAR of 3.5 [160]. While the optimal DAR will depend not only on the payload class, but also the exact payload that is being used, the decades of research on mostly small molecule toxin ADCs have given us insight into the relationship between DAR and pharmacokinetics and pharmacodynamics. It has become clear, that the payload will therefore heavily influence the in vivo behaviour of and confer its own characteristics and specificities to the whole ADC complex.
In the next chapter, we will give an overview over currently established small molecule toxins as well as other alternative payloads that are currently under investigation (Fig. 4 and Table 3 (Ref. [3, 4, 7, 161, 162, 163, 164, 165])).
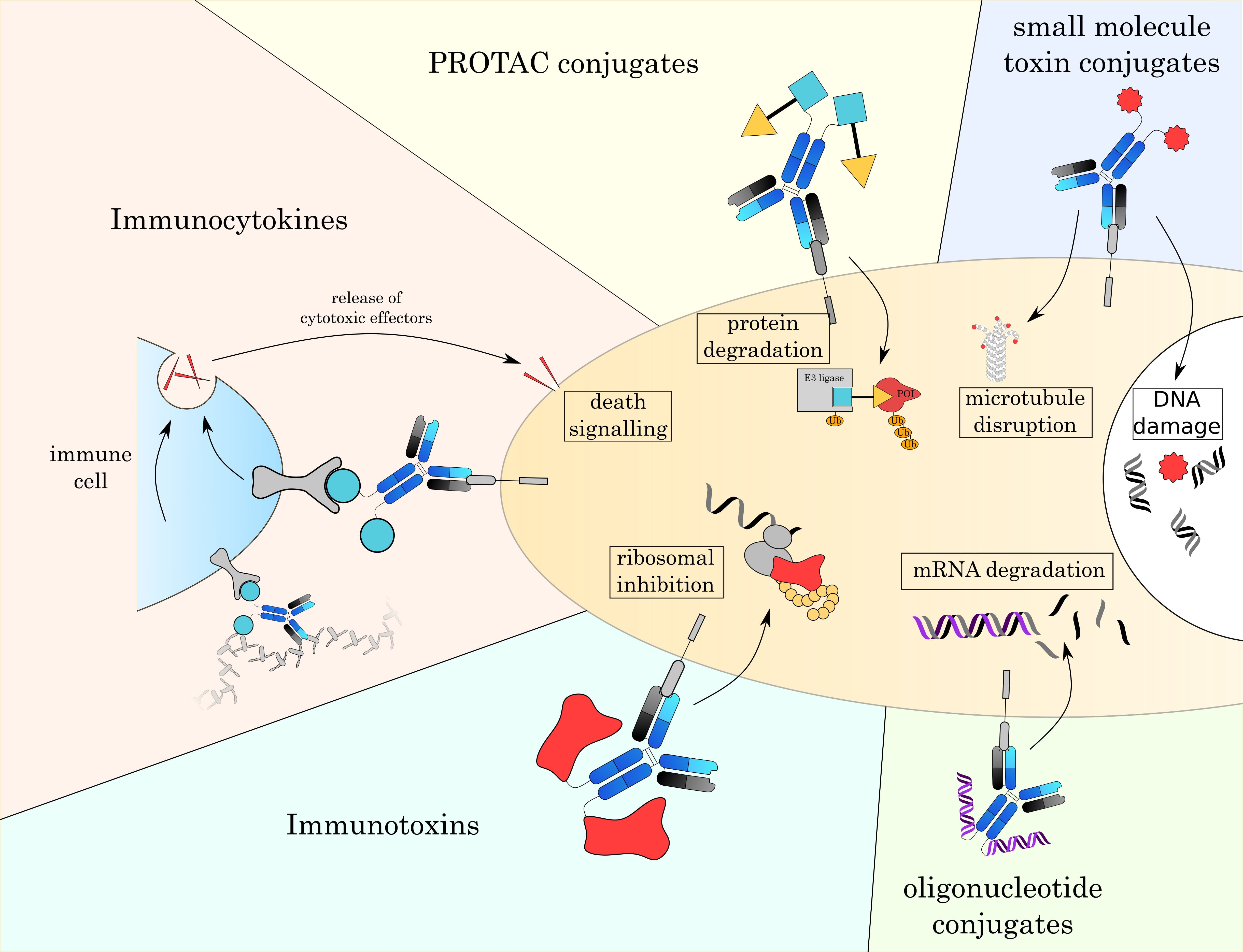 Fig. 4.
Fig. 4.Overview over different formats of ADCs. With classic small toxin molecule ADCs being limited to mostly the disruption of microtubules or damaging DNA, the development of novel payloads allows to attack a broader range of intracellular targets. These new modes of action will enable new therapeutic strategies and overcome resistance mechanisms to currently approved ADCs.
| Type | Site of action | Cargo release mechanism | Mode of action | Conjugation method | Stage of development | Common DAR ranges | Hydrophobicity | Notable examples |
| Small molecule toxin | cytoplasmic | Ab digestion, acidification, proteases | DNA damage, microtubule inhibition | Chemical conjugation | FDA approval | 2–8 | high | Gemtuzumab ozogamicin [3], Brentuximab vedotin [4] |
| Protein toxin | Depending on toxin | Proteases | Ribosomal inhibition | genetic fusion | FDA approval | 1–2 | low | Moxetumomab pasudotox [7] |
| Cytokine | extracellular | Proteases | Activation of immune cells (T cells, NK cells, macrophages) | genetic fusion | Clinical studies | 1–2 | low | L19-IL2 [161], F16-IL2 [162] |
| Oligonucleotide | cytoplasmic | Ab digestion, acidification, proteases | Gene knock-down (siRNA), DNA damage & microtubule inhibition (toxin) | Chemical conjugation | Pre-clinical studies | 1–2 (chemical conjugation) | very low | aKRAS-siRNA [163], F105-P [164] |
| PROTAC | cytoplasmic | Ab digestion, acidification, proteases | Targeted protein degradation | Chemical conjugation | Clinical studies | 2–8 | high | BRD4 degrader [165] |
10 out of 12 currently approved and most of the clinically investigated toxins can be categorized into two main classes: Microtubule inhibitors such as auristatins, maytansinoids and tubulysins on one side and DNA damaging agents such as calicheamicin, duocarmycin and pyrrolobenzodiazipines on the other (Fig. 5). These toxins are small but usually lipophilic agents that negatively impact the stability and the binding profile of the conjugate especially at high DARs [166, 167]. One major difference between both toxin classes is that microtubule inhibitors, while very potent, primarily act on dividing cells. Due to the high proliferative rate of cancer cells compared to slowly dividing healthy cells, this creates another layer of specificity. On the other hand, slowly dividing cancer stem cells are harder to address with this class of toxin and can cause a relapse of the tumor. In contrast, DNA damaging agents are generally better able to attack non-dividing cells [168]. In the class of microtubule inhibitors, auristatins inhibit cell division by preventing GTP hydrolysis and therefore inhibit depolymerization and reorganization of the microtubules. The two most commonly used auristatins are MMAE and MMAF. The main difference between the two is the strongly reduced membrane permeability of MMAF due to the addition of a phenylalanine residue. In contrast to auristatins, maytansines and tubulysins dysregulate microtubules by promoting depolymerization [169, 170]. One of the most well-known maytansinoids is DM-1, which is used in the ADC trastuzumab emtansine (Kadcyla) and exhibits no bystander effect due to its positive charge and inability to cross membranes. Tubulysins, while acting functionally similar to maytansines, have more recently emerged as interesting toxins due to their ability to kill tumors that have shown multi drug resistance before [171, 172].
