


1 Department of Internal Medicine, University of Kansas Medical Center, Kansas City, KS 66160, USA
2 Department of Internal Medicine, Division of Medical Oncology, University of Kansas Medical Center, Kansas City, KS 66205, USA
Academic Editor: Roberto Bei
Abstract
Utilizing viruses in the treatment of cancer, or oncolytic viral therapy (OVT), began in the 1950s with the idea that viruses could invade and destroy cancer cells. Barriers to this approach included a lack of specificity towards cancer cells and intolerable toxicities. However, it was discovered that OVT increases cytokines such as interferon gamma and interleukins within the tumor microenvironment. This “priming” of the tumor microenvironment can lead to an improved innate immunologic response to tumor cells. An “OVT-as-monotherapy” approach has led to modest tumor response rates that have unfortunately not translated well in clinical trials. Currently, only one OVT agent—talimogene laherparevec (TVEC)—has been approved by the FDA for unresectable melanoma with limited visceral metastases. Further advancements in immunotherapy combined with improved viral engineering over the last decade have paved the way for a renewed focus on OVT. For example, various viruses have been modified to infiltrate and upregulate PD-L1 signaling within tumor cells. Upregulation of PD-L1 on tumor cells can increase tumor cell response to immunotherapies that utilize the interaction between PD-L1 on tumor cells and PD-1 on lymphocytes to allow for immune cell destruction of cancer cells. Combining OVT and immunotherapy offers more promise than OVT as monotherapy. Currently, several are actively investigating the combinatorial approach of OVT and immunotherapy in treating non-small cell lung cancer (NSCLC), colorectal cancer (CRC), breast cancer, melanoma, pancreatic cancer, multiple myeloma, and head and neck squamous cell carcinoma. In this review, we will discuss the history of OVT including its limitations as a monotherapy. We will also discuss the background of combining OVT and immunotherapy including possible benefits and pitfalls of this approach. Lastly, we will review current clinical trials investigating OVT and immunotherapy in multiple cancers.
Keywords
- oncolytic viral therapy
- immunotherapy
- immune checkpoint inhibition
The idea of using viruses to treat cancer cells has been around nearly as long as the discovery of viruses themselves. In 1892, Dmitri Ivanovsky [1] described an agent which was “non-bacterial” affecting the tobacco plant, naming this the tobacco mosaic virus. Six years later, “foot and mouth virus” was not only filtered out by scientists but was directly implicated in causing animal disease [2]. Three years later, in 1901, the yellow fever virus became the first virus discovered by scientists causing disease in humans [3]. Finally, in 1931, Max Knoll and Ernst Ruska obtained the first images of viruses with the invention of electron microscopy [4].
During the forty years between Ivanovsky’s discovery and Knoll and Ruska’s images, several case reports were published detailing patients with cancers who magically improved after becoming infected with viral illnesses. In 1897, roughly 5 years after the Ivanovsky’s discovery of the tobacco mosaic virus, George Dock [5] published a case report of a 42-year-old woman with leukemia who became afflicted with influenza. Notably, this was roughly 4 decades before it was known that a virus was responsible for influenza. Dock reported that after his patient developed “tonsillitis and coryza” as well as severe fatigue and weakness, her white blood cell count decreased roughly 70-fold with a concordant reduction in her hepatosplenomegaly as well. She was able to live for roughly one more year after this infection, a remarkable achievement for a patient with acute leukemia at that time. In 1951, there was another case report involving a four year-old boy with profound hepatosplenomegaly and lymphocytic leukemia with a white blood cell count of 300,000, hemoglobin of 5.6 grams per deciliter, and a platelet count of 150,000 [6]. He was treated with intravenous iron for 33 days with no effect and discharged home. About 12 days later, he had a fever of 38.6 degrees Celsius with new, enlarged cervical lymph nodes and vesicles on his face, scalp, and neck consistent with varicella. After seven days, his white blood cell count decreased to 7500 and he no longer had any hepatosplenomegaly. This “remission” lasted roughly 1 month, and he unfortunately died shortly afterwards from “acute lymphatic leukemia”. While the ultimate outcome of these case reports of patients with mostly hematologic malignancies typically ended in death after a few months, they offered a possible window into new cancer therapeutics. This window could be summarized with the following principle: In mostly hematologic malignancies, which tended to have the most rapid cell growth and produce the most depleted immune systems in patients, certain viruses under the right contexts could induce short remissions.