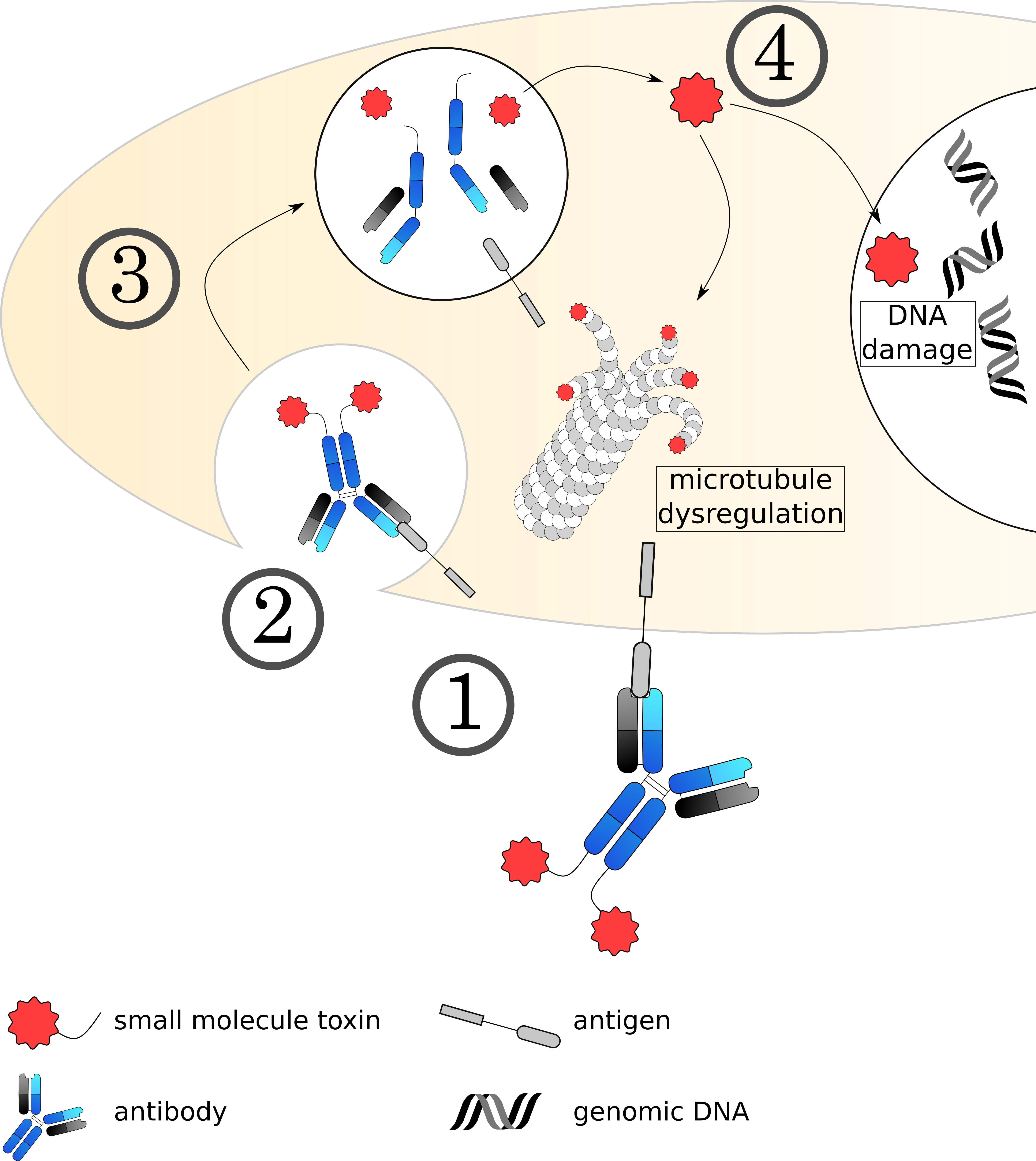 Fig. 5.
Fig. 5.Mechanisms of small molecule toxin ADCs. (1) The ADC binds to its target on the cell surface via the antibody, followed by (2) internalization of the complex. (3) The early endosome acidifies, matures and fuses with a lysosome. (4) The acidic, protease rich environment leads to a breakdown of the ADC, releasing the toxin to the cytoplasm. Currently used toxins induce cell death usually by dysregulating microtubule formation or damaging the DNA.
The second common class of toxins are DNA damaging agents that use four main ways to achieve apoptosis: (1) DNA alkylation (pyrrolobenzodiazepine, duocarmycin), (2) DNA strand breaks (calicheamicin), (3) Intercalation (anthracycline) and inhibition of topoisomerases that reduce shearing stress on the DNA (camptothecin derivatives). Alkylating agents can be further modified to act as DNA crosslinkers. Inhibitors of topoisomerase I or topoisomerase II cause DNA strand breaks by preventing re-ligation and trapping the DNA-enzyme complex after cleavage via a mechanism termed “interfacial inhibition” [173]. Topoisomerase inhibitors have seen a growing interest with two ADCs based on these toxins being recently approved. A big promise of many DNA-damaging agents is their ability to target resting cancer stem cells [174], although not all ADCs based on DNA damaging agents managed to fulfil this promise [175]. Some agents such as the alkylator duocarmycin exhibit very high potency in the pM range, allowing the killing of cells that express low levels of the antigen [176]. For more in-depth information about these two toxin classes, we recommend these reviews: [177, 178, 179].
While many different antibodies against various antigens have been developed into ADCs, comparatively few toxins have been established due to strict requirements regarding their potency, solubility, stability against metabolism and last but not least the ability to include a chemical handle for conjugation without disrupting the often intricate structure of the toxin. Due to the overall relatively narrow selection of available toxins, the development of toxins and agents attacking novel targets within the cell would vastly improve the options of ADC treatment, which is further enhanced by resistance mechanisms of cancers to currently used toxins and ADCs.
Amatoxins (reviewed in detail here [180]), in the ADC field mostly known for
Spliceosome inhibitors target the cell’s ability to generate functional mRNA and thereby impair protein expression. Thailanstatin A and Spliceostatins are currently under investigation. Studies could show effectiveness on HER2-expressing cells and gastric cancer xenograft models using a construct based on Trastuzumab [187, 188]. However, since the current number of studies on these molecules is rather scarce, it remains to be seen if they can be translated into powerful new ADCs.
Antibody oligonucleotide conjugates (AOC) combine two of life’s most important molecule classes: proteins and nucleic acids. Using oligonucleotides (ON) as a “drug” enables a multitude of new applications, ranging from technical methods like microscopy to therapeutic applications such as modulation of gene expression by siRNA and antisense RNA. ONs offer high customizability in their design and function, selectivity towards their target, and can interfere in a vast array of cellular processes since they are already a natural component of the cell. This approach allows targeting the expression of a broad selection of genes on the mRNA level and thereby protein expression, offering an interesting and powerful alternative to small molecule toxins (Fig. 6).
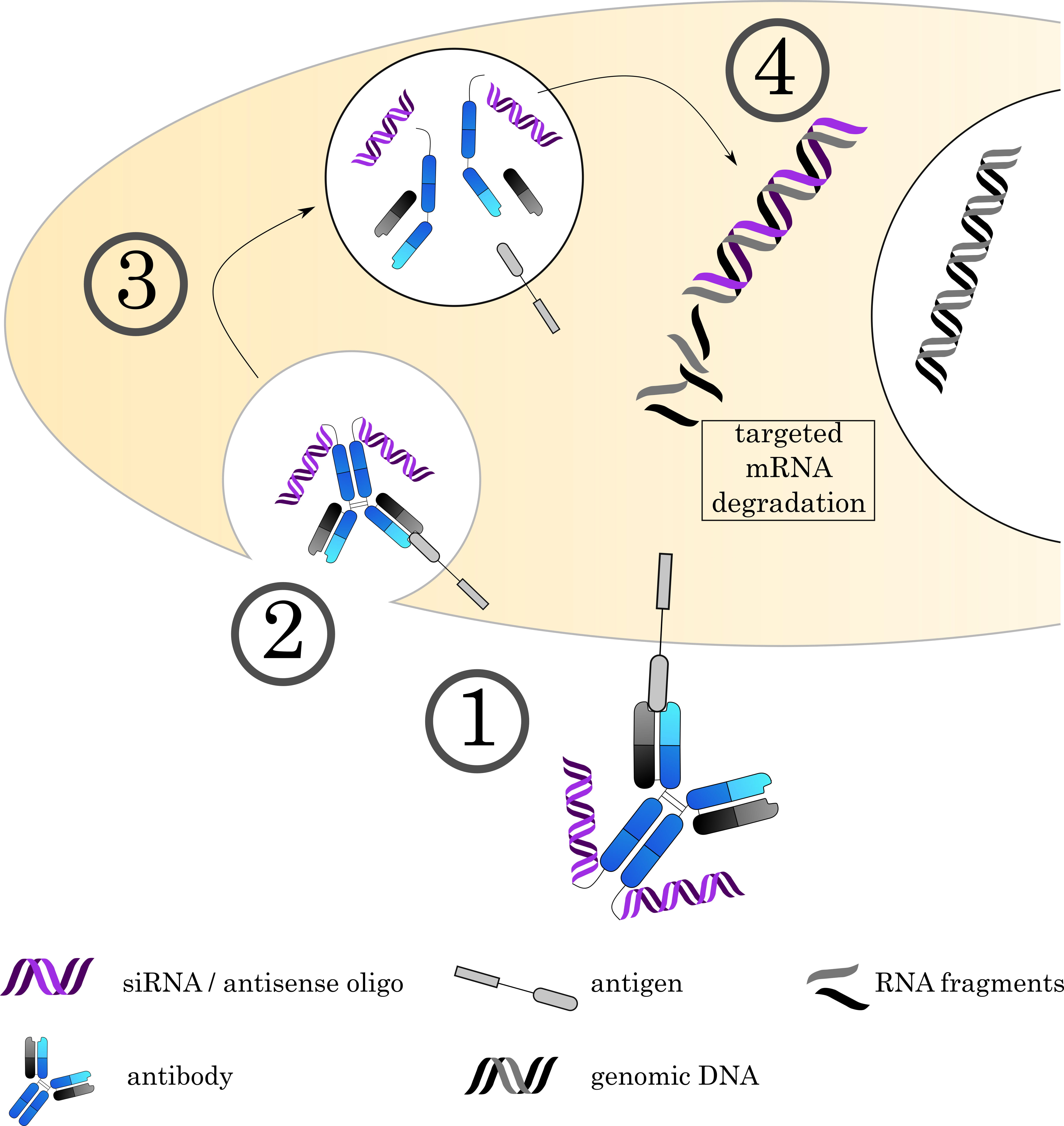 Fig. 6.
Fig. 6.Mechanisms of Antibody-oligonucleotide conjugates. (1) The AOC (depicted here is an AOC derived by chemical conjugation. Alternatively, carrier molecules like proteamine can be used as discussed in the text) binds to its target on the cell surface via the antibody, followed by (2) internalization of the complex. (3) The early endosome acidifies, matures and fuses with a lysosome. (4) The acidic, protease rich environment leads to a breakdown of the ADC, releasing the toxin to the cytoplasm. Current research focuses on siRNA and antisense oligos to achieve specific degradation of mRNA. However, alternative approaches also use the DNA as a carrier for small molecule toxins.
However, the use of ONs comes at a price: The large nature of ONs compared to more traditional small molecule toxins drastically changes the way the whole conjugate behaves. Even short ONs have a comparatively large weight and carry a strong negative charge, granting the conjugate pronounced characteristics of the ON itself even at low ratios of ON per antibody. Furthermore, both RNA and DNA exhibit low stability in serum, necessitating further development of more stable conjugates. While the high charge of the ON improves solubility of the conjugate, the ON itself cannot penetrate membranes unlike many small molecule toxins, impairing lysosomal escape to reach the target in the cytoplasm. This problem often necessitates the use of endosomal escape agents that destabilize the endosome and allow for more efficient release into the cytosol. Commonly employed agents, especially in the field of siRNA delivery, are disruptive peptides, which have the advantage of being easy to incorporate into the antibody as long as the antibody sequence is known, compounds that cause the protonic sponge effect to rupture the endosomal membrane and buffering additives such as chloroquine [189, 190, 191, 192, 193, 194]. Regarding the targeted antigen, the requirements are similar to those of small molecule toxin ADCs.