In 1947, Sidney Farber [7] published the results of his study utilizing
aminopterin—often considered the first successful chemotherapy agent—to
induce remissions in children with leukemia from his basement lab in Boston. Two
years later, alongside the infancy of chemotherapy, a clinical trial was
presented at the American Association for Cancer Research in which 22 patients
with Hodgkin’s lymphoma were “treated” with the hepatitis B virus [8]. In this
study, 7 out of 22 patients had a demonstrable clinical response with 4 out of 22
patients experiencing a reduction in tumor size. Unfortunately, 14 out of 22
patients developed hepatitis. Over the next 30 years, there were at least three
other clinical trials in which cancer patients were infected with viruses. In
1952, 34 patients with various cancers were infected with Egypt Virus 101 in
which only 4 patients demonstrated tumor regression [9]. In 1956, 30 patients
with cervical cancer were infected with adenovirus with no tumor responses seen
[10]. In 1974, 90 patients with various cancers were treated with the mumps virus
in which 37 patients experienced either a complete regression or tumor shrinkage
of
Owing to both the lack of a positive signal and improved ethical guidelines in clinical trials in the 1970s, there was a pause on treating cancer patients with non-attenuated viruses. Instead, the ability to develop ex vivo culture of human cells allowed researchers to use rodent models to further test oncolytic viral therapies. One study utilized various cancer rodent models and infected them with very large doses of the Russian Far East encephalitis virus producing complete regressions [12]. Unfortunately, in each case, all rodents died of encephalitis shortly afterwards. Clearly, viruses showed great potential although their unpredictability and pathogenicity continued to prevent any meaningful progress until viral engineering became available in the 1990s.
Historically, oncolytic viral therapy (OVT) focused on direct entry and replication of viruses within rapidly dividing tumor cells causing their destruction. However, this paradigm has changed, and more promising potential lies in OVT transforming the tumor microenvironment from an immunosuppressed environment into an immunostimulatory environment in combination with checkpoint inhibitor therapies (see Fig. 1). In normal cells, protein fragments are expressed on major histocompatibility (MHC) class I molecules on the surface of the cell [13]. Cytotoxic T cells (CTL) then recognize either healthy or viral/tumoral antigens via an interaction between T cell receptors (TCRs) and MHC class I molecules. This interaction causes the CTL to either spare the cell (if healthy) or kill the cell (if viral/tumoral). However, tumor cells manipulate this interaction in many ways including by evading recognition by the CTL or suppressing antigen presentation [14]. Either through viral engineering or naturally occurring mechanisms, OVT offers a therapeutic strategy that circumvents this problem.
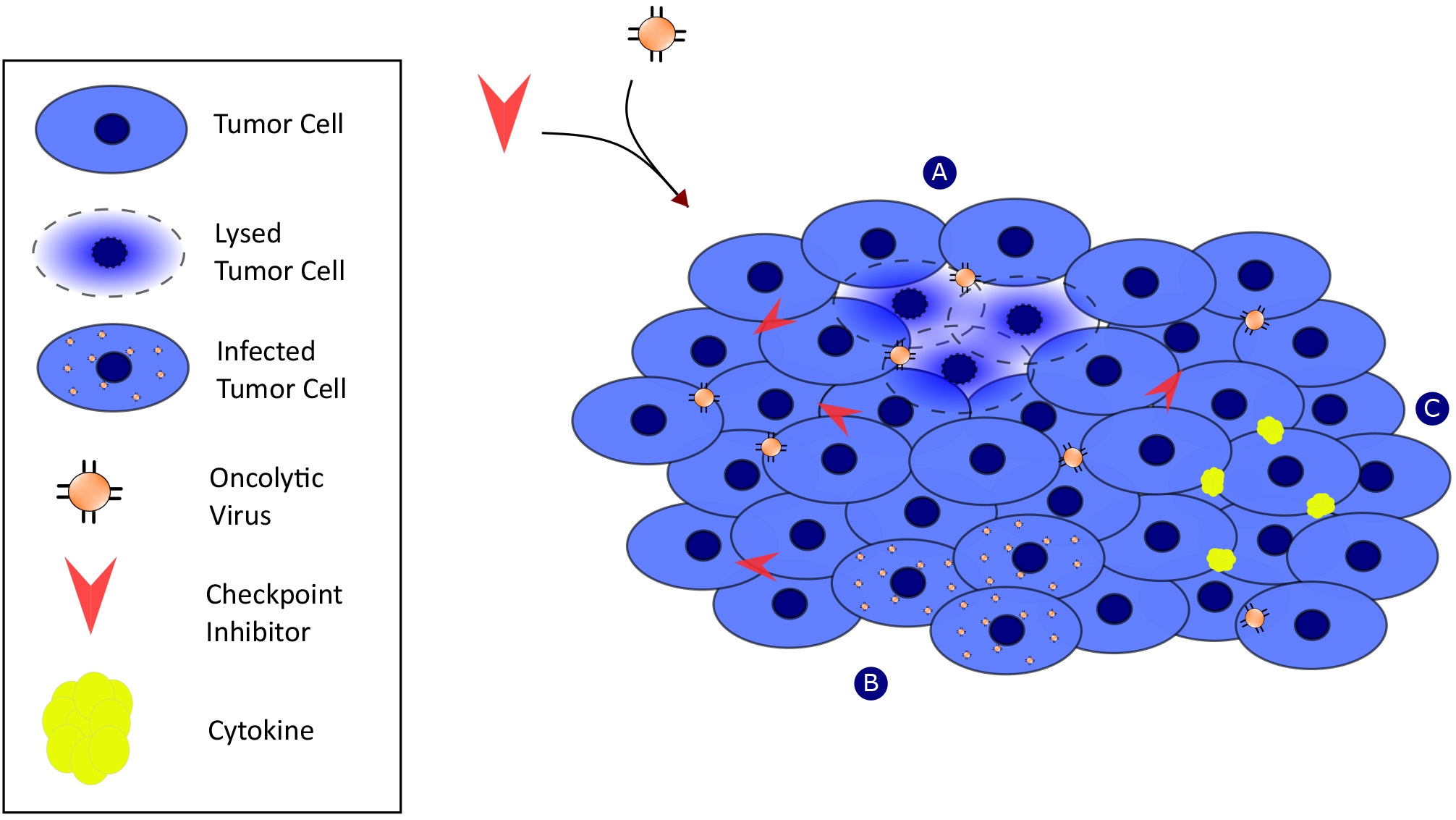 Fig. 1.
Fig. 1.Oncolytic viruses mediate antitumor response synergistically with checkpoint inhibitor activity through multiple mechanisms of action. Mechanism-A: viral replication within tumor cells. Mechanism-B: Induction of tumor cell death via Mechanism-A. Mechanism-C: release of cytokines and initiation of innate immunity.
The first OVT approved by the FDA in 2015 was Talimogene laherparepvec (T-VEC), an intralesional therapy for unresectable melanoma with limited visceral disease [15]. T-VEC is an attenuated herpes simplex virus I (HSV-1) that is genetically modified to specifically infect and lyse cancer cells and preserve normal cells. Normal cells block viral replication via the enzyme protein kinase R (PKR) which phosphorylates eukaryotic initiation factor 2 (eIF2). Activated eIF2 causes the disruption of viral protein synthesis [16]. HSV-1 contains infected cell protein 34.5 (ICP34.5) which dephosphorylates eIF2 and allows viral protein synthesis and subsequent viral replication to succeed [17]. Infected cell protein 47 (ICP47) is another protein produced by herpes viruses that inhibits antigen presentation to CTSs and allow for further viral proliferation [18]. In T-VEC, the genes for both ICP34.5 and ICP47 are deleted and replaced with human granulocyte-macrophage colony stimulating factor (GM-CSF) [19]. In cancer cells, the PKR-eIF2 defense mechanism against viruses is defective [16]. Therefore, T-VEC may safely infect normal cells without replicating within and destroying them and instead target cancer cells causing their destruction. This feat of viral engineering ultimately led to a phase III clinical trial of 436 patients with Stage IIIb–IV unresectable melanoma in which participants were enrolled in a two-to-one ratio to T-VEC and GM-CSF [15]. While T-VEC did not demonstrate an overall survival benefit, patients who were treated with T-VEC had a higher durable response rate compared to CM-CSF, which led to FDA approval.