The strong charge of ONs presents an interesting opportunity: Loading the AOC via ionic interactions of the negative ON with a positive binding partner. To this end, proteamine [163, 195] and poly-arginine [196] are commonly used carriers to provide positive charges for the loading of ONs. A main advantage of this method is that the ONs do not need a functional group for conjugation, as well as the possibility to apply this strategy to a broad variety of different types of ONs without the need for chemical modification of the ON. Depending on the chosen antibody format and carrier molecule, both can be expressed together as one fusion protein. Additionally, the introduction of a moiety with strong positive charges is assumed to facilitate endosomal release of the ON due to the protonic sponge effect [194]. The first proof-of-concept for antibody-mediated delivery of siRNA dates back to 2005. In this study, Song et al. [164] used proteamine-Fab fusion protein for the delivery of siRNA and successfully targeted the HIV-1 capsid gene and thereby impaired virus replication, showing not only that antibodies can be efficient carriers of functional ONs, but also that loading via ionic interactions is strong enough for targeted delivery. Bäumer et al. [163] could show that their protamine-based AOC was able to slow the growth of colon cancer in vivo by delivering anti-KRAS siRNA via an anti-EGFR antibody.
However, while multiple groups reported effective use of AOCs created by this strategy, the reversible nature of ionic interactions raises concerns about stability of the complex. These concerns are especially pronounced in vivo. Dissociation of the ON from its carrier molecule would not only diminish the effectiveness of the conjugate itself but also likely lead to unwanted side effects. Further research is needed to improve our understanding of the stability, uptake and metabolism of AOCs relying on ionic interactions if this type of AOC should be developed for use in clinical trials.
One highly utilized strategy to generate AOCs is the use of lysine [197, 198] or cysteine [199] residues for protein surface conjugation. Antibody and ON are linked by bifunctional linkers, with one side targeting a surface feature of the antibody (lysine, cysteine), while the other functional group reacts with the ON. This forces the use of chemically modified ONs but enables the usage of a wide range of different established coupling strategies, as long as the chemical groups are inert towards the ON. Analogous to traditional small molecule ADCs, these strategies allow utilization of cleavable and non-cleavable linkers for improved control over the liberation of the ON.
Using direct conjugation of ON and antibody, multiple groups have reported promising results for the reduction of gene expression when loading the antibody with siRNA or antisense ONs. Arnold et al. [200] reported downregulation of a gene in glioblastoma stem cells that promotes cancer invasion. By using an anti-CD22 antibody coupled to an antisense ON targeting MXD3, Zhang et al. [201] could show increased survival in B-ALL xenograft mouse models. While full length antibodies still remain in the main focus of current research, other formats such as nanobodies see increased usage as well [201, 202]. In a very intricate study, Yu et al. [203] used a combination of antibodies and siRNA conjugated via a multi-functional peptide to deliver siRNA in vivo to a solid tumor and achieved a notable knock-down in their GFP-model.
Another particularly creative approach is the use of double stranded DNA not as the functional component itself, but as a carrier molecule for the DNA-intercalating toxin doxorubicin [204, 205]. The highly hydrophilic DNA can counteract the high hydrophobicity of commonly used toxins, which could provide one avenue to improve stability of the overall conjugate.
Although double stranded ONs such as siRNAs are most commonly used, a third important class of AOCs involves the coupling of single stranded ONs to the antibody and thereby creating another highly specific binding site for the conjugate. This strategy has seen extensive use in technical applications such as microscopy [110], immuno-PCR [206] and multiplexed protein detection [197]. However, single stranded ONs have also been utilized for toxin delivery. Dovgan et al. [207] provided proof-of-concept that single stranded ON-Antibody conjugates can be used as a platform to generate antibody-DNA-toxin conjugates, by hybridizing the complementary DNA strand fused to MMAE.
Proteolysis targeting chimeras (PROTACS) are at the forefront of targeted protein degradation. By binding both to the target protein and an E3 ligase, they promote poly-ubiquitinylation of the target by spatial proximity. The target protein is subsequently degraded via the ubiquitin-proteasomal pathway.
PROTACs are a highly interesting moiety, since — unlike commonly used small molecules — the catalytic mode of action enables effective action at sub-stoichiometric levels, promising high efficacy and potentially reduced side effects due to lower dosage of the drug. On top of that, PROTACs allow the targeting of proteins that have been deemed “hard to target” or even undruggable. As all novel technologies, PROTACs face larger hurdles for therapeutic use. Namely, due to their comparatively high mass, PROTACs exhibit despite improvements still low cellular uptake. Systemic administration also leads to poor tissue selectivity and increased side effects. Like antibodies are restricted to targeting surface proteins, PROTACs are restricted to proteins available in the cytosol.
PROTACs were conceptualized two decades ago. Sakamoto et al. [208, 209] developed a peptide that promotes the degradation of proteins by ubiquitinylation via the SCF complex. Further developments moved the PROTAC concept from peptides to small molecules [210] thereby improving cellular uptake compared to the previously used peptide-based PROTACs. However, cellular uptake remains a current problem for PROTACS. Although E3 ligases are a highly diverse class of proteins, the currently most frequently investigated E3 ligases in the PROTAC field are the Von-Hippel-Lindau ligase (VHL) [211, 212, 213] and Cereblon [214, 215]. The PROTAC concept has since then seen novel developments such as homo-PROTACS, consisting of two identical domains for degradation of E3 ligases [216, 217, 218], and photo-PROTACS for light induced activation [219, 220]. Related technologies, including lysosome-targeting chimeras, autophagy-targeting chimeras and autophagosome-tethering compounds have seen a rise in interest and present novel strategies, each featuring their own obstacles and advantages [221].
While most research has focused on the direct use of PROTAC molecules, the combination of PROTACs and antibodies has been a more recent development. Their potentially advantageous profile regarding dosage and enhanced range of targets make PROTACs a highly interesting moiety for use in ADC technology. With the antibody conferring higher tissue selectivity and the PROTAC high efficacy, both molecule classes complement each other well. Two main approaches have developed: Antibody-PROTAC conjugates that are similar in structure to classical ADCs. After internalization of the PROTAC-antibody conjugate, the PROTAC molecule is released and penetrates into the cytosol (Fig. 7). Maneiro et al. [165] used this strategy to specifically target the transcriptional regulator BRD4 in HER2+ breast cancer cell lines. Antibody-coated nanoparticles for the delivery of PROTACs also showed in vitro efficacy as well [222]. Dragovich et al. [223, 224] conducted a large scale study using different antibodies and linkers. In addition to yielding important insights into linker and conjugate design, they could also show in vivo efficacy of their conjugate when targeting BRD4 to VHL for degradation.
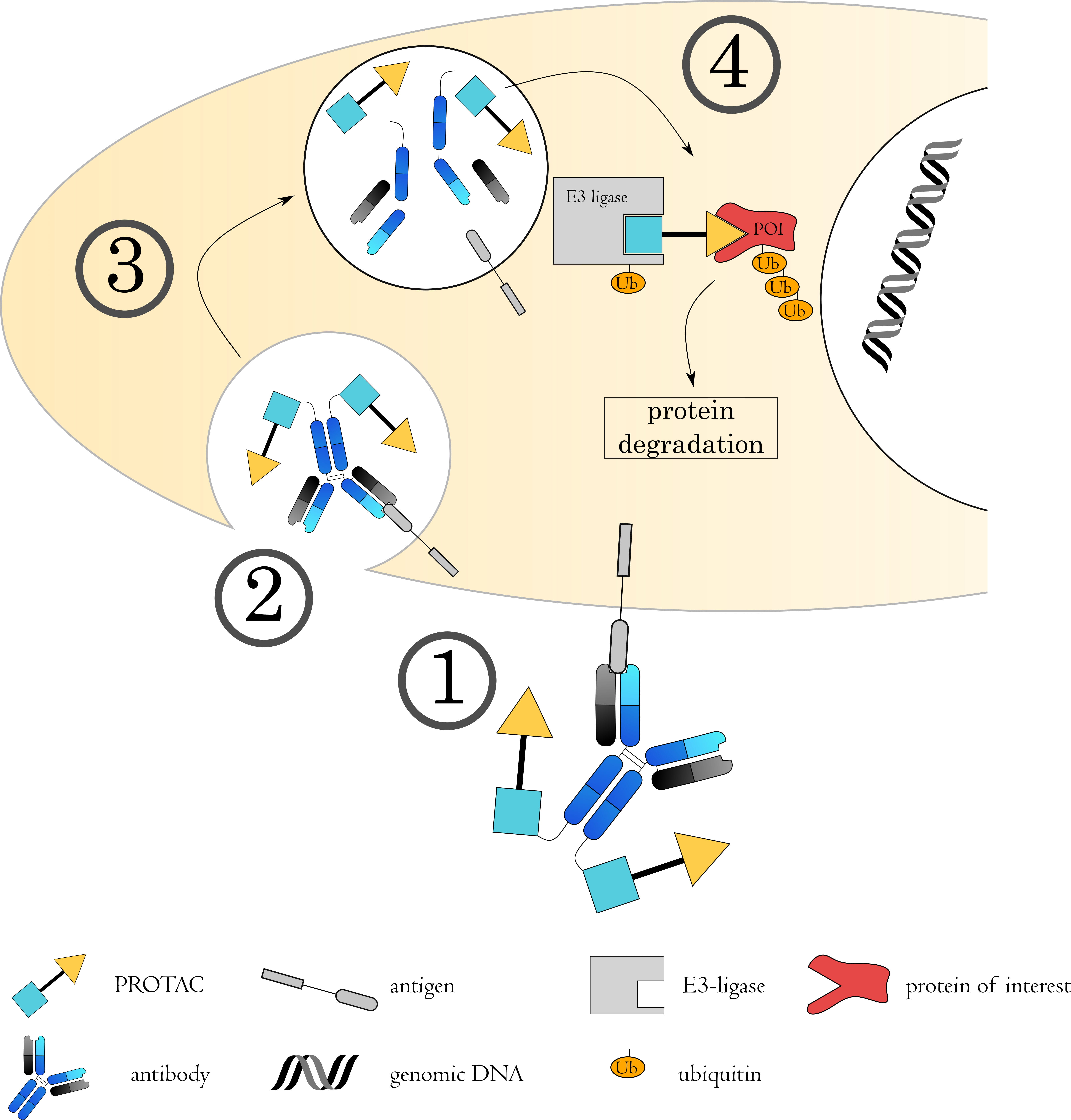 Fig. 7.
Fig. 7.Mechanisms of Antibody-PROTAC conjugates. (1) The antibody-PROTAC conjugate binds to its target on the cell surface via the antibody, followed by (2) internalization of the complex. (3) The early endosome acidifies, matures and fuses with a lysosome. (4) The acidic, protease rich environment leads to a breakdown of the ADC, releasing the toxin to the cytoplasm. Here, the PROTAC binds to both the protein of interest (POI) as well as an E3 ubiquitin ligase. This spatial proximity induces ubiquitin transfer to the POI, from where it will be targeted to the proteasome for degradation.
Although not strictly an ADC, a different mode of action is employed by antibody-based PROTACs (AbTACs). Here, a bispecific antibody fulfills both functions of the PROTAC with no small molecule necessary: The antibody binds both the protein of interest as well as the E3 ligase to generate close proximity of both proteins. Cotton et al. [225] reported this approach for the first time. Utilizing a bispecific antibody directed against both ring finger protein 43, a transmembrane E3 ligase, and programmed death ligand 1 (PD-L1) as the target protein, PD-L1 was successfully degraded.
The advancements of PROTACs in general with some molecules entering clinical trials and their use in combination with antibodies offer an exciting field of research and candidate for therapy. The combination of PROTACS with antibodies, or fragments thereof, as well as the use of AbTACs, can offer a new array of opportunities for the field of ADCs due to the high symbiotic potential of both moieties. Nevertheless, compared to other ADC approaches, antibody-PROTAC conjugates are still in their infancy, and as such suffer from many unknowns in novel technologies. Future studies have to investigate their factors like their specificity, stability, safety profile, pharmacokinetics and pharmacodynamics in detail.
An alternative to chemical drug conjugation is the recombinant fusion of antibodies with cytotoxic proteins (Fig. 8). Such constructs, known as immunotoxins, have the advantage of uniform batch production and hence circumvent the issues of DAR heterogeneity and resistance associated with conventional ADCs. While the utilization of immunotoxins is a principle that has been present for decades, much refinement to the original iterations of immunotoxins in the 1990s had to be made before they could approach clinical relevance. In following section, we examine the strides made over the last decade in antibody-immunotoxin development and application.
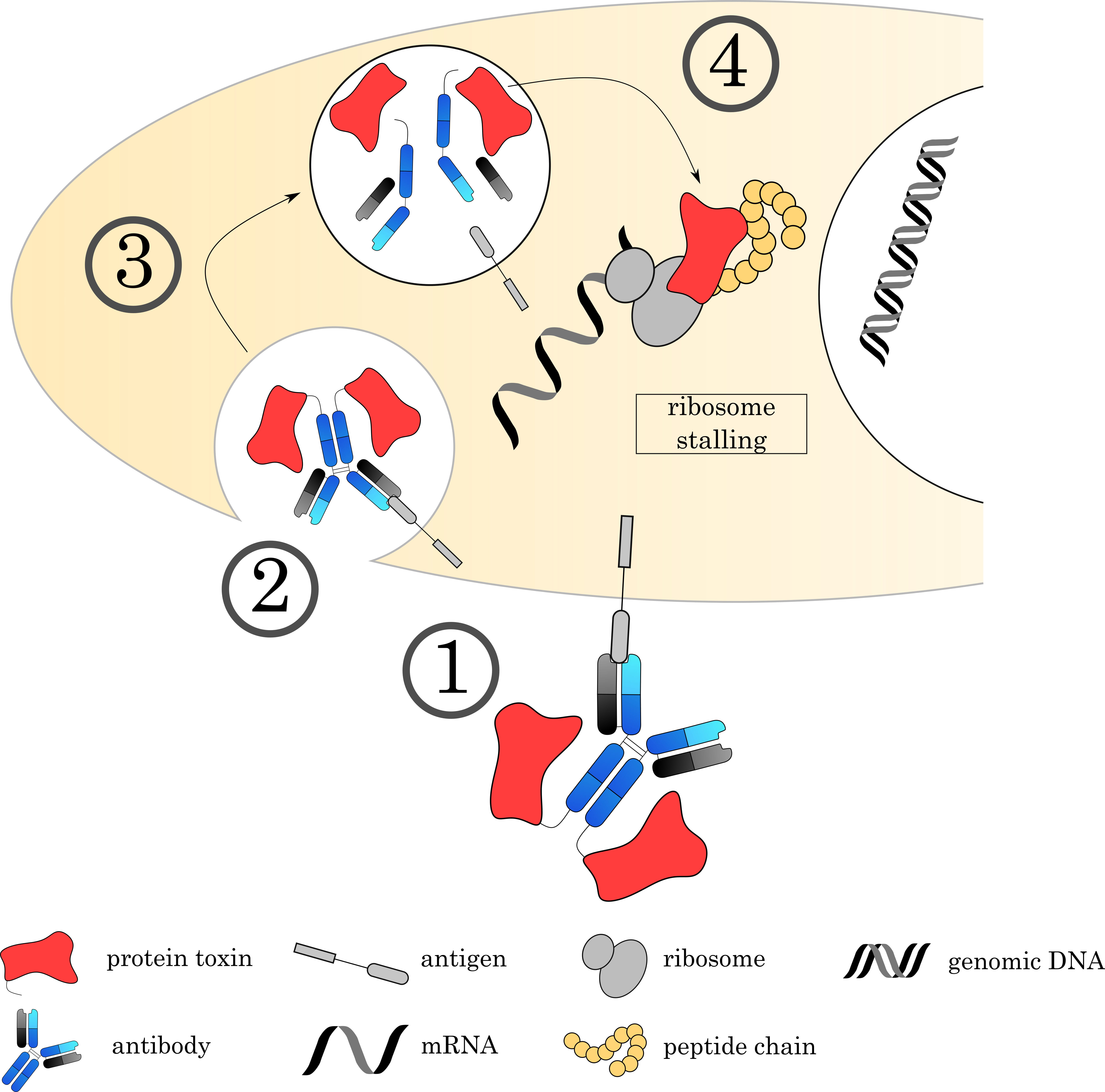 Fig. 8.
Fig. 8.Mechanisms of Immunotoxins. (1) The Immunotoxin binds to its target on the cell surface via the antibody, followed by (2) internalization of the complex. (3) The early endosome acidifies, matures and fuses with a lysosome. (4) Proteases digest a recognition sequence in the linker region, liberating the ADC and releasing the toxin to the cytoplasm. Current research focuses on immunotoxins that bind to EF2, an important factor for ribosomal translocation. This stalls the ribosome and inhibits protein synthesis.
The dearth of naturally-occurring cytotoxic proteins gives way to a wide pool of candidates for cytotoxic fusion partners. Derivatives of Pseudomonas exotoxin A (PE) are the most commonly used proteins for immunotoxin construction in the current landscape. PE’s mechanism of action involves its three domains: domain I facilitates cell surface binding and endocytosis via CD91, domain II aids cytoplasmic translocation, and domain III contains the catalytic ADP-ribosyltransferase site as well as a C-terminal endoplasmic localization sequence (REDL). Once endocytosed, PE is separated from the binding domain via furin cleavage within domain II and localizes to the endoplasmic reticulum. After translocation to the cytosol it ADP-ribosylates elongation factor 2 (EF2). This abrogates peptide translation along ribosomes and causes cell death through mass stalling of protein synthesis [226]. PE’s attractiveness as an immunotoxin comes from its efficiency - less than 1000 internalized PE molecules have been demonstrated as sufficient for cell death [227]. Truncated versions of PE are normally used in immunotoxins, wherein the binding domain is completely eliminated along with different lengths of domain II. Namely, PE40, PE38, and PE24 (numbers indicative of molecular weight in kDa) are currently being used clinically and experimentally.
Moxetumomab pasudotox, is the only FDA-approved antibody-based immunotoxin cancer treatment to date. Originally designed in 1997 as an anti-CD22 scFv fused to PE38, it pioneered the use of PE as an immunotoxin. Following the development of an affinity-matured version in 2003 and its outstanding efficacy in preclinical models, FDA approval was granted to moxetumumab pasudotox (Lumoxiti) in 2018 for the treatment of hairy cell leukemia [7]. Its progression to the market signified a milestone for antibody-based immunotoxins and dispelled apprehensions associated with the use of bacterial, potentially immunogenic, toxins for human therapy. Another well-advanced agent is oportuzumab monatox, an scFv-PE38 fusion against the TAA epithelial cell adhesion molecule (EpCAM) [228]. The excellent preclinical performance of the agent has led to its deployment in clinical trials under the brand name Vicineum (Sesen Bio) for the treatment of BCG-refractory urothelial carcinoma, where it is currently in phase III [229]. A number of scFv PE-based agents are also showing promising performances in earlier clinical trials, such as SS1P and LMB-2 for the treatment of mesothelioma and adult T-cell lymphoma, respectively [230, 231].
While the recent positive trajectory of PE-based immunotoxin translation is
obvious, concerns still exist regarding these agents’ immunogenicity. The
occurrence of antidrug antibodies during clinical studies due to the bacterial
origin of PE is commonplace [232]. Recent research has therefore been focused on
the modification of existing immunotoxins to achieve significantly lower
immunogenicities while maintaining comparable efficacies as the parent construct.
The process of systemically de-immunizing PE immunotoxins is being spearheaded by
the Laboratory of Molecular Biology (LMB) at the National Cancer Institute, who
had originally developed moxetumumab pasudotox, SS1P, and LMB-2 among others. The
group first significantly improved tolerability by removing most of domain II in
PE38, thus generating PE24 [233]. By removing the lysosomal protease-sensitive
domain II whilst maintaining its 11-amino acid furin cleavage site needed for
functionality, PE’s antigen processing and therefore immunogenicity was reduced
whilst maintaining the impressive cytotoxic efficacy of its parent construct.
Systemic identification of B- and T-cell epitopes allowed the group to produce a
variant of SS1P (termed LMB-T14) using a de-immunized mutant PE24, which
maintained comparable in vitro and in vivo efficacies to native
SS1P [234]. This likely signifies a watershed moment in immunotoxin technology,
wherein present and future constructs can be designed to minimize immunogenicity
and hence ease translation into the clinic. Alongside LMB-T14, de-immunized PE24
immunotoxins against hepatoma and HER2
Diphtheria toxin (DT) is another popular immunotoxin fusion protein, second only to PE. The mechanism of DT, similarly to PE, involves the stalling of protein synthesis by ADP-ribosylation of EF2. The DT structure is divided into two subunits, wherein domain A contains the catalytic site and facilitates endosomal escape, while domain B facilitates cell entry/endocytosis via HBEGF on the cell surface. The toxin is also further processed after endocytosis through furin-mediated cleavage and allowed to translocate to the cytosol, resulting translation stalling and highly efficient cell death. In another parallel to PE, immunotoxins generally utilize truncated versions of DT where domain B is replaced by the targeting moiety [226]. However, DT-based immunotoxins have not quite made the same clinical headway as their PE-based counterparts
DT2219, originally developed in 2005 by Vallera et al. [46], is a
bispecific construct composed of DT390 (amino acids 1-390 of full DT) linked to
sequential scFvs specific to CD19 and CD22. The bispecific design of DT2219 was
intended for the simultaneous treatment of CD19 and CD22-expressing cancer cells
in B-cell lymphoma. The agent succeeded experimentally, producing durable
antitumor responses in mouse xenografts positive for both antigens and
outperforming individually dosed single-target agents. DT2219 has therefore been
brought to the clinic, with a phase I-II trial having been concluded in 2019
[47]. Inspired by DT2219, a number of similar constructs have more recently been
developed. The same group engineered another bispecific DT390 construct targeting
epithelial cell adhesion marker (EpCAM) and HER2 [238]. The agent, termed
DTEpCAM23, produced picomolar IC
Beyond PE and DT, a wide array of experimental trials has been conducted with
alternative cytotoxin payloads. One example is gelonin, a plant-derived
ribosome-inactivating protein. Pirie et al. [241] demonstrated the
viability of gelonin-based immunotoxins against CEA and EGFR, producing
IC
Saporin, like gelonin also a ribosome inhibitor, has been shown to have
anti-tumoral effects in vitro as a free toxin [245]. Giansanti
et al. [246] further fused saporin to the CDZ domain of human
calcium/calmodulin-dependent serine protease to generate a molecule that binds
extracellularly to CD96. This construct showed toxicity on glioblastoma cell
lines. Saporin also showed anti-tumoral activity as an anti-Her3 ADC in melanoma
cell lines as well as a reduction of metastasis in a murine lung cancer model
[247]. Further studies have shown activity against non-Hogdkin’s lymphoma in
xenograft mouse models after conjugation to an
Given the inherent risks of using foreign proteins in therapy, the utilization of human-derived cytotoxins as payloads is a natural point of attention. To this end, toxins such as granzyme B and tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) have previously been fused to targeting moieties to facilitate targeted, cell death without the risks of immunogenicity [251]. While tumor growth control has been documented, the field has yet to take off due to these toxins’ lower effectiveness in comparison with the aforementioned bacterial and plant-derived proteins. However, recent studies have demonstrated nanomolar cytotoxicities in vitro using granzyme B and TRAIL-linked scFvs [252, 253]. Whether in vivo performances similar to non-human toxins can be obtained remains to be seen, but would represent a monumental step in the development of safe and effective immunotoxins.
One important feature of immunotoxins is that the antibody can be fused on the genetic level to the desired toxin and expressed as one amino acid chain. This abolishes the need for chemical conjugation and complicated purification methods to remove excess compounds and thereby simplifies the downstream processing significantly. The product is highly homogenous regarding the DAR but depending on the nature of the toxin and the format, high DARs can be difficult to achieve using genetic fusions only. This approach also requires cloning of individual candidates before they can be investigated, which can prove to be a laborious process in contrast to chemical conjugation methods that allow rapid screening of the most promising combination of antibody and drug.
Cytokines are a biologically and functionally very diverse group of proteins.
They play an important role as signaling molecules and modulators of the immune
system and response to an infection. Application of cytokines therefore allows
direct interaction with and stimulation of immune cells to strengthen favorable
therapeutic effects. While studies reported efficacy of the treatment for some
cancers and many cytokine products have either received approval (e.g.,
IFN
Antibody-cytokine fusions (immunocytokines, Fig. 9) are a promising strategy to improve the clinical performance of cytokines. Similar to the concept of small molecule toxin ADCs, the antibody-based carrier molecule allows for tissue specific delivery of the cytokine payload, thereby allowing for higher concentration within the tumor tissue and decreasing systemic effects. The increased molecular weight of the fusion protein also leads to slower clearance and longer half-life of the therapeutic compared to the small cytokines, presenting another advantage for the immunocytokine concept. Generally, the immunocytokine binds to the tumor via the antigen binding site of the antibody, while the cytokine recruits and activates an immune cell in close proximity. Immunocytokines have been a field of research that has been studied for decades. Covering all constructs in detail would breach the scope of this review. Therefore, we will focus on discussing key design concepts and considerations. For further reading and in-depth analyses of single cytokines and immunocytokines, we strongly recommend the following reviews: [257, 258, 259, 260, 261].
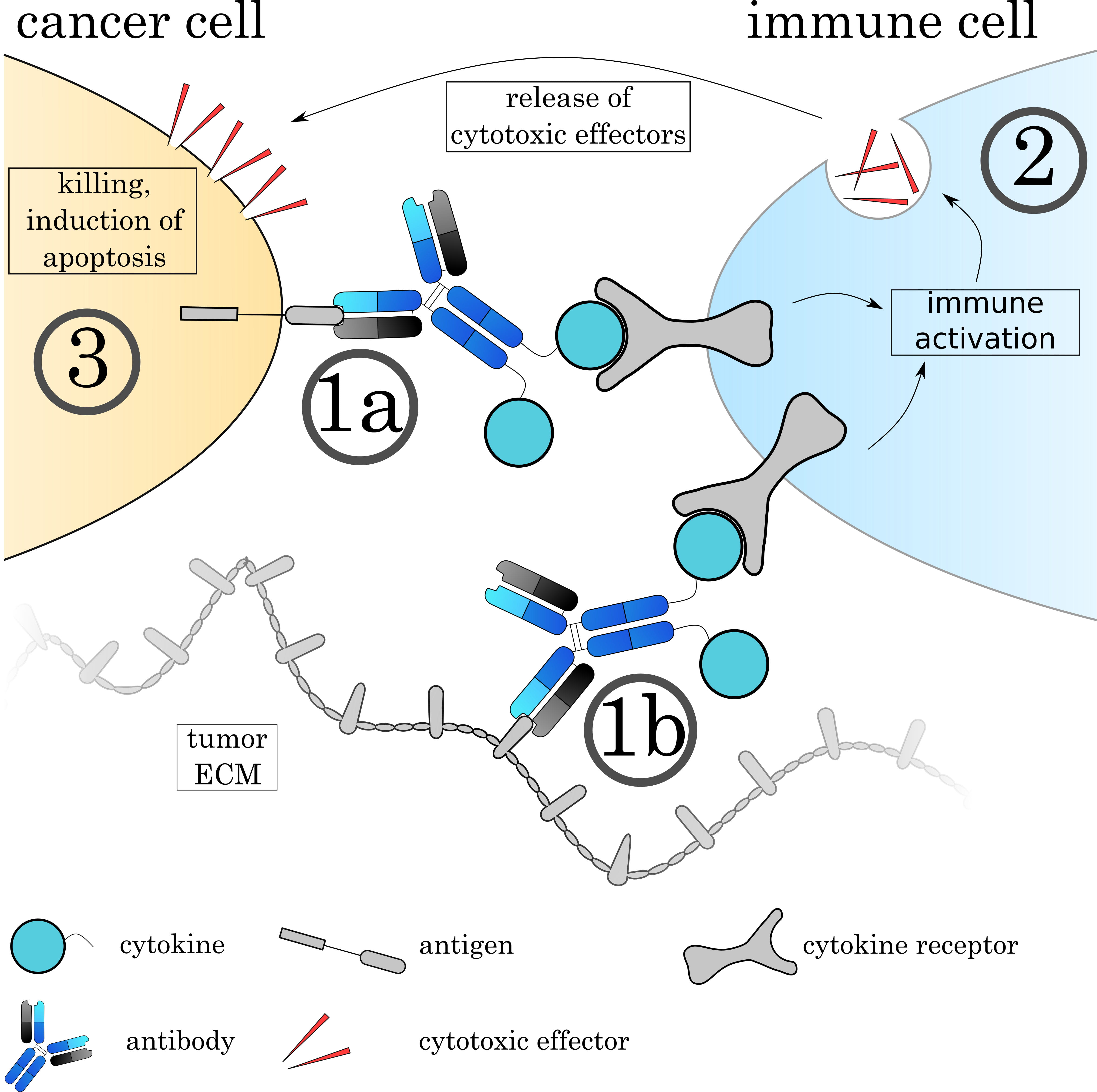 Fig. 9.
Fig. 9.Mechanisms of Immunocytokines. The Immunocytokine binds to its target either on the cell surface (1a) or on the tumoral extracellular matrix (1b) via the antibody. (2) Immune cells recognize the attached cytokine via their respective cytokine receptor, leading to activation of the cell. The effect varies depending on the cytokine and immune cell. (3) Many immune cells such as cytotoxic T cells and natural killer cells release cytotoxic agents to induce apoptosis on the target cell.
One important consideration for immunocytokine design is obviously the choice of cytokine. The cytokine will define the mechanism of action and thereby the activated downstream pathways, as well as the targeted cell type. Cytokines can have a range of effects ranging from “immune-activating” cytokines that are best suited for the therapy of cancers to “immune-suppressing” cytokines that can help to alleviate auto-immune diseases and chronic inflammation. Depending on the cytokine, effects can include activation and/or expansion of T-cells, NK-cells or macrophages, directly inhibit tumor growth or angiogenesis or mature antigen presenting cells [258]. The therapeutic aim must be designed depending on susceptibilities and resistances of the cancer or disease. Additionally, it is important to consider that cytokines not only act as monomers (e.g., IFNa and IL-2), but others also as hetero-dimers (IL-12), homo-dimers (IFNy) or trimers (TNFa). The multimerization of the cytokine chains will therefore introduce additional antibody molecules to the complex and lead to multivalent binding. Due to the fact that many immune cells carry cytokine receptors on their surface, one problem that immunocytokines face is cytokine-mediated binding while in circulation to healthy cells. As shown by Tzeng et al. [262], the cytokine can even overrule the specificity of the antibody. In an optimal case, the cytokine should be functionally ‘caged’ during circulation and functionality restored only at the target site.
Multiple antibody formats have been used for immunocytokine generation, ranging from smaller formats like scFv to and full-length antibodies, mostly of the IgG type, as well as mono-, di- and trivalent constructs. The format size comes with the previously discussed trade-off between improved tissue penetration and extravasation for smaller formats on one side and faster clearance from the system on the other. While the antibody itself does not necessarily have to exhibit anti-tumoral functionality, full-length-antibodies offers a second vector of attack by ADCC [263] and CDC [264]. Therefore, the construct can utilize two distinct modes of action for maximized therapeutic effects. Current targets of immunocytokines include cellular tumor markers such as EpCAM and CD20 [162, 265, 266] as well as proteins of the tumor stroma and tumor ECM proteins such as splice isoforms of fibronectin and tenascin-C [161, 267, 268, 269] or the targeting of DNA/Histone complexes [270]. Unlike small molecule toxin ADCs, immunocytokines act without internalization. Similar to immunotoxins, the possibility to fuse the antibody and cytokine on a genetic level comes with both the advantages of a defined product and simplified purification, but at the cost of low throughput during screening.
In summary, antibody-cytokine fusions are a highly promising approach to circumvent common problems of current free cytokine therapy that, despite having has shown promising results in combinatorial therapy, but still struggles from often narrow therapeutic windows due to unspecific tissue distribution and thereby side effects.
ADCs have emerged as one of the most likely pillars in the future of oncology—a fact emphatically highlighted by the slew of recent FDA approvals, as well as the numerous agents currently in late-stage clinical trials. The strides made in the field since the initial approval of Mylotarg in 2000 has been a testament to the growth capacity of ADC technology, be it in conjugation chemistry, linker technology, or payload development. The industry, however, still remains relatively young. The inherently vast modularity of ADCs affords room for continuous innovation, not least of which has been seen within the scope of novel experimental payloads. In this sense, the term “drug” is likely to be reimagined in the context of ADCs, and far exceed its traditional association to small molecule toxins within the coming years.
JS, MA and AS conceived the manuscript. MA wrote the introduction, section 2, section 4.4 and designed Fig. 2. AS wrote section 3 and designed Figs. 1,3. JS wrote section 4 (except 4.4) and designed Figs. 5,6,7,8,9. JS, MA and AS were involved in corrections and revisions of the manuscript and figures. JS did the final formatting and proof reading.
Not applicable.
We would like to thank Heinrich Leonhardt for his support.
This work was funded by grants from the German federal ministry for education and research (Bundesministerium für Bildung und Forschung, grant no. 16GW0358) to Heinrich Leonhardt (Ludwig-Maximilians-Universität München, Department of Biology II, Human Biology and Bioimaging, Munich, Germany).
The authors declare no conflict of interest.
Publisher’s Note: IMR Press stays neutral with regard to jurisdictional claims in published maps and institutional affiliations.