While T-VEC may be limited as a monotherapy, several trials are evaluating the immunostimulatory action of T-VEC within the tumor microenvironment and combining it with immune checkpoint inhibitors including pembrolizumab and atezolizumab for hepatocellular carcinoma, breast cancer, and other gastrointestinal cancers (see Table 1). As checkpoint inhibitors specifically act to restore normal immune function by blocking immunoinhibitory proteins, combining these therapies with T-VEC may create a powerful synergistic treatment strategy. In 2016, a clinical trial combining T-VEC with the CTLA-4 inhibitor, ipilimumab, in previously untreated and resectable Stage IIIb–IV melanoma revealed a response rate of 50% [20]. This is an improvement from previous response rates with ipilimumab monotherapy (11%) and T-VEC monotherapy (26%) [20]. Other possible combinatorial therapies with OVT include PD-L1 and PD-1 inhibitors [21]. As OVTs like T-VEC specifically attract T-cells to the tumor microenvironment and upregulate tumor expression of PD-L1, for example, combining T-VEC with pembrolizumab offers a mechanism to enhance treatment response of an already effective therapy [22]. One of the limitations of anti-PD-1 therapy is that it is most effective against tumors which express a high amount of PD-L1. If PD-L1 levels were artificially enhanced by OVTs like T-VEC, then deeper responses may be observed [22]. Currently, there are several clinical trials evaluating the efficacy of T-VEC monotherapy or in combination with immune checkpoint inhibitors against melanoma, non-melanoma skin cancer, pancreatic cancer, unresectable gastrointestinal or ovarian cancer, angiosarcoma, sarcoma, rectal cancer, liver cancer, and breast cancer (see Table 1).
| Study title | Identifier | Interventions | Condition | Phase | Status |
| Neo-adjuvant T-VEC + nivolumab combination therapy for resectable early metastatic (Stage IIIB/C/D–IV M1a) melanoma with injectable disease | NCT04330430 | Talimogene laherparepvec (TVEC) | Stage III melanoma | Phase II | Recruiting |
| Stage IV melanoma | |||||
| Study of talimogene laherparepvec (T-VEC) in pancreatic cancer | NCT03086642 | Talimogene laherparepvec (TVEC) | Pancreatic cancer | Phase I | Recruiting |
| Talimogene laherparepvec for the treatment of peritoneal surface malignancies | NCT03663712 | Talimogene laherparepvec (TVEC) | Stage IV Peritoneal Surface Dissemination From Gastrointestinal or Recurrent, Platinum-resistant Ovarian Cancer That Cannot be Completely Resected | Phase I | Recruiting |
| Combination of talimogene laherparepvec with atezolizumab in early breast cancer | NCT03802604 | Talimogene laherparepvec (TVEC) | Breast cancer | Phase I | Recruiting |
| Atezolizumab | |||||
| A study of talimogene laherparepvec (T-VEC) in combination with pembrolizumab in patients with metastatic and/or locally advanced sarcoma | NCT03069378 | Talimogene | Sarcoma | Phase II | Recruiting |
| Laherparepvec (TVEC) | Epitheloid sarcoma | ||||
| Cutaneous angiosarcoma | |||||
| Talimogene laherparepvec in combination with neoadjuvant chemotherapy in triple negative breast cancer | NCT02779855 | Talimogene | Breast cancer | Phase I/II | Active, not recruiting |
| Laherparepvec (TVEC) | Ductal carcinoma | ||||
| Paclitaxel | Invasive breast carcinoma | ||||
| Invasive ductal breast carcinoma | |||||
| Talimogene laherparepvec with pembrolizumab in melanoma following progression on prior anti-PD-1 based therapy (MASTERKEY-115) (Mk-3475-A07/KEYNOTE-A07) | NCT04068181 | Talimogene | Melanoma | Phase II | Active, not recruiting |
| Laherparepvec (TVEC) | |||||
| Pembrolizumab | |||||
| Talimogene laherparepvec, nivolumab and trabectedin for sarcoma | NCT03886311 | Talimogene | Sarcoma | Phase II | Recruiting |
| Laherparepvec (TVEC) | |||||
| Nivolumab | |||||
| Trabectedin | |||||
| Talimogene laherparepvec, chemotherapy, and radiation therapy before surgery in treating patients with locally advanced or metastatic rectal cancer | NCT03300544 | Talimogene Laherparepvec (TVEC) | Locally advanced rectal adenocarcinoma | Phase I | Recruiting |
| Capecitabine | Metastatic rectal adenocarcinoma | ||||
| Fluorouracil | Stage IIIA–IVB rectal adenocarcinoma | ||||
| Leucovorin | |||||
| Oxaliplatin | |||||
| Radiation therapy | |||||
| Trial to evaluate the safety of talimogene laherparepvec injected into tumors alone and in combination with systemic pembrolizumab MK-3475-611/Keynote-611 | NCT02509507 | Talimogene Laherparepvec (TVEC) | Hepatocellular carcinoma | Phase I/II | Active, not recruiting |
| Pembrolizumab | Liver metastases | ||||
| Study of talimogene laherparepvec with atezolizumab for triple negative breast cancer and colorectal cancer with liver metastases | NCT03256344 | Talimogene Laherparepvec (TVEC) | Metastatic triple negative breast cancer | Phase I | Active, not recruiting |
| Atezolizumab | Metastatic colorectal cancer with liver metastases | ||||
| Ipilimumab, nivolumab, and talimogene laherparepvec before surgery in treating participants with localized, triple-negative or estrogen receptor positive, HER2 negative breast cancer-deleted | NCT04185311 | Talimogene Laherparepvec (TVEC) | Estrogen receptor positive, HER2 negative localized breast cancer | Phase I | Active, not recruiting |
| Ipilimumab | Triple negative localized breast cancer | ||||
| Nivolumab | |||||
| Phase I study of TBI-1401(HF10) plus chemotherapy in patients with unresectable pancreatic cancer | NCT03252808 | HF10 | Stage III pancreatic cancer | Phase I | Active, not recruiting |
| Gemcitabine | Stage IV pancreatic cancer | ||||
| Nab-paclitaxel | |||||
| TS-1 | |||||
| Study of pembrolizumab and M032 (NSC 733972) | NCT05084430 | M032 | Glioblastoma multiforme | Phase I/II | Not yet recruiting |
| Pembrolizumab | Anaplastic astrocytoma | ||||
| Gliosarcoma | |||||
| Genetically engineered HSV-1 Phase 1 study for the treatment of recurrent malignant glioma | NCT02062827 | M032 | Recurrent Glioblastoma multiforme | Phase I | Recruiting |
| Progressive glioblastoma multiforme | |||||
| Anaplastic astrocytoma | |||||
| Gliosarcoma | |||||
| HSV G207 in children with recurrent or refractory cerebellar brain tumors | NCT03911388 | G207 | Recurrent or progressive cerebellar neoplasms of the brain | Phase I | Recruiting |
| HSV G207 alone or with a single radiation dose in children with progressive or recurrent supratentorial brain tumors | NCT02457845 | G207 | Recurrent or progressive supratentorial neoplasms of the brain | Phase I | Active, not recruiting |
| OH2 oncolytic viral therapy in pancreatic cancer | NCT04637698 | OH2 | Pancreatic cancer | Phase I/II | Recruiting |
| OH2 oncolytic viral therapy in solid tumors | NCT03866525 | OH2, with or without irinotecan or HX008 | Solid tumors | Phase I/II | Recruiting |
| Gastrointestinal cancer | |||||
| OH2 injection in solid tumors | NCT04386967 | OH2 | Solid tumors | Phase I/II | Recruiting |
| Pembrolizumab | Melanoma |
While T-VEC is the only FDA-approved OVT, there are several other herpes-based
OVTs that are under investigation as monotherapies, in combination with immune
checkpoint inhibitors, or in combination with chemotherapies. HF-10 is a
naturally occurring HSV-1 OVT which has deletions in UL56 and
LAT and increased expression of UL53 [23]. While the ultimate
purpose of these natural genetic mutations is unknown, HF-10 has demonstrated
antitumor responses in murine models of melanoma [24]. M032 is another
herpes-based OVT that has been reengineered with a deletion of the
Like herpes-based OVTs, adenovirus-based OVTs are modified to include deletions in genes that promote virulence in healthy cells. The first adenovirus-based OVT was H101, which includes E1B and E3 gene partial deletions. H101 was approved by the China Food and Drug Administration Department in 2005 for nasopharyngeal carcinoma [28]. One of the more successful trials of H101 was a study of 165 patients with unresectable hepatocellular carcinoma who were treated with TACE and with or without H101. TACE with H101 showed an overall survival benefit of 12.8 months (TACE with H101) versus 10.49 months (TACE without H101) (p = 0.046) [29]. H101, along with other adenovirus-based OVTs, is currently under investigation for several cancers including intratumoral injections for gynecologic malignancies, hepatocellular carcinoma, and cholangiocarcinoma (see Table 2).
| Study title | Identifier | Interventions | Condition | Phase | Status |
| Intra-tumor injection of oncolytic viruses H101 combined with or without radiotherapy in refractory/recurrent gynecological malignancies | NCT05051696 | H101 | Female genital neoplasms | Phase I | Recruiting |
| Intraperitoneal injection of oncolytic viruses h101 for patients with refractory malignant ascites | NCT04771676 | H101 | Refractory malignant ascites | Phase II | Recruiting |
| HAIC plus H101 vs HAIC alone for unresectable HCC at BCLC A-B | NCT03780049 | Hepatic artery infusion therapy (HIAC) with FOLFOX | Unresectable hepatocellular carcinoma | Phase III | Recruiting |
| H101 | |||||
| Oncolytic adenovirus DNX-2401 in treating patients with recurrent high-grade glioma | NCT03896568 | DNX-2401 | Recurrent anaplastic astrocytoma | Phase I | Recruiting |
| Therapeutic conventional surgery | Recurrent glioblastoma | ||||
| Recurrent gliosarcoma | |||||
| Recurrent malignant glioma | |||||
| Oncolytic adenovirus, DNX-2401, for naive diffuse intrinsic pontine gliomas | NCT03178032 | DNX-2401 | Brainstem glioma | Phase I | Active, not recruiting |
| Neoadjuvant therapy | |||||
| A randomised phase II open-label study with a phase Ib safety lead-in cohort of ONCOS-102, an immune-priming GM-CSF coding oncolytic adenovirus, and pemetrexed/cisplatin in patients with unresectable malignant pleural mesothelioma | NCT02879669 | ONCOS-102 | Unresectable malignant pleural mesothelioma | Phase I/II | Active, not recruiting |
| Pemetrexed | |||||
| Cisplatin/Carboplatin | |||||
| Cyclophosphamide | |||||
| A phase 1/2 study to investigate the safety, biologic and anti-tumor activity of ONCOS-102 in combination with durvalumab in subjects with advanced peritoneal malignancies | NCT02963831 | ONCOS-102 | Colorectal cancer | Phase I/II | Active, not recruiting |
| Durvalumab | Platinum-resistant ovarian cancer | ||||
| Appendiceal cancer |
Another adenovirus-based OVT is DNX-2401, which is currently being investigated in clinical trials against recurrent glioma [30]. DNX-2401 specifically deletes E1A, a gene responsible for its virulence, and inserts an RGD-4C motif which allows it to specifically infect tumor cells [31]. Further trials are underway investigating its efficacy against recurrent gliomas in combination with other therapies, including checkpoint inhibitors (see Table 2). In one active phase II trial, DNX-2401 is injected directly into a recurrent glioblastoma or gliosarcoma followed by pembrolizumab every 3 weeks for up to 2 years or until disease progression (NCT02798406).
One last adenovirus-based OVT currently being utilized alongside checkpoint inhibitors is ONCOS-102, which is like H101 and DNX-2401 but is also armed with GM-CSF [32]. The first indication of a positive synergistic effect of ONCOS-102 and checkpoint inhibitor therapy was in a melanoma huNOG mouse model [33]. This paved the way for a phase I trial in which patients with checkpoint inhibitor-refractory, unresectable melanoma were given ONCOS-102 in combination with pembrolizumab [34]. Objective responses were observed in 35% of patients. The FDA has now fast tracked this combination to allow for phase II/III studies for checkpoint inhibitor-refractory, unresectable melanoma. ONCOS-102 is also being utilized in combination with another checkpoint inhibitor, durvalumab, in a current Phase I/II study in patients with peritoneal disease with epithelial ovarian or metastatic colorectal cancer [35] (see Table 2).
Newcastle disease virus (NDV) is often thought of as an ideal candidate for OVT
as it does not cause serious illness in humans [36]. MEDI9253, an NDV-based OVT,
has emerged as a safe anti-cancer therapy currently being studied with checkpoint
inhibitor therapies for several metastatic solid tumors (see Table 3). Like
ONCOS-102, MEDI9253 carries a gene for GM-CSF, however, it also encodes an
IL-12 gene, which induces the production of IFN-
| Study title | Identifier | Interventions | Condition | Phase | Status |
| A study of MEDI9253 in combination with durvalumab in select solid tumors | NCT04613492 | MEDI9253 | Solid tumors | Phase I | Recruiting |
| Durvalumab | |||||
| A study of metronomic CP and JX-594 in patients with advanced breast cancer and advanced soft-tissue sarcoma (METROmaJX) | NCT02630368 | JX-594 and Cyclophosphamide | Solid tumors | Phase I/II | Recruiting |
| Cyclophosphamide | Soft tissue sarcoma | ||||
| Avelumab, JX-594, and cyclophosphamide | Breast cancer | ||||
| A study of recombinant vaccinia virus in combination with cemiplimab for renal cell carcinoma | NCT03294083 | Pexastimogene Devacirepvec (JX-594) | Metastatic or unresectable renal cell carcinoma | Phase I/II | Recruiting |
| Cemiplimab | |||||
| A phase I/II study of Pexa-vec oncolytic virus in combination with immune checkpoint inhibition in refractory colorectal cancer | NCT03206073 | Pexastimogene Devacirepvec (JX-594) | Refractory colorectal cancer | Phase I/II | Active, not recruiting |
| Tremelimumab | |||||
| Durvalumab | |||||
| Immunization strategy with intra-tumoral injections of Pexa-vec with ipilimumab in metastatic/advanced solid tumors | NCT02977156 | Pexastimogene Devacirepvec (JX-594) | Advanced and metastatic solid tumors | Phase I | Active, not recruiting |
| Ipilimumab |
There is an abundance of trials utilizing OVT, with most of the promising
studies utilizing a synergistic effect with checkpoint inhibitors against solid
tumors. One common barrier is the limitation of drug delivery and the challenges
of evading immune responses to OVT [39]. While there are trials that are trying
to utilize novel viral engineering to deliver OVT intravenously, almost all have
poor bioavailability and must be injected intratumorally or locoregionally. This
presents a challenge for metastatic cancers with a large tumor burden. One
strategy that is being explored involves coating OVTs with biocompatible polymers
to block antibodies from binding [40]. Another strategy is to pre-treat patients
with a complement-inhibiting peptide [40]. Moreover, due to the intricate
engineering process involved with OVT, there is a high cost associated with
production and utilization of OVT that may not be practical in real world
clinical settings. The estimated cost of T-VEC plus ipilimumab for advanced
unresectable melanoma, the most promising OVT thus far, was
Perhaps the most promising trend with OVT lies in the combination therapy with checkpoint inhibitors, specifically in “cold tumors” with low expressions of PD-L1 on the surface. OVTs promote immunologic responses including type I interferons which increase PD-L1 expression on cancer cells [42]. This would allow for tumors with previously low levels of PD-L1 expression which would have been poor targets for checkpoint inhibition to potentially be ideal targets for checkpoint inhibition.
JR — drafting of the manuscript; SH — drafting of the manuscript, creation of figure; AS — manuscript revision; All — final approval of the manuscript submitted.
Not applicable.
Not applicable.
This research received no external funding.
AS reports research grants (to institution) from AstraZeneca, Bristol Myers Squibb, Merck, Exelixis, Clovis, KAHR medical, Actuate therapeutics, Incyte corporation, Daiichi sankyo, and advisory board fees from AstraZeneca, Daiichi Sankyo, Bristol Myers Squibb, Pfizer and Exelixis. The remaining authors report no relevant COIs